

Abstract
This study assessed the contribution of L-type Ca2+ channels and other Ca2+ entry pathways to Ca2+ store refilling in choroidal arteriolar smooth muscle.
Voltage-clamp recordings were made from enzymatically isolated choroidal microvascular smooth muscle cells and from cells within vessel fragments (containing < 10 cells) using the whole-cell perforated patch-clamp technique. Cell Ca2+ was estimated by fura-2 microfluorimetry.
After Ca2+ store depletion with caffeine (10 mm), refilling was slower in cells held at -20 mV compared to -80 mV (refilling half-time was 38 ± 10 and 20 ± 6 s, respectively).
To attempt faster refilling via L-type Ca2+ channels, depolarising steps from -60 to -20 mV were applied during a 30 s refilling period following caffeine depletion. Each step activated L-type Ca2+ currents and [Ca2+]i transients, but failed to accelerate refilling.
At -80 mV and in 20 mm TEA, prolonged caffeine exposure produced a transient Ca2+-activated Cl− current (ICl(Ca)) followed by a smaller sustained current. The sustained current was resistant to anthracene-9-carboxylic acid (1 mm; an ICl(Ca) blocker) and to BAPTA AM, but was abolished by 1 μm nifedipine. This nifedipine-sensitive current reversed at +29 ± 2 mV, which shifted to +7 ± 5 mV in Ca2+-free solution. Cyclopiazonic acid (20 μm; an inhibitor of sarcoplasmic reticulum Ca2+-ATPase) also activated the nifedipine-sensitive sustained current.
At -80 mV, a 5 s caffeine exposure emptied Ca2+ stores and elicited a transient ICl(Ca). After 80 s refilling, another caffeine challenge produced a similar inward current. Nifedipine (1 μm) during refilling reduced the caffeine-activated ICl(Ca) by 38 ± 5 %. The effect was concentration dependent (1-3000 nm, EC50 64 nm). In Ca2+-free solution, store refilling was similarly depressed (by 46 ± 6 %).
Endothelin-1 (10 nm) applied at -80 mV increased [Ca2+]i, which subsided to a sustained 198 ± 28 nm above basal. Cell Ca2+ was then lowered by 1 μm nifedipine (to 135 ± 22 nm), which reversed on washout.
These results show that L-type Ca2+ channels fail to contribute to Ca2+ store refilling in choroidal arteriolar smooth muscle. Instead, they refill via a novel non-selective store-operated cation conductance that is blocked by nifedipine.
Smooth muscle cells can produce a sustained contraction in the depolarised state through activation of voltage-dependent L-type Ca2+ channels (Missiaen et al. 1992). Receptor activation can also generate contraction without depolarisation by releasing Ca2+ from intracellular stores. Contraction cannot be sustained without replenishment of the stores from the extracellular medium. Store refilling can occur via L-type Ca2+ channels in smooth muscle (Ganitkevich & Isenberg, 1992; Gagov et al. 1994; McCarron et al. 2000).
Many cells possess Ca2+ entry channels that do not depend on depolarisation but are activated by depletion of Ca2+ stores (Parekh & Penner, 1997; Gibson et al. 1998; Lewis, 1999). In some non-excitable cells depolarisation-dependent Ca2+ channels are absent and the cells are thus entirely dependent on these store-operated channels. These include the smooth muscle cells from small metarterioles of the rat retina where L-type Ca2+ channels are absent (Scholfield & Curtis, 2000). Ca2+ channels that are not dependent on depolarisation may also be activated by agonists acting through second messenger pathways (Broad et al. 1999; Bolsover et al. 1999).
Several types of Ca2+ channel that are activated by store depletion have been described and include transient receptor potential (TRP) proteins (Birnbaumer et al. 1996; Montell, 1998). Store-filling channels appear to be opened following Ca2+ release into the cytoplasm via activation of IP3 or ryanodine receptors. The link between the stores and the store-operated channels has remained elusive, but may occur through a second messenger pathway or via a direct interaction of the stores with the plasma membrane (Ma et al. 2000). As well as store filling, these channels may also provide a direct source of Ca2+ for muscle contraction (Gibson et al. 1998).
The store-filling channels have been distinguished by blockade with heavy metal ions as well as somewhat more selective agents such as SK&F96365 and LOE908 (Gibson et al. 1998; Iwamuro et al. 1999). The dihydropyridines are generally regarded as specific blockers of the L-type Ca2+ channel. However, they also show some blockade of other voltage-dependent Ca2+ channels (see McDonald et al. 1994). Recently, there has been some evidence that these compounds also block store-filling pathways that are not depolarisation dependent. Thus, in cultured skeletal muscle cells held at -100 mV, two novel dihydropyridines blocked single channel Ca2+ current (Hopf et al. 1996). In vascular smooth muscle, a Ca2+ leak current was blocked by efonidipine (Matsuoka et al. 1997). Other workers claim that nifedipine can block Ca2+ influx or reduce store filling via a pathway which does not involve L-type Ca2+ channels (Gillo et al. 1993; Willmott et al. 1996; Lenz & Kleineke, 1997; Krutetskaia et al. 1997; Wu et al. 1997).
