

Abstract
Hypotonic stress induces ATP release followed by Ca2+ oscillations in bovine aortic endothelial cells (BAECs). We have investigated the cellular mechanism of the hypotonic stress-induced ATP release.
Hypotonic stress induced tyrosine phosphorylation of at least two proteins, of 110 and 150 kDa. Inhibition of tyrosine kinase by the tyrosine kinase inhibitors herbimycin A and tyrphostin 46 prevented ATP release and ATP-mediated Ca2+ oscillations induced by hypotonic stress.
ATP release was also inhibited by the pretreatment of the cells with botulinum toxin C3, and augmented by lysophosphatidic acid. Furthermore, pre-treating the cells with Y-27632, a selective inhibitor of Rho-kinase, also suppressed the hypotonic stress-induced ATP release and Ca2+ oscillations, indicating that Rho-mediated activation of Rho-kinase may be involved in the hypotonic ATP release.
Hypotonic stress also induced a transient rearrangement of the actin cytoskeleton, which was suppressed by the tyrosine kinase inhibitors Y-27632 and cytochalasin B. However, pretreatment of the cell with cytochalasin B inhibited neither the hypotonic stress-induced ATP release nor the Ca2+ oscillations.
These results indicate that tyrosine kinase and the Rho-Rho-kinase pathways are involved in hypotonic stress-induced ATP release and actin rearrangement, but actin polymerization is not required for ATP release in BAECs.
Mechanical stress stimulates the production of biologically active mediators, gene expression and cellular alignment in vascular endothelial cells (for a review see Davies, 1995). In particular, the ‘immediate’ endothelial responses to mechanical stress (Takahashi et al. 1997), including alterations in cellular messengers, such as Ca2+ transient (Oike et al. 1994a) and the production of nitric oxide (NO) (Korenaga et al. 1994), have a significant importance for the real-time regulation of vascular tonus.
We have used hypotonic stress as an in vitro example of mechanical stress in this study. Though hypotonic stress does not correspond to a particular in vivo mechanical stress loaded onto the endothelium, hypotonic cell swelling would generate some stress on the cell membrane. Actually, reported hypotonic stress-induced endothelial responses share some common characteristics with shear stress-induced ones. For instance, both of these stresses induce ATP release in endothelium (Bodin et al. 1991; Oike et al. 2000). Transient reorganization of actin cytoskeleton induced by hypotonic stress (Oike et al. 1994b) is quite similar to that induced by shear stress (Knudsen & Frangos, 1997). Furthermore, shear stress has been reported in aortic endothelium to activate chloride current (Barakat et al. 1999), which is similar to hypotonic stress-activated volume-regulated anion channel (VRAC) current (Nilius et al. 1996). Therefore, we consider that investigation of hypotonic stress-induced responses would provide significant information about endothelial mechanosensitivity.
We have observed that hypotonic stress induced Ca2+ oscillations in bovine aortic endothelial cells (BAECs), which were inhibited by phospholipase C inhibitors (neomycin or U-73122), P2 antagonist (suramin) and ATP-hydrolysing agent (apyrase). We showed that ATP release of 91.5 amol cell−1 (10 min)−1 was induced by 40 % hypotonic stress (Oike et al. 2000). We have also reported that the hypotonic stress-induced, ATP release-mediated Ca2+ transient leads to NO production in BAECs (Kimura et al. 2000), thereby suggesting the potential role of this mechanism in the regulation of vascular tonus. However, the cellular mechanism by which ATP is released by hypotonic stress from endothelium has not been clarified yet. Tyrosine kinase has been reported to regulate hypotonic stress-induced activation of VRACs in bovine pulmonary endothelium by Voets et al. (1998). The same group also reported the involvement of Rho, a small G-protein, and Rho-activated protein kinase (Rho-kinase), a serine/threonine kinase, in the activation of VRACs (Nilius et al. 1999). Hypotonic stress has also been reported in epithelial cells to activate phosphatidylinositol 3-kinase (PI 3-kinase) (Tilly et al. 1996a) and mitogen-activated protein (MAP) kinase (Tilly et al. 1996b). Therefore, it would be of interest to examine the possible involvement of these intracellular signalling pathways in hypotonic stress-induced ATP release.
Furthermore, the mechanism by which hypotonic stress is sensed by endothelium has not been fully clarified yet. Oike et al. (1994b) have shown the possibility that hypotonic stress-induced generation of arachidonic acid is related to the rearrangement of actin cytoskeleton in human umbilical cord endothelial cells (HUVECs). The cytoskeleton, especially actin filaments, is involved in various cellular signalling in many cell types (for a review, see Janmey, 1998). Therefore, the possible role of the actin cytoskeleton as a stress sensor in hypotonic stress-induced ATP release should be considered as well.
In this study, we examined the signalling pathways that are involved in hypotonic stress-induced ATP release in BAECs. Furthermore, the possible contribution of the actin cytoskeleton to these signalling pathways was also investigated. The results show for the first time that hypotonic stress-induced ATP release is mediated by the tyrosine kinase and Rho-Rho-kinase pathways.
METHODS
Culture of bovine aortic endothelial cells (BAECs)
Bovine thoracic aortas were obtained from the local slaughter house. Endothelial cells were scraped off with a razor blade and cultured in Dulbecco's modified Eagle's medium with 10 % fetal bovine serum as previously described (Oike & Ito, 1997). The cells of the second subculture were used for the present experiments.