L-type Ca2+ channel antagonists, including nifedipine, are commonly used to treat cardiovascular disease, particularly hypertension and angina pectoris. They are predominantly vasodilators, acting particularly at the level of the microcirculation (van Zwieten & Pfaffendorf, 1993). Thus, if these antagonists do block store-filling channels, it may be that vasodilatation occurs via this mechanism rather than L-type Ca2+ channels, which are sparse, at least in small ocular microvessels (Scholfield & Curtis, 2000).
In the present study, we aim to show that in microvascular smooth muscle, store filling is nifedipine sensitive at a membrane potential well negative of the L-type Ca2+ channel activation voltage at physiological Ca2+ concentrations. To accomplish this, we firstly demonstrate that even when active, Ca2+ flowing through L-type Ca2+ channels fails to contribute to store filling in choroidal smooth muscle. We then show that at -80 mV, these cells possess a nifedipine-sensitive Ca2+ influx pathway, stimulated by store depletion via activation of ryanodine or IP3 receptors. A preliminary report of some of this work has been published previously (Curtis & Scholfield, 2000a).
METHODS
Tissue isolation
Rabbits were killed by pentobarbitone overdose (80 mg kg−1; injected into an ear vein) and the choroids of the eyes removed as a single sheet. These were chopped into 1 mm squares and placed in Ca2+-free solution with collagenase (0.2 mg ml−1). The suspension was stirred at 34 °C for 25 min and the tissue broken up by trituration using a fire-polished Pasteur pipette. Subsequently, the homogenate was centrifuged at 1000 r.p.m. for 1 min and the supernatant discarded. The suspension was then stored at 4 °C in 100 μm Ca2+ medium until needed. Homogenates contained cells and vessel fragments that remained usable for up to 6 h under these conditions. In most experiments, recordings were made from single cells still embedded in vessel fragments. These fragments comprised up to 10 functional cells as judged by their larger input capacitance compared to single cells (20-70 pF for fragments compared with 3-8 pF for isolated myocytes). The isolated myocytes were identified by their semilunar shape similar to those of microvascular smooth muscle in situ and were around 4 μm in width and 35 μm in length.
Electrophysiology
Homogenate was placed in a 2 ml recording bath on the stage of an inverted microscope for 10 min. Preheated medium was then allowed to flow into one end of the bath and withdrawn from the other at 2 ml min−1. The solution passed through a heat exchanger such that the temperature in the recording bath was 36 °C. The system for the delivery of drugs consisted of a manifold (volume 0.3 μl) leading from seven separate reservoirs each controlled by a valve. The flow of solution from the manifold into the bath was through a single tube (350 μm in diameter, 6 mm in length, 0.2 μl volume) long enough to allow temperature equilibration with the solution flowing through the bath. Its outlet was positioned 200 μm away from the vessel fragment of interest. The flow into the bath and over the vessels was streamlined and the delay time for new solution to reach the preparation was 1.5 s as measured by switching over to a solution containing fura-2.
Membrane current was recorded using the whole-cell perforated patch-clamp technique (Horn & Marty, 1988) in voltage-clamp mode with an Axopatch-1D patch-clamp amplifier (Axon Instruments, USA). Electrodes (1-2 MΩ in free bathing solution) were pulled from filamented borosilicate glass capillaries (1.5 mm o.d. × 1.17 mm i.d., Clark Electromedical Instruments, UK). Internal pipette solutions were K+ based with amphotericin B as the perforating agent (see solutions below). Recordings were delayed until full perforation of the membrane patch had been achieved, which usually took 3-5 min. Liquid junction potentials (< 2 mV) were compensated electronically. Series resistance (9-15 MΩ) and cell capacitance were usually uncompensated. For experiments using square step protocols, leakage currents were subtracted on-line. From a given holding potential, the correction signal was obtained by averaging three hyperpolarising steps (-30, -20 and -10 mV from the holding voltage), a procedure that did not activate ion channels, but allowed measurement of passive membrane properties and leak. Recordings were low pass filtered at 0.5 kHz and sampled at 2 kHz by a National Instruments PC1200 interface using software provided by J. Dempster (University of Strathclyde, UK).