Measurement of intracellular Ca2+ concentration
[Ca2+]i was measured by using an Attofluor digital fluorescence microscopy system (Atto Instruments, Rockville, MD, USA). Cells grown on coverslips were loaded with fura-2 (fura-2 AM, Dojindo, Kumamoto, Japan) as previously described (Oike & Ito, 1997) and mounted on an inverted-microscope (Axiovert 135, Carl Zeiss, Jena, Germany). The fura-2 fluorescence images, containing images from 20-30 cells, were recorded into a rewritable optical disk recorder (LQ-4100A, Panasonic, Osaka, Japan) at a rate of approximately 1 Hz. Fluorescence images of each cell were converted to apparent Ca2+ concentration as previously described (Oike & Ito, 1997). All experiments were performed at room temperature (20-25 °C).
Luciferin-luciferase bioluminescence assay
The extracellular ATP concentration ([ATP]o) was measured by using luciferin-luciferase bioluminescence. Cells were seeded on 96-well plates, and cultured for 2 days before use. Each well contained 5000 cells on average before the experiment. After carefully removing the culture medium, 50 μl of isotonic or hypotonic Krebs solution containing 10 mg ml−1 luciferase-luciferin (Wako Co., Osaka, Japan) was added to each well. Then the plate was immediately put in a dark box and photons were counted for an appropriate period by a luminescence detection system (Argus-50/2D luminometer, Hamamatsu Photonics, Hamamatsu, Japan). Data were analysed with Argus-50 software (Hamamatsu Photonics). To convert the photon counting into [ATP]o, photon-[ATP] relationships were obtained by measuring the chemiluminescence of homogeneous ATP solutions containing 10 mg ml−1 luciferase-luciferin. It should be noted, however, that the local ATP concentration just above the ATP-releasing cells may be much higher than that of the solution surface, and therefore the calculated [ATP]o values might be lower than the actual local ATP concentration above the cell.
ECL Western blot analysis
Cellular tyrosine kinase activity was assessed by enhanced chemiluminescence (ECL) Western blotting by using a commercial kit (ECL phosphorylation detection system, Amersham Pharmacia Biotech, Uppsala, Sweden). Cells were exposed to hypotonic stress for 1, 2, 5 and 10 min, and cell lysates were prepared. Western blot analysis for phosphotyrosine was performed by using the anti-phosphotyrosine antibody clone PY20 (Glenney et al. 1988). Emitted chemiluminescence was then detected and analysed by a lumino image analyser (FAS-1000, Toyobo, Osaka, Japan).
Immunological staining of endothelial F-actin
Rearrangement of F-actin by hypotonic stress was examined immunologically by using rhodamine-conjugated phalloidin (Molecular Probes, Eugene, OR, USA) according to the previously reported method (Knudsen & Frangos, 1997).
Solutions and drugs
The standard extracellular solution was a modified Krebs solution (1.5 mm Ca2+ solution) containing (mm): 132 NaCl, 5.9 KCl, 1.2 MgCl2, 1.5 CaCl2, 11.5 glucose, 11.5 Hepes; pH adjusted to 7.3 with NaOH. Hypotonic solutions were made by adding appropriate volumes of distilled water to normal Krebs solution. We confirmed in supplementary experiments that the reduction of the ionic concentrations in these hypotonic solutions did not influence the [Ca2+]i responses (Oike et al. 2000).
Tryphostin 46 was obtained from Biomol Research Laboratory Inc. (Plymouth Meeting, PA, USA), suramin from Bayer (Leverkusen, Germany) and herbimycin A from Kyowa (Tokyo, Japan). Y-27632 was kindly provided from Yoshitomi Pharmaceutical Industries, Ltd (Osaka, Japan). All other drugs were purchased from Sigma.
Data analysis
Pooled data are given as means ± standard error of the mean, and statistical significance was determined using Student's unpaired t test. Probabilities less than 5 % (P < 0.05) were regarded as significant.
RESULTS
Hypotonic stress-induced Ca2+ oscillations and ATP release in BAECs
Firstly we confirmed our previous findings that hypotonic stress induces ATP release followed by Ca2+ oscillations (Oike et al. 2000). Perfusion and solution exchange of isotonic solution did not induce any changes in [Ca2+]i at least up to 15 min in BAECs (n = 5, Fig. 1A). On the other hand, 40 % hypotonic solution elicited Ca2+ oscillations (Fig. 1b), which were inhibited by inhibitors of phospholipase C and the P2 receptor and ATP-hydrolysing agents (Oike et al. 2000).
Figure 1. Effects of hypotonic stress on the intracellular Ca2+ concentration and extracellular ATP concentration ([ATP]o) in bovine aortic endothelial cells (BAECs).

A, perfusion of isotonic Krebs solution and the solution exchange procedure did not induce any Ca2+ responses at least up to 15 min. Solution was changed to a new perfusate at the arrows. B, hypotonic stress (40 %) induced Ca2+ oscillations in BAECs. C, time-dependent change in [ATP]o. a, the luciferin bioluminescence images of released ATP. b, luciferin bioluminescence was measured for the indicated period. Columns and symbols show the rate and accumulated change in [ATP]o, respectively. Open columns and circles are values from isotonic solution, and hatched columns and filled circles are from hypotonic solution. **P < 0.01 vs. isotonic solution. D, [ATP]o measured for 10 min after starting application of isotonic and hypotonic solutions, and the effects of suramin (100 μm), reactive blue 2 (100 μm), apyrase (2 units ml−1) and hexokinase (5 units ml−1) on hypotonic stress-induced [ATP]o elevation. **P < 0.01 compared with hypotonic stress alone; ††P < 0.01.
We detected the released ATP by using luciferin- luciferase (Fig. 1C and D). ATP release was observed from shortly after starting the hypotonic challenge and persisted for at least 20 min (Fig. 1Cb). ATP release was also observed in isotonic Krebs solution, but the amount was significantly smaller than in hypotonic solution (Fig. 1C). [ATP]o was elevated to 9.27 ± 0.50 (n = 26) and 2.67 ± 0.28 nm (n = 20) at 10 min in response to 40 % hypotonic and isotonic solutions, respectively (P < 0.01, Fig. 1D). As summarized in Fig. 1D, hypotonic stress-induced [ATP]o elevation was augmented by inhibitors of ectoATPase (suramin and reactive blue 2) due to the inhibition of ATP degradation, and reduced by ATP-hydrolysing agents (apyrase and hexokinase).
We have previously confirmed that hypotonic stress (40 %) did not induce significant cell disruption or microlysis in BAECs, measured by lactate dehydrogenase (LDH) leakage or ethidium bromide uptake (Oike et al. 2000), thereby suggesting that hypotonic stress-induced ATP release was not due to non-specific leakage of ATP.
Effects of modulators of intracellular signalling pathways on hypotonic stress-induced responses in BAECs
To investigate the cellular mechanism of these hypotonic stress-induced responses, we examined the effects of inhibitors of some intracellular signalling pathways on the Ca2+ transients and ATP release. The cellular Ca2+ response to hypotonic stress was assessed by the percentage of cells in a single microscopic field (containing 20-30 cells) that show a Ca2+ transient.
Western blotting showed that hypotonic stress induced tyrosine phosphorylation of at least two proteins, of 110 and 150 kDa, and the former had higher phosphotyrosine activity (Fig. 2A). The densitometric analysis of these phosphotyrosine bands revealed that the maximal activation of tyrosine kinase was obtained at 2 min (Fig. 2b), which was similar to the time course of ATP release (Fig. 1C). Therefore, we firstly examined the effects of tyrosine kinase inhibitors on hypotonic stress-induced Ca2+ oscillations. Tyrosine kinase was inhibited either by pretreatment of the cell with 1 μm herbimycin A for 12 h at 37 °C or by the continuous perfusion of 100 μm tyrphostin 46 throughout the experiment. These agents significantly inhibited hypotonic stress-induced Ca2+ transients (Fig. 3A and b). This was not due to a reduction of the cellular responsiveness to ATP, because exogenously applied ATP (300 nm) evoked Ca2+ oscillations in herbimycin A- (Fig. 3C and D) and tyrphostin 46-treated cells (Fig. 3D). Herbimycin A and tyrphostin 46 did not affect the basal level of [ATP]o (Fig. 3F), but significantly suppressed hypotonic stress (40 %)-induced [ATP]o elevation (herbimycin A: 5.22 ± 0.17 nm, n = 12; tyrphostin 46: 4.92 ± 0.42 nm, n = 16; both P < 0.01 vs. control, Fig. 3F) without changing the time course of ATP release (Fig. 3E). Total cellular ATP content was assessed by measuring [ATP]o for 15 min after perforating the cell membrane with 1 % Triton X-100. The value was not different between control and herbimycin A-treated cells (control, 1.13 ± 1.1 mm, n = 7; herbimycin A, 1.21 ± 0.11 mm, n = 7, P > 0.05), thereby suggesting that the inhibitory effects of tyrosine kinase inhibitors on ATP release were not due to the reduction of cellular ATP content. These results suggest that tyrosine kinase is involved in the hypotonic stress-induced ATP release.
Figure 2. Hypotonic stress induced tyrosine phosphorylation of cellular proteins in BAECs.

A, of several bands observed in Coomassie Brilliant Blue stain (CBB), ECL Western blotting with anti-phosphotyrosine antibody clone PY20 revealed that 110 and 150 kDa proteins were tyrosine phosphorylated by hypotonic stress (Western blotting). Each lane was loaded with 200 μg protein. The results shown are representative of four experiments. B, time course of tyrosine kinase activity was quantitatively analysed by densitometry. Summation of the band densities of 110 and 150 kDa bands was expressed as a fold increase compared to its basal level in isotonic solution. Values were taken from four experiments.
Figure 3. Possible involvement of tyrosine kinase in endothelial responses to hypotonic stress.

A, herbimycin A (1 μm) abolished hypotonic stress (40 %)-induced Ca2+ oscillations. B, percentage of cells showing Ca2+ oscillations in a microscopic field. Both herbimycin A and tyrphostin 46 (100 μm) suppressed hypotonic stress-induced Ca2+ oscillations significantly. **P < 0.01. C, exogenous application of 300 nm ATP induced Ca2+ oscillations in herbimycin A-treated cells. D, herbimycin A and tyrphostin 46 did not alter cellular responsiveness to exogenously applied ATP (300 nm). n.s., P > 0.05 vs. control. E, effects of herbimycin A (▪) and tyrphostin 46 (▴) on hypotonic stress (40 %)-induced [ATP]o elevation. Time course of [ATP]o elevation is shown. **P < 0.01 vs. control (•). F, effects of tyrosine kinase inhibitors on [ATP]o measured for 10 min after starting hypotonic challenge. **P < 0.01 vs. hypotonic stress alone; n.s., P > 0.05 vs. isotonic solution; ††P < 0.01.