Ca2+ measurements
Subsarcolemmal and global cell Ca2+ were measured using different methods. The local subsarcolemmal concentration was measured indirectly by monitoring the amplitude of the Ca2+-activated Cl− current. This technique gives an accurate and reliable indication of subsarcolemmal Ca2+ since (i) there is a linear relationship between the amplitude of the Ca2+-activated Cl− current and [Ca2+]i (Pacaud et al. 1992; Large & Wang, 1996), and (ii) Ca2+-activated Cl− channels continuously report the Ca2+ concentration with no indication of adaptation (Gomez-Hernandez et al. 1997). For global cell Ca2+, the choroidal cells were incubated in 5 μm fura-2 AM (with 5 μm Pluronic F-127) for 1 h at 4 °C in 100 μm Ca2+ medium. They were subsequently washed in 100 μm Ca2+ medium and stored at 4 °C until required. These cells were placed into a similar recording bath mounted on the stage of an inverted microscope and cells patch clamped as described above. The Ca2+ microfluorimetry system consisted of a dual monochromator passing 340 nm/380 nm light (5 nm band width), a light chopper (PTI DeltaScan, NJ, USA), and an inverted microscope with an oil immersion objective (× 40, NA 1.3). The emission side of the microscope comprised an adjustable rectangular window, a filter (510 nm) and a photon counting photomultiplier tube (PMT) in the light path. Fluorescence equipment was controlled by PTI Felix software, which also performed the storage and analysis of the fluorescence data. Before experimentation, cells were superfused with normal solution for 10 min before patching. At the end of an experiment, the bathing medium was changed to Ca2+-free/4 mm EGTA to obtain the 340 nm/380 nm ratio Rmin and then equilibrated with 5 mm Mn2+ in Ca2+-free solution to obtain the background. Rmax was determined separately in five cells using a 10 mm Ca2+ solution with 200 μg ml−1 amphotericin B to permeabilise the cell to Ca2+. These values were used to calculate [Ca2+]i using the relationship of Grynkiewicz et al. (1985). In experiments where fura-2 was loaded in microvessel fragments, an area equivalent to three to four cells was selected on the emission side.
Space clamping
One concern when recording from a cell electrically coupled to neighbouring cells is the adequacy of the space clamp. It was important to verify that all the smooth muscle cells within a fragment would be clamped at the same voltage. This problem was addressed by making simultaneous patch-clamp and intracellular Ca2+ recordings from arteriolar segments approximately 100 μm in length (50 μm longer than those used for most of the experiments). Single smooth muscle cells were initially patched at one end of the vessel and [Ca2+]i was recorded from three to four cells in this region during a 1 s depolarising voltage pulse from -80 to -20 mV. [Ca2+]i from an equal area of cells at the opposite end of the vessel was then measured with the patch electrode still attached. At both sites, the voltage steps evoked [Ca2+]i transients, through L-type Ca2+ channel activation (see Results), which were of a similar amplitude (12 ± 3 nm at the patched cell and 11 ± 2 nm at the other end; n = 6; P > 0.05; Student's paired t test). To further test the adequacy of the space clamp, voltage ramps from -100 to +40 mV were applied over 50 s. At the patched cell [Ca2+]i rose linearly beginning at -48 ± 4 mV, the potential for the half-maximal increase in [Ca2+]i was +5 ± 3 mV and peak [Ca2+]i at +40 mV was 28 ± 3 nm (n = 4). Respective values for cells 100 μm away were not significantly different (-49 ± 3 mV; +2 ± 5 mV; 30 ± 6 nm; P > 0.05 for each data set; paired t test). Assuming a constant L-type Ca2+ channel density among the smooth muscle cells of the vessel, these results indicate that segments of up to 100 μm in length can be homogeneously space clamped. This concurs with the long space constants (between 600 and 900 μm) reported for other microvessels (Beach et al. 1996; Hirst et al. 1997).
Source of fluorescence
In isolated retinal arterioles, we have previously shown that endothelial cells do not make a contribution to (i) the responses produced by test agents on the microvascular smooth muscle and (ii) the overall fluorescence signal originating from fura-2 (Scholfield & Curtis, 2000). The experiments described below were designed to confirm that this was also the case for isolated choroidal arterioles.
It is well known that many agents work indirectly on vascular smooth muscle by stimulating endothelial cells to release substances which then act on the adjacent smooth muscle cells. To evaluate this possibility, acetylcholine (10 μm) and bradykinin (1 μm), which are known to act only through endothelial cells, were tested. Neither had any effect on the fluorescence ratio of fura-2-loaded choroidal vessels (n = 6 per treatment). This suggests that either (i) these drugs could not gain access to endothelial cells or (ii) the endothelial cells were not viable. Thus, the effects of the agents detailed in the Results were most likely to be through a direct action on the arteriolar smooth muscle cells. Some key experiments were performed or repeated with isolated single cells to further confirm that there was no contribution from endothelial cells.
Experiments were conducted to test whether endothelial cells contributed to the total fluorescence signal. Some of the vessels had segments along their length stripped of arteriolar smooth muscle, yet endothelial cells were retained. Along these sections, the fluorescence ratio was less than twice the background and the signal was unaffected by 10 mm caffeine, 70 mm KCl, 10 nm endothelin-1 or 0.2 mg ml−1 saponin (n = 4 per treatment group). Similar findings were attained using capillaries (< 8 μm in diameter) derived from the choriocapillaris, which are composed mainly of endothelial cells (n = 6). In both denuded arterioles and capillaries, Mn2+ failed to quench the fluorescence signal, indicating that the fluorescence was not accessible to Mn2+ or did not originate from fura-2. The above results suggest that the endothelial cells did not load with fura-2, and thus did not contribute to the total fluorescence signal.
Solutions
The bathing solution contained (mm): 120 NaCl; 5 KCl; 5 d-glucose; 2 CaCl2; 1.3 MgCl2; 10 Hepes (pH adjusted to 7.3 with NaOH). In Ca2+-free solution, the CaCl2 was omitted and low Ca2+ solutions were made by adding the appropriate amount of CaCl2. For perforated-patch recordings the pipette contained (mm): 133 KCl; 1 MgCl2; 0.5 EGTA; 10 Hepes (pH adjusted to 7.2 using NaOH) to which 200 μg ml−1 amphotericin B was added. In all experiments, outward K+ currents were blocked by 20 mm tetraethylammonium chloride (TEA) (Curtis & Scholfield, 2000b).