Tilly et al. reported that hypotonic stress activates PI 3-kinase (Tilly et al. 1996a) and Hsp27 phosphorylation (Tilly et al. 1996b) in epithelial cells. Thus, we then examined the effects of 10 μm wortmannin, an inhibitor of PI 3-kinase and MAP kinase, on hypotonic stress-induced Ca2+ transients. However, cells treated with wortmannin showed normal hypotonic stress-induced Ca2+ transients (Fig. 4A and C). Furthermore, hypotonic stress-induced Ca2+ oscillations were not affected significantly in cells incubated at 42 °C for 2 h to induce heat shock protein (Fig. 4B and C). These suggest that PI 3-kinase, MAP kinase and Hsp are not involved in hypotonic stress-induced Ca2+ transients.
Figure 4.

A, pretreatment with 10 μm wortmannin for 15 min did not inhibit hypotonic stress-induced Ca2+ oscillations. B, pre-incubation of cells at 42 °C for 2 h also did not inhibit hypotonic stress-induced Ca2+ oscillations. C, percentage of the cells showing Ca2+ oscillations is shown. n.s., P > 0.05 vs. untreated cells.
Possible involvement of Rho cascade in hypotonic stress-induced endothelial responses
We then examined the possible involvement of the Rho-Rho-kinase pathway, which plays a role in the activation of VRACs in pulmonary endothelium (Nilius et al. 1999), in hypotonic stress-induced ATP release.
Hypotonic stress (40 %)-induced [ATP]o elevation in cells pre-incubated for 24 h with 5 μg ml−1 botulinum C3 toxin, which is known to inhibit Rho selectively (Mackay & Hall, 1998), was significantly reduced (5.59 ± 0.60 nm, n = 8, P < 0.01 vs. untreated cells, Fig. 5Aa). In contrast, C3 did not alter the basal level of ATP release in isotonic solution (Fig. 5Aa). Hypotonic stress-induced [ATP]o elevation was augmented by ectoATPase inhibitors in C3-treated cells (suramin, 5.93 ± 0.19 nm, n = 6; reactive blue 2, 10.53 ± 0.72 nm, n = 8), but was still lower than in untreated cells (P < 0.01, see Fig. 1D for comparison). The effect of C3 was not due to the reduction of cellular ATP content, since [ATP]o elevation after cell lysis was not different between control and C3-treated cells (1.13 ± 1.1 and 1.23 ± 0.8 mm in control and C3-treated cells, both n = 7, P > 0.05, Fig. 5Ab). Furthermore, C3 did not induce significant cell death, assessed by LDH leakage, during 24 h of incubation (not shown). Therefore, we suppose that the inhibitory effect of C3 on hypotonic stress-induced ATP release was not due to non-specific cellular damage or ATP degradation.
Figure 5. Effects of botulinum C3 toxin and lysophosphatidic acid (LPA) on hypotonic stress-induced ATP release.

A, cells were pre-incubated with 5 μg ml−1 C3-containing culture medium for 24 h. a, [ATP]o was measured for 10 min in isotonic and 40 % hypotonic solutions. b, cells were lysed with 1 % Triton X-100, and [ATP]o was measured for 15 min as an indicator of total cellular ATP content. B, [ATP]o was measured for 10 min in the presence and absence of 1 μm LPA in isotonic and hypotonic solutions. *P < 0.05; **P < 0.01; n.s., P > 0.05.
In contrast, lysophosphatidic acid (LPA, 1 μm), which is known to activate Rho (Moolenaar, 1995), significantly increased basal [ATP]o (control 2.67 ± 0.28 nm, n = 20; LPA 3.91 ± 0.26 nm, n = 5, P < 0.05, Fig. 5b) and augmented 20 % hypotonic stress-induced [ATP]o elevation (control 5.60 ± 0.71 nm, n = 16; LPA 8.52 ± 1.31 nm, n = 8, P < 0.05, Fig. 5b), though it did not affect 40 % hypotonic stress-induced [ATP]o elevation (Fig. 5b). The augmentative action of LPA on 20 % hypotonic stress-induced [ATP]o elevation was reversed to the control level by washing out LPA for 20 min (5.70 ± 1.01 nm, n = 5, P > 0.05 vs. untreated control). These results suggest the involvement of Rho in the hypotonic stress-induced ATP release.
Next, we examined the effects of Y-27632, a selective inhibitor of Rho-kinase (Narumiya et al. 1997), on hypotonic stress-induced ATP release. Hypotonic stress (40 %) did not induce Ca2+ oscillations (Fig. 6A and b), but externally applied ATP induced Ca2+ oscillations (Fig. 6C) in Y-27632-treated cells. Furthermore, Y-27632 inhibited hypotonic stress (40 %)-induced [ATP]o elevation (Fig. 6D, 2.41 ± 0.40 nm, n = 10, P < 0.01 vs. control) without affecting the basal [ATP]o in isotonic solution (Fig. 6D) or total cellular ATP content (Fig. 6E).
Figure 6. Effects of Y-27632 on hypotonic stress-induced responses in BAECs.
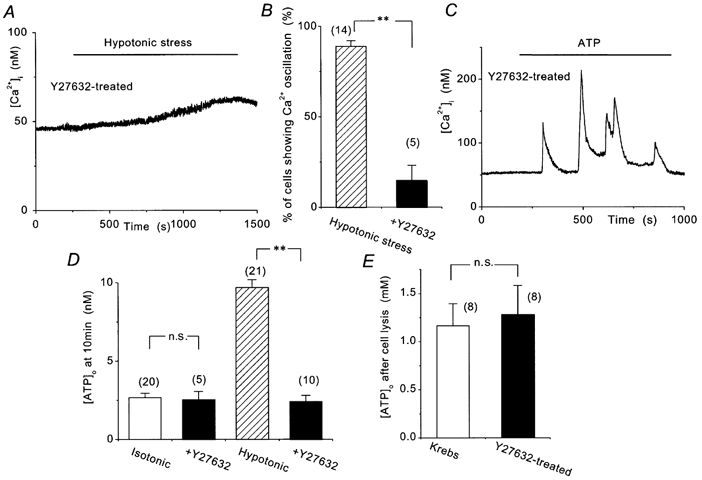
A, hypotonic stress failed to induce Ca2+ oscillations in cells pretreated for 20 min with 10 μm Y-27632. B, percentage of cells showing hypotonic stress-induced Ca2+ oscillations. **P < 0.01. C, exogenously applied ATP (300 nm) induced a normal Ca2+ response in the Y-27632-treated cell. D, [ATP]o measured for 10 min in isotonic and hypotonic (40 %) solution in control and Y-27632-treated cells. **P < 0.01; n.s., P > 0.05. E, [ATP]o was measured for 15 min after cell lysis.
These observations suggest that activation of the Rho- Rho-kinase pathway is involved in the hypotonic stress-induced ATP release.
Effects of cytochalasin B on hypotonic stress-induced responses in BAECs
It is well known that Rho-kinase activity is closely related to the rearrangement of actin filaments (Narumiya et al. 1997). So we then examined the effect of a disruption of the actin filaments on hypotonic stress-induced ATP release. As shown in Fig. 7A, hypotonic stress-induced Ca2+ oscillations were preserved when actin polymerization was inhibited by the pretreatment of the cells with 30 μm cytochalasin B for 20 min. Furthermore, cells pretreated with cytochalasin B also showed normal [ATP]o elevation in both isotonic and 40 % hypotonic solutions (Fig. 7b).
Figure 7. Effects of cytochalasin B on hypotonic stress-induced Ca2+ oscillations and ATP release.
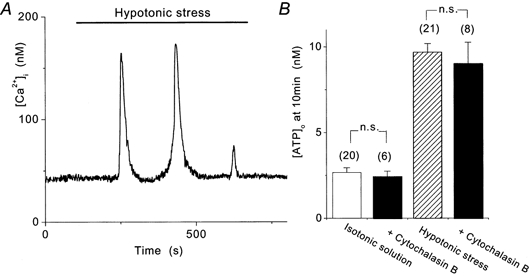
A, hypotonic stress induced Ca2+ oscillations in 30 μm cytochalasin B-treated cells. B, hypotonic stress-induced [ATP]o elevation as well as basal level of [ATP]o in isotonic solution was not different from control. n.s., P > 0.05.
These results suggest that actin polymerization is not involved in the cellular mechanism of hypotonic stress-induced ATP release.
Effects of herbimycin A, Y-27632 and cytochalasin B on hypotonic stress-induced rearrangement of F-actin
To provide further evidence for the lack of correlation between actin rearrangement and ATP release, we finally examined the effect of hypotonic stress on the actin cytoskeleton in BAECs.
As shown in Fig. 8A, 40 % hypotonic stress induced a transient rearrangement of actin filaments in control cells. A small amount of F-actin was present in isotonic solution (Fig. 8A, upper picture). Two minutes after the application of hypotonic stress, actin stress fibres were formed (middle picture), which were redistributed in 15 min into the focal adhesion complex (lower picture). These changes were not observed in herbimycin A (1 μm)-treated cells (Fig. 8b) and Y-27632 (10 μm)-treated cells (Fig. 8C), thereby suggesting that activation of tyrosine kinase and Rho-kinase is involved both in hypotonic stress-induced ATP release and in the rearrangement of actin filaments.
Figure 8. Transient rearrangement of F-actin cytoskeleton by hypotonic stress.

Cells were stimulated with 40 % hypotonic stress for the indicated time and stained with rhodamine-conjugated phalloidin. Control cells show some stress fibres before stimulation (0 min) and marked actin bundle formation shortly after stimulation (2 min), which was rearranged into focal adhesion complex in 15 min (A). Cells treated with 1 μm herbimycin A (b), 10 μm Y-27632 (C) or 30 μm cytochalasin B (D) lack such a rearrangement of F-actin.
In contrast, formation of actin stress fibres induced by hypotonic solution was completely abolished in a cell pretreated for 20 min with 30 μm cytochalasin B (Fig. 7D). This result is in contrast with the effect of cytochalasin B on ATP release, and supports the idea that the rearrangement of actin filaments is not related to ATP release.