Amphotericin B, caffeine, EGTA, nifedipine, anthracene-9-carboxylic acid (9-AC), collagenase type 1A and TEA were purchased from Sigma (Poole, UK). Endothelin-1 (human, porcine) was obtained from Tocris (Bristol, UK) and American Peptide Co. (CA, USA).
Data are presented as means ±s.e.m. Paired comparisons were made using Student's t test and multiple comparisons were made using two-way analysis of variance (ANOVA) with replication. The Kolmogorov-Smirnov two-sample test was used to examine differences between L-type Ca2+‘window’ currents before and after store depletion (Sokal & Rohlf, 1995).
RESULTS
Since the amount of Ca2+ in stores is difficult to measure reliably, store content was estimated by the amount of Ca2+ that could be discharged from the store into the cytosol. Ca2+ distribution is heterogeneous (Jaggar et al. 2000) so we assessed discharge by measuring both global cell Ca2+, by microfluorimetry, and the local subsarcolemmal concentration, by its effect on the Ca2+-activated chloride current.
L-type Ca2+ current (ICa(L))
The aim of the first series of experiments was to identify the voltage range for the L-type Ca2+‘window’ current in choroidal arterioles. This ‘window’ current represents the two competing influences of depolarisation-dependent activation and inactivation of the channels to produce a maximal steady-state current. It was predicted by examining the overlap between steady state inactivation and activation curves (McDonald et al. 1994). Ca2+ currents were recorded in a bathing solution with Ca2+ (2 mm) as the major charge carrier. To isolate Ca2+ current the bathing solution contained 20 mm TEA and 1 mm 9-AC to block K+ and Cl− currents, respectively. Vessel segments were held at -80 mV and subjected to 500 ms test pulses ranging from -100 to +20 mV. Steps to potentials more positive than -60 mV evoked a fast inward current, which peaked within 10 ms (Fig. 1Aa). The current was maximal between -20 and -10 mV (Vpeak) and reversed at +20 mV (Fig. 1Ab). In vessel fragments, the current was abolished after bath application of 1 μm nifedipine (Fig. 1Aa and b) and on washing out, the inward current returned to the same magnitude. Thus this current appeared to be carried through L-type Ca2+ channels. ICa(L) was too small to resolve in single microvascular smooth muscle cells.
Figure 1. L-type Ca2+ current in choroidal arteriolar smooth muscle cells.
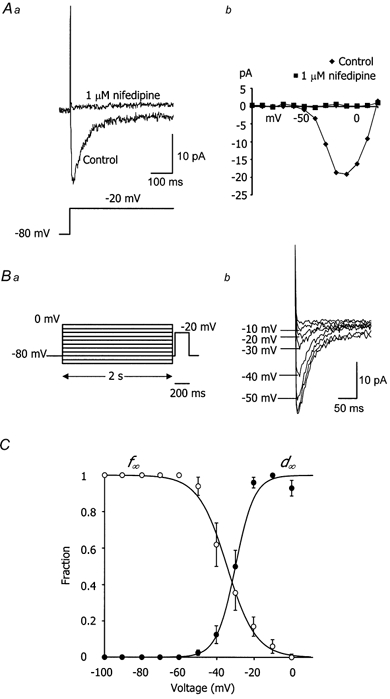
Aa, upper traces show the currents recorded from a choroidal arteriole in response to a voltage step from -80 to -20 mV (lower trace), first in the absence and then in the presence of 1 μm nifedipine. b, I-V relationships for the Ca2+ current before and after 1 μm nifedipine. B, voltage dependence of inactivation of ICa(L). a, inactivation protocol where the vessel was stepped to conditioning potentials ranging from -100 to 0 mV before stepping to a test potential of -20 mV. b, currents recorded during the test steps and responses following conditioning pulses of -50, -40, -30, -20 and -10 mV. C, inactivation (○) and activation curves (•). Open circles show the normalised peak amplitudes of the inward currents evoked using the inactivation protocol in B. Currents were normalised to the maximal current detected with a conditioning potential of -100 mV. Filled circles show the normalised peak amplitudes of the inward currents evoked by stepping from a holding potential of -80 mV to various test potentials. Currents were normalised to the maximal inward current detected with a voltage step to either -10 or -20 mV. Error bars represent s.e.m. Inactivation (f∞) and activation (d∞) curves were fitted with Boltzmann functions (see text for further explanation). Between -60 and 0 mV, the activation and inactivation curves overlap revealing the ‘window’ of current for the L-type Ca2+ channels.
The voltage dependence of inactivation was studied by using a double-pulse protocol applied to vessel fragments (Fig. 1b). A variable conditioning pulse was applied for 2 s immediately before a fixed test pulse to -20 mV. The test step of -20 mV was chosen since this was where maximal ICa(L) was observed. Figure 1B shows a typical record where it is clear that the current available during the test step depended on the previous conditioning potential. Figure 1C is a summary of eight such experiments where the normalised peak current during the test step is plotted against the previous conditioning potential. The data were fitted with (continuous line, Fig. 1C) a Boltzmann distribution of the form:
| (1) |
where V0.5 is the voltage at which half the channels are inactivated, and k is the slope factor at this voltage. In 2 mm[Ca2+]o, V0.5 was -34.5 mV and k was -7.7 mV.