DISCUSSION
Shear stress-stimulated ATP release has been reported repeatedly in endothelium (Bodin et al. 1991; Hassessian et al. 1993). We showed that hypotonic stress also induced ATP release in BAECs, which then evoked Ca2+ oscillations (Fig. 1) (Oike et al. 2000) and NO production (Kimura et al. 2000). Though endogenous ectoATPase supposedly degraded the released ATP (Fig. 1D), the released ATP gradually accumulated in the extracellular space at least up to 30 min (Fig. 1Cb). This might be partly because ectoATPase activity was suppressed in our experimental condition of room temperature, but the rate of ATP degradation by endothelium has been reported to be gradual (Slakey et al. 1990; Yagi et al. 1991). For instance, Yagi et al. (1991) reported that more than half of the ATP which was applied to BAECs exogenously remained after 30 min at 37 °C. Therefore, in spite of the endogenous ectoATPase activity of endothelium, we consider that the hypotonic stress induces the significant accumulation of ATP in the extracellular space. Though no Ca2+ transient was observed in isotonic solution (Fig. 1A), a significant amount of ATP release was also observed in isotonic solution (Fig. 1C and D). This may be an inevitable artifact due to the experimental procedure (Grygorczyk & Hanrahan, 1997) or physiological basal ATP release. Even if some of the basal ATP release were physiological, its mechanism would probably be different from the hypotonic stress-induced one, because it was not inhibited significantly by any treatments in the present experiment (Figs 3F, 5Aa, 6D and 7b). Furthermore, since ATP induces Ca2+ transients in an all-or-none manner above threshold in BAECs (Kimura et al. 1998), the basal ATP release would not induce any Ca2+ responses if it were lower than the threshold concentration.
This study provides the first evidence that the Rho-Rho-kinase pathway is involved in the hypotonic stress-induced ATP release in vascular endothelium (Fig. 5 and 6). The Rho inhibitor C3 toxin inhibited hypotonic ATP release (Fig. 5Aa), but this was not due to non-specific cell damage since intracellular ATP content (Fig. 5Ab) and cell viability were not altered in C3-treated cells. Furthermore, LPA, which is a multifunctional phospholipid and induces receptor-mediated activation of intrinsic Rho (Moolenaar, 1995), significantly augmented both the basal level of ATP release and 20 % hypotonic stress-induced ATP release (Fig. 5b). LPA, however, did not augment 40 % hypotonic stress-induced [ATP]o elevation, probably because maximal ATP release or Rho activation was obtained already by 40 % hypotonic stress. Therefore, this evidence as a whole strongly suggests that Rho is involved in hypotonic ATP release. Several molecules, such as p160ROCK and p140mDia, are known to be downstream targets for Rho (for a review, see Narumiya et al. 1997). We observed that hypotonic ATP release was also suppressed by Y-27632, an inhibitor of Rho-kinase (Fig. 6D). However, since Y-27632 does not discriminate between p160ROCK/ROCK-I and Rho-kinase/ROCKII (Narumiya et al. 1997), we could not identify the isoform of Rho-kinase that is involved in ATP release. Involvement of Rho-kinase has been reported recently in the activation of VRACs (Nilius et al. 1999). Therefore, though the Rho-Rho-kinase pathway was originally described as a regulator of the formation of actin stress fibre (Narumiya et al. 1997), it may play a critical role in the hypotonic stress-induced responses in vascular endothelium.
We showed that F-actin is transiently rearranged in response to hypotonic stress in BAECs (Fig. 8A). Tilly et al. also reported similar results in epithelial cells (Tilly et al. 1996a). The involvement of Rho in the formation of actin stress fibres is now generally accepted (Hall, 1998). Based on their observations in HeLa cells, Narumiya et al. (1997) have suggested that the actin stress fibre is formed by the coordination of two distinct Rho-mediated pathways, i.e. an unidentified Rho effector-mediated actin polymerization and a Rho-kinase-mediated actin bundling. We confirmed the involvement of both pathways in hypotonic stress-induced formation of actin stress fibres in BAECs, since both cytochalasin B and Y-27632 inhibited it (Fig. 8C and D). However, it was also clearly shown in this study that the rearrangement of the actin cytoskeleton is not related to the hypotonic stress-induced ATP release, because the disruption of action cytoskeleton with cytochalasin B did not affect ATP release and Ca2+ oscillations (Fig. 7). Furthermore, the hypotonic stress-induced rearrangement of actin filaments was not suppressed by inhibiting ATP-induced Ca2+ transients with the P2 antagonist suramin (data not shown). Therefore, though the formation of actin stress fibres and ATP release share at least the Rho-Rho-kinase pathway, they are basically independent phenomena in BAECs.
We observed that hypotonic stress induced tyrosine phosphorylation of at least two proteins (Fig. 2A). The time course of tyrosine phosphorylation (Fig. 2b) was similar to that of ATP release (Fig. 1C), and tyrosine kinase inhibitors suppressed the hypotonic stress-induced ATP release (Fig. 3E and F) and the formation of actin stress fibres (Fig. 8). Therefore, we suppose that tyrosine kinase is also involved in hypotonic stress-induced responses in BAECs. Ishida et al. (1996) reported in HUVECs that laminar flow increased the phosphotyrosine activity of 60-80, 110, 125-150 and 180-190 kDa proteins, and they identified ‘125-150 kDa protein’ as focal adhesion kinase (FAK). However, tyrosine phosphorylation of FAK is probably not involved in hypotonic stress-induced responses in BAECs, since we could not observe the tyrosine phosphorylation of 125 kDa protein (Fig. 2A). Though the ‘110 kDa protein’ was tyrosine phosphorylated both by hypotonic stress (present study) and fluid shear stress (Ishida et al. 1996), we could not identify this molecule in this study. Furthermore, the relationship between tyrosine kinase and the Rho-Rho-kinase pathway is not clear from the present results. We also could not reveal whether ATP release and actin reorganization require the same tyrosine kinase activity or not. Further investigations are needed to elucidate the precise role and identification of the tyrosine kinase(s) that is (are) involved in ATP release and actin rearrangement.
The pathway through which ATP is released from endothelium by hypotonic stress and shear stress has not been clarified yet. The evidence shown in this study would give an important clue for the investigation of the ATP-releasing pathway in endothelium. It has been reported that the activation of tyrosine kinase (Voets et al. 1998) and Rho-Rho-kinase (Nilius et al. 1999) are also required for the activation of VRACs, thereby suggesting the possibility of a close relationship between ATP release and VRAC activation. However, there is a report arguing the absence of a correlation between them in an epithelial cell line (Hazama et al. 1999). Whether the ATP-releasing pathway is related to VRACs in BAECs will be the subject of future investigations.
Hypotonic stress was used as an example of in vitro mechanical stress in this study. However, it should be noted that hypotonic stress is not a physiological mechanical stress in endothelium, and therefore further studies are needed to clarify whether these cellular mechanisms are also involved in shear stress-induced ATP release or not. In conclusion, we showed that hypotonic stress-induced ATP release is mediated by tyrosine kinases and the Rho-Rho-kinase cascade, and the rearrangement of actin cytoskeleton is not related to ATP release in BAECs.
Acknowledgments
The authors thank Drs B. Nilius and G. Droogmans for critical reading of the manuscript. This study was carried out as part of ‘Ground Research Announcement for Space Utilization’ promoted by National Space Development Agency of Japan and Japan Space Forum, and a part of ‘Fund for Basic Experiments Oriented to Space Station Utilization’ promoted by the Institute of Space and Astronautical Science of the Ministry of Education of Japan. This study was also supported in part by a grant-in-aid from the Japan Society for the promotion of Science.
References
- Barakat AI, Leaver EV, Pappone PA, Davies PF. A flow-activated chloride-selective membrane current in vascular endothelial cells. Circulation Research. 1999;85:820–828. doi: 10.1161/01.res.85.9.820. [DOI] [PubMed] [Google Scholar]
- Bodin P, Bailey D, Burnstock G. Increased flow-induced ATP release from isolated vascular endothelial cells but not smooth muscle cells. British Journal of Pharmacology. 1991;103:1203–1205. doi: 10.1111/j.1476-5381.1991.tb12324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies PF. Flow-mediated endothelial mechanotransduction. Physiological Reviews. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenney JR, Jr Zokas L, Kamps MP. Monoclonal antibodies to phosphotyrosine. Journal of Immunological Methods. 1988;109:277–285. doi: 10.1016/0022-1759(88)90253-0. [DOI] [PubMed] [Google Scholar]
- Grygorczyk R, Hanrahan JW. CFTR-independent ATP release from epithelial cells triggered by mechanical stimuli. American Journal of Physiology. 1997;272:C1058–1066. doi: 10.1152/ajpcell.1997.272.3.C1058. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hassessian H, Bodin P, Burnstock G. Blockade by glibenclamide of the flow-evoked endothelial release of ATP that contributes to vasodilatation in the pulmonary vascular bed of the rat. British Journal of Pharmacology. 1993;109:466–472. doi: 10.1111/j.1476-5381.1993.tb13592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazama A, Shimizu T, Ando-Akatsuka Y, Hayashi S, Tanaka S, Maeno E, Okada Y. Swelling-induced, CFTR-independent ATP release from a human epithelial cell line: lack of correlation with volume-sensitive Cl− channels. Journal of General Physiology. 1999;114:525–533. doi: 10.1085/jgp.114.4.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T, Peterson TE, Kovach NL, Berk BC. MAP kinase activation by flow in endothelial cells. Role of β1 integrins and tyrosine kinases. Circulation Research. 1996;79:310–316. doi: 10.1161/01.res.79.2.310. [DOI] [PubMed] [Google Scholar]