Because of the rapid de-activation of ICa(L) (around 10 ms) it was not possible to separate the tail currents from capacitative transients and thus the voltage dependence of activation was calculated from the peak current- voltage relationship. In the same eight vessels, peak conductance was normalised and plotted against the test potential (Fig. 1C). The sigmoidal curve was fitted by a Boltzmann function of the form:
| (2) |
where V0.5 was -30.4 mV and the slope was 4.3 mV.
The activation and inactivation curves overlap substantially between -40 and -15 mV revealing a relatively large ‘window’ of current for the L-type Ca2+ channels within this voltage range.
Ca2+ store refilling is slower at -20 mV than at -80 mV
In the next series of experiments, the role of membrane potential in store filling was assessed.
Initially, the effect of membrane potential on the time course for refilling of the Ca2+ stores was examined. In Fig. 2Aa, arterioles were voltage clamped at -80 mV, well away from the activation voltage for L-type Ca2+ channels in a normal 2 mm Ca2+ solution. They were challenged with 10 mm caffeine to deplete Ca2+ from the intracellular stores and the amount of Ca2+ released was assessed by the amplitude of the resulting inward transient Ca2+-activated Cl− current (Curtis & Scholfield, 2000b). After varying times following the washout of caffeine, the vessels were again tested with caffeine in order to determine the amount of refilling (Fig. 2Aa). With periods ≥ 80 s, caffeine evoked a reproducible Ca2+-activated Cl− current indicating that the stores had refilled fully during this time. Periods shorter than 80 s reduced the second response (Fig. 2Aa), suggesting that only partial refilling had occurred. This procedure was then repeated in the same vessel at -20 mV, where L-type Ca2+‘window’ current was considerable (12 % of the maximal transient Ca2+ current at Vpeak). Experiments were carried out in a randomised fashion (for 3 out of the 7 vessels tested recordings were started at -20 mV rather than -80 mV). The amplitudes of the caffeine-induced currents at the two potentials were taken as a percentage of their respective controls (first caffeine dose at -80 or at -20 mV) and plotted against time (Fig. 2Ab). The percentages were significantly less at -20 mV compared to -80 mV, indicating that store refilling was slower at the depolarised potential (P < 0.01; two-way ANOVA with replication; n = 7). The half-time (t50) for refilling was 20 ± 6 s at -80 mV compared to 38 ± 10 s at -20 mV.
Figure 2. Effect of membrane potential on the time course for Ca2+ store refilling.
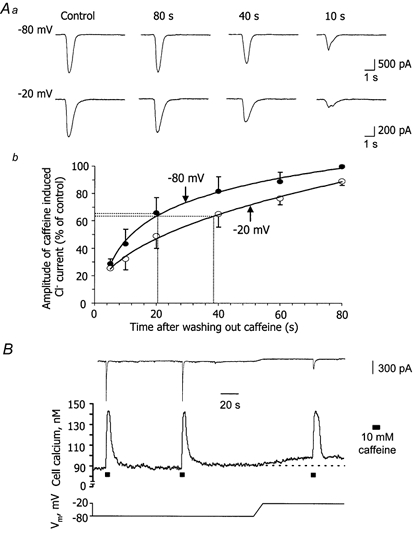
Aa, measurement of store-refilling times at -80 and -20 mV. A vessel segment was initially held at -80 mV (or -20 mV) and 10 mm caffeine applied for 5 s to release Ca2+ from intracellular stores. The amount of Ca2+ released was monitored from the amplitude of the resulting Ca2+-activated Cl− current. After varying times following the washout of caffeine, the vessel was once more tested with caffeine. This procedure was then repeated in the same vessel at -20 mV (or -80 mV). The currents at the two potentials are scaled differently so that they are presented as the same size such that the extent of refilling at the different washout times can be visually compared. Note, however, that the control current is much smaller at -20 mV (compare scale bars) because the vessel is much closer to ECl. b, in 7 vessels, the amplitudes of the caffeine-induced Cl− currents were taken as a percentage of their respective controls and plotted against time. Bars are s.e.m. and dotted lines show the half-time for refilling of 20 s at -80 mV and 38 s at -20 mV. B, voltage dependence of Ca2+ release. Simultaneous [Ca2+]i (middle trace) and current record (upper trace) for a vessel initially clamped at -80 mV. The Ca2+ stores were dumped with 10 mm caffeine then refilled during an 80 s washout period and a second dose of caffeine applied. Afterwards, the stores were again allowed to refill fully but this time the holding potential was adjusted (lower trace; over 10 s) to -20 mV. Once basal [Ca2+]i had steadied, a further dose of caffeine was added. Ca2+ release at the two potentials was similar.
The results above imply that, at -20 mV, where vessels are closer to the calcium equilibrium potential, store refilling times seem to be influenced more by the reduced driving force for Ca2+ influx than any increased Ca2+ entry through L-type Ca2+ channels. A further possibility that might explain the attenuated refilling at -20 mV is the voltage dependence of Ca2+ release. For instance, if there were more Ca2+ released with 10 mm caffeine at the depolarised potential, then intuitively, longer refilling times might be expected. It was impossible to test this hypothesis by patch clamping alone, since the amplitudes of control Ca2+-activated Cl− currents with caffeine were considerably smaller at -20 mV than at -80 mV (Fig. 2Aa). This was a consequence of the cells being closer to the chloride equilibrium potential (ECl), which was set to 0 mV in these experiments by the internal pipette solution. Figure 2B shows an original voltage-clamp and [Ca2+]i tracing for a choroidal arteriole, where the voltage dependence of Ca2+ release has been studied. At first, the arteriole segment was held at -80 mV and the Ca2+ stores were dumped by application of 10 mm caffeine for 5 s. This resulted in a [Ca2+]i transient that peaked within 4 s and had declined to the resting level after 30 s. After the first application of caffeine, the Ca2+ stores were allowed to refill fully over 80 s and a second dose was applied. A similar [Ca2+]i transient was evoked. The stores were again allowed to refill fully at -80 mV, but in this case, the holding potential was adjusted to -20 mV. Once the rise in the basal [Ca2+]i level had stabilised (8 ± 1 nm; n = 6), 10 mm caffeine was applied. From the new basal [Ca2+]i level, a slightly smaller [Ca2+]i transient was produced when compared with the second dose of caffeine at -80 mV. Although this pattern was observed in the six cells tested, there was no significant difference in Ca2+ release between the two potentials (at -80 mV Ca2+ release was 58 ± 8 nm and at -20 mV it was 55 ± 7 nm; P > 0.05; paired t test; n = 6). This accords with previous observations (Kamishima & McCarron, 1997).
Depolarising voltage steps fail to accelerate Ca2+ store refilling
In the next series of experiments, an attempt was made to hasten refilling rates via L-type Ca2+ channels by applying depolarising voltage steps. Figure 3A shows an example of six experiments where to start with vessel fragments were held at -60 mV (see legend) and exposed to 10 mm caffeine. After washout of caffeine, the Ca2+ stores were allowed to refill over a period of 30 s and a second dose of caffeine was applied. The second [Ca2+]i transient with caffeine was approximately half the size of the first, indicating that the Ca2+ stores had only partially refilled over the 30 s. The fragments were then given 2 min for the stores to refill fully. The above protocol was then repeated, but this time with four 1 s depolarising voltage steps from -60 to -20 mV during the 30 s refilling period. The step to -20 mV was selected since this is where the maximal transient ICa(L) was observed (see above). Each individual voltage step evoked a similar sized peak in inward L-type Ca2+ current and ensuing [Ca2+]i transient. The peak of the [Ca2+]i transients lagged behind the L-type Ca2+ currents by around 1 s. There was also a small summative increase in basal [Ca2+]i following each voltage step. Notwithstanding this, at the end of the four pulses, [Ca2+]i fell back to the resting level before the second dose of caffeine was applied. For six cells, the Ca2+ currents and resultant [Ca2+]i transients averaged -17 ± 1 pA and 13 ± 2 nm, respectively. When voltage steps were not present during the 30 s refilling phase, the mean amplitude of the second caffeine-induced [Ca2+]i transient was 51 ± 3 nm. The corresponding [Ca2+]i peak with the depolarising voltage pulse regime (51 ± 2 nm) was not significantly different (P > 0.05; paired t test). These results suggest that stimulation of Ca2+ influx through L-type Ca2+ channels does not accelerate Ca2+ store refilling in choroidal arteriolar smooth muscle.
Figure 3. Depolarising voltage steps during Ca2+ store refilling.
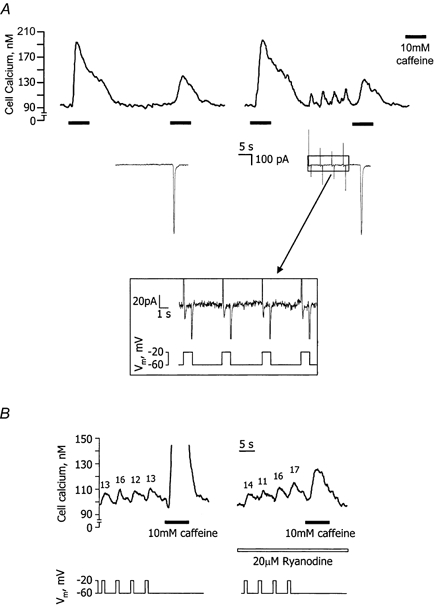
A, synchronised [Ca2+]i (upper trace) and current record (middle trace; only pertinent sections of holding current are shown) for a vessel fragment held at -60 mV. To start with, 10 mm caffeine was used to deplete [Ca2+]i stores. The stores were then allowed to refill for 30 s and a second dose of caffeine was applied. The vessel then had 2 min for the stores to refill fully and this protocol was repeated but with four 1 s depolarising voltage steps from -60 to -20 mV during the 30 s refilling period. Each voltage step evoked an L-type Ca2+ current (expanded lower panel) and consequent [Ca2+]i transient. Despite the increased Ca2+ influx during refilling with the depolarising voltage pulse regime, the second response to caffeine was not different from that seen when the membrane potential remained constant. Prolonged periods at depolarised potentials reduce the store-filling rate in choroidal smooth muscle (see Fig. 2Ab) and the protocol above was designed to lessen this effect. This was achieved by (i) keeping the period spent at -20 mV short (only 4 s out of 30 s) and (ii) ensuring that the change in membrane potential (-40 mV) was minimised by applying voltage steps from -60 mV rather than -80 mV. B, [Ca2+]i record (upper trace) demonstrating the effect of 20 μm ryanodine on the [Ca2+]i transients elicited by four 1 s depolarising voltage steps from -60 to -20 mV (lower trace) (current record not shown). Labels above each transient indicate the increase in [Ca2+]i (in nm) with each step. Caffeine (10 mm) was applied at the end of the voltage steps to test the effectiveness of ryanodine action.
A possible flaw in the test above was that Ca2+-induced Ca2+ release (CICR) might have been occurring during each depolarising voltage step. This would effectively deplete some Ca2+ from the stores during the 30 s refilling phase and in doing so attenuate the subsequent caffeine response. To ensure that this was not the case the [Ca2+]i transients associated with the depolarising voltage steps were examined in the presence and absence of the specific CICR blocker, ryanodine (20 μm). Any decrease in the size of the [Ca2+]i transients with ryanodine would be indicative of CICR (Shmigol et al. 1998). Caffeine (10 mm) added at the end of the voltage steps was used to monitor the efficacy of ryanodine action. Figure 3B shows a typical record with the stores fully loaded. Under control conditions the [Ca2+]i transients in five vessels averaged 10 ± 1 nm, while in the same cells, following 5 min exposure to ryanodine, they were similar (11 ± 1 nm; P > 0.05; paired t test; note that two 8 s pulses of 10 mm caffeine were applied, one at the beginning and the other 2.5 min into the ryanodine pre-incubation to ensure open channel block of ryanodine receptors). Ryanodine worked effectively to block CICR during these trials since the caffeine-induced [Ca2+]i transients were reduced by 80 ± 5 % in the presence of this drug. Thus, Ca2+ entry through L-type Ca2+ channels in the earlier experiments did not induce CICR.
Nifedipine-sensitive store-operated cation current
The evidence so far indicates that store refilling was faster at -80 than at -20 mV and that further enhancement of Ca2+ influx through L-type Ca2+ channels failed to increase the filling rate. Preliminary experiments suggested store filling to be nifedipine sensitive and the remaining experiments sought to explore this further. Our first objective was to establish whether choroidal microvascular smooth muscle cells possess Ca2+ current that could sustain store filling, and if so, whether this can be blocked by nifedipine. In large smooth muscle cells the store-operated current is known to be extremely small (˜-3 pA; Wayman et al. 1996), and hence it was not surprising that it could not be detected in much smaller single choroidal microvascular cells (for example, see the failure to show an inward current in the single cell shown in Fig. 6Aa). Nonetheless, we did find that electrically coupled multiple cells of arteriolar fragments produced a summation of the currents to a value that could be measured.
Figure 6. Effect of nifedipine and the removal of extracellular Ca2+ on the Et-1-induced sustained increase in [Ca2+]i in cells held at -80 mV.
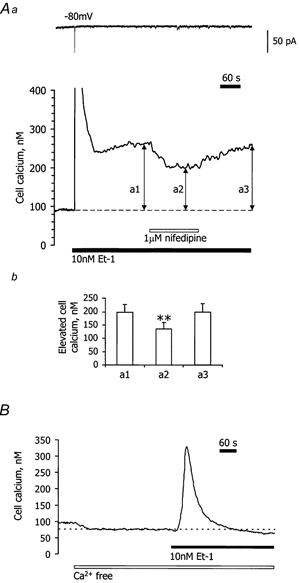
Aa, simultaneous voltage-clamp (upper panel) and [Ca2+]i (lower panel) records for a single choroidal microvascular smooth muscle cell. The cell was held at -80 mV throughout. During the period marked by the filled bar, 10 nm Et-1 was added to the bathing solution. This produced a transient increase in [Ca2+]i (peak truncated) followed by a steady level of elevated [Ca2+]i. A single Ca2+-activated Cl− current coincided with the transient rise in [Ca2+]i, but no other measurable changes in the holding current were observed since in single cells the store-operated current fell below the limit of detection. Nifedipine (1 μm) was added during the period marked by the open bar. b, a histogram from 7 similar experiments where [Ca2+]i above the resting level was compared during the sustained phase of Et-1 action before adding nifedipine (a1), during its action (a2) and after recovery from nifedipine (a3). **P < 0.01 for a2 compared to a1 (paired t test) and the error bars refer to s.e.m.B, [Ca2+]i record for a different cell held at -80 mV (holding current not shown) and bathed in Ca2+-free solution (open bar). Et-1 (10 nm) was present during the period marked by the filled bar. Note that the plateau phase of the Et-1 response is abolished by removal of extracellular Ca2+.
Vessel segments up to 100 μm in length were voltage clamped at -80 mV and in contrast to the preceding experiments, caffeine (10 mm) was applied for periods of > 10 s. Under these conditions, caffeine activated a biphasic inward current. This consisted of the initial Ca2+-activated Cl− current described above, followed by a much smaller sustained current (Fig. 4Aa). The sustained current had an amplitude of -12 ± 1 pA and persisted for up to 5 min with caffeine still present (n = 10). The transient current was reduced by 82 ± 4 % with the Cl− channel blocker 9-AC (1 mm; n = 6; Fig. 4Ab), and was absent in vessels pre-incubated for 30 min with the intracellular Ca2+ chelator BAPTA AM (50 μm; n = 7; Fig. 4b). In contrast, the sustained current was observed both in the presence of 9-AC (Fig. 4Ab; current amplitudes were -12 ± 2 pA in caffeine and -11 ± 4 pA in caffeine with 9-AC) and in vessels loaded with BAPTA AM (Fig. 4b). These results indicated that the sustained current was not a Cl− conductance or Ca2+ dependent.
Figure 4. Caffeine activates a nifedipine-sensitive, store-depletion-dependent, non-selective cation current.
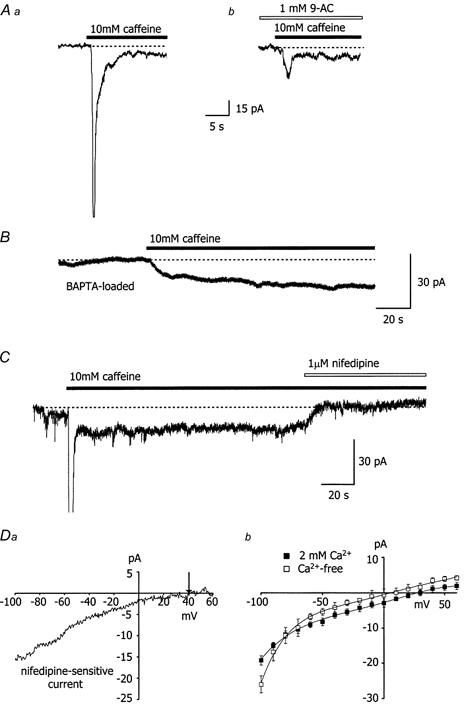
Aa, 10 mm caffeine activates a transient and sustained inward current in choroidal arterioles held at -80 mV. Caffeine was applied as indicated by the filled bar and the dotted line represents zero current. In all of the experiments in this series, the holding current was reset to zero prior to recording. b, the transient current activated by 10 mm caffeine was inhibited by the chloride channel blocker anthracene-9-carboxylic acid (1 mm), whilst the sustained current was unaffected. B, only the sustained component of the caffeine-induced current was observed when vessels (held at -80 mV) were loaded with BAPTA AM (50 μm) to chelate intracellular Ca2+. C, the sustained current activated by 10 mm caffeine could be completely blocked by 1 μm nifedipine in vessels held at -80 mV. Caffeine and nifedipine were applied as indicated by the filled and open bars, respectively. Da, I-V relationship for the nifedipine-sensitive current activated by 10 mm caffeine. In this arteriole, the reversal potential for the nifedipine-sensitive current was around +40 mV (denoted by the arrow). b, mean I-V relationships for the nifedipine-sensitive current in the presence (n = 5) and absence (n = 7) of 2 mm extracellular Ca2+.
The sustained inward current activated by 10 mm caffeine at -80 mV was completely inhibited by 1 μm nifedipine (n = 6; Fig. 4C; current amplitudes were -10 ± 2 pA in caffeine and -1 ± 3 pA in caffeine with nifedipine). In the absence of caffeine, nifedipine had no effect on the resting current (6 vessels). In five other vessels, a 10-fold higher concentration of ethanol (0.1 %), the vehicle for nifedipine, had no effect on the sustained inward current. The I-V relationship for the nifedipine-sensitive sustained current was assessed by ramping the membrane potential from -100 mV to +60 mV over 200 ms, first in the presence of caffeine and then in caffeine plus nifedipine. The I-V relationship for the nifedipine-sensitive current (constructed by deducting the caffeine + nifedipine I-V from the I-V for caffeine alone) showed some inward rectification and reversed at +29 ± 2 mV (n = 5; Fig. 4Da and b), well positive of ECl, which was set to 0 mV. Further evidence that this current is not carried by Cl− ions is derived from the fact that the I-V relationship of the Cl− conductance in choroidal arteriolar smooth muscle exhibits marked outward rectification (Curtis & Scholfield, 2000b). The nifedipine-sensitive sustained current was maintained in Ca2+-free solution, but the reversal potential shifted significantly less positive to +7 ± 5 mV (n = 7) and the inward rectification became more pronounced (Fig. 4Db).
In vessels held at -80 mV, depletion of the Ca2+ stores was also achieved using the sarcoplasmic reticulum Ca2+-ATPase inhibitor cyclopiazonic acid (CPA; 20 μm). CPA produced a large transient Ca2+-activated Cl− current (-146 ± 40 pA), albeit slower than caffeine. This was succeeded by a smaller sustained