- Janmey PA. The cytoskeleton and cell signaling: component localization and mechanical coupling. Physiological Reviews. 1998;78:763–781. doi: 10.1152/physrev.1998.78.3.763. [DOI] [PubMed] [Google Scholar]
- Kimura C, Koyama T, Oike M, Ito Y. Hypotonic stress-induced NO production in endothelium depends on endogenous ATP. Biochemical and Biophysical Research Communications. 2000;274:736–740. doi: 10.1006/bbrc.2000.3205. [DOI] [PubMed] [Google Scholar]
- Kimura C, Oike M, Ito Y. Acute glucose overload abolishes Ca2+ oscillation in cultured endothelial cells from bovine aorta: a possible role of superoxide anion. Circulation Research. 1998;82:677–685. doi: 10.1161/01.res.82.6.677. [DOI] [PubMed] [Google Scholar]
- Knudsen HL, Frangos JA. Role of cytoskeleton in shear stress-induced endothelial nitric oxide production. American Journal of Physiology. 1997;273:H347–355. doi: 10.1152/ajpheart.1997.273.1.H347. [DOI] [PubMed] [Google Scholar]
- Korenaga R, Ando J, Tsuboi H, Yang W, Sakuma I, Toyo-oka T, Kamiya A. Laminar flow stimulates ATP- and shear stress-dependent nitric oxide production in cultured bovine endothelial cells. Biochemical and Biophysical Research Communications. 1994;198:213–219. doi: 10.1006/bbrc.1994.1030. [DOI] [PubMed] [Google Scholar]
- Mackay DJ, Hall A. Rho GTPases. Journal of Biological Chemistry. 1998;273:20685–20688. doi: 10.1074/jbc.273.33.20685. [DOI] [PubMed] [Google Scholar]
- Moolenaar WH. Lysophosphatidic acid, a multifunctional phospholipid messenger. Journal of Biological Chemistry. 1995;270:12949–12952. doi: 10.1074/jbc.270.22.12949. [DOI] [PubMed] [Google Scholar]
- Narumiya S, Ishizaki T, Watanabe N. Rho effectors and reorganization of actin cytoskeleton. FEBS Letters. 1997;410:68–72. doi: 10.1016/s0014-5793(97)00317-7. [DOI] [PubMed] [Google Scholar]
- Nilius B, Eggermont J, Voets T, Droogmans G. Volume-activated Cl− channels. General Pharmacology. 1996;27:1131–1140. doi: 10.1016/s0306-3623(96)00061-4. [DOI] [PubMed] [Google Scholar]
- Nilius B, Voets T, Prenen J, Barth H, Aktories K, Kaibuchi K, Droogmans G, Eggermont J. Role of Rho and Rho kinase in the activation of volume-regulated anion channels in bovine endothelial cells. Journal of Physiology. 1999;516:67–74. doi: 10.1111/j.1469-7793.1999.067aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oike M, Droogmans G, Nilius B. Mechanosensitive Ca2+ transients in endothelial cells from human umbilical vein. Proceedings of the National Academy of Sciences of the USA. 1994a;91:2940–2944. doi: 10.1073/pnas.91.8.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oike M, Ito Y. Dynamic regulation of intracellular Ca2+ concentration in aortic endothelial cells. European Journal of Pharmacology. 1997;319:291–298. doi: 10.1016/s0014-2999(96)00846-1. [DOI] [PubMed] [Google Scholar]
- Oike M, Kimura C, Koyama T, Yoshikawa M, Ito Y. Hypotonic stress-induced dual Ca2+ responses in bovine aortic endothelial cells. American Journal of Physiology. 2000;279:H630–638. doi: 10.1152/ajpheart.2000.279.2.H630. [DOI] [PubMed] [Google Scholar]
- Oike M, Schwarz G, Sehrer J, Jost M, Gerke V, Weber K, Droogmans G, Nilius B. Cytoskeletal modulation of the response to mechanical stimulation in human vascular endothelial cells. Pflügers Archiv. 1994b;428:569–576. doi: 10.1007/BF00374579. [DOI] [PubMed] [Google Scholar]
- Slakey LL, Gordon EL, Pearson JD. A comparison of ectonucleotidase activities on vascular endothelial and smooth muscle cells. Annals of the New York Academy of Sciences. 1990;603:366–378. doi: 10.1111/j.1749-6632.1990.tb37686.x. discussion 378–379. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Ishida T, Traub O, Corson MA, Berk BC. Mechanotransduction in endothelial cells: temporal signaling events in response to shear stress. Journal of Vascular Research. 1997;34:212–219. doi: 10.1159/000159225. [DOI] [PubMed] [Google Scholar]
- Tilly BC, Edixhoven MJ, Tertoolen LG, Morii N, Saitoh Y, Narumiya S, de Jonge HR. Activation of the osmo-sensitive chloride conductance involves P21rho and is accompanied by a transient reorganization of the F-actin cytoskeleton. Molecular Biology of the Cell. 1996a;7:1419–1427. doi: 10.1091/mbc.7.9.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilly BC, Gaestel M, Engel K, Edixhoven MJ, de Jonge HR. Hypo-osmotic cell swelling activates the p38 MAP kinase signalling cascade. FEBS Letters. 1996b;395:133–136. doi: 10.1016/0014-5793(96)01028-9. [DOI] [PubMed] [Google Scholar]
- Voets T, Manolopoulos V, Eggermont J, Ellory C, Droogmans G, Nilius B. Regulation of a swelling-activated chloride current in bovine endothelium by protein tyrosine phosphorylation and G proteins. Journal of Physiology. 1998;506:341–352. doi: 10.1111/j.1469-7793.1998.341bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi K, Shinbo M, Hashizume M, Shimba LS, Kurimura S, Miura Y. ATP diphosphohydrolase is responsible for ecto-ATPase and ecto-ADPase activities in bovine aorta endothelial and smooth muscle cells. Biochemical and Biophysical Research Communications. 1991;180:1200–1206. doi: 10.1016/s0006-291x(05)81323-3. [DOI] [PubMed] [Google Scholar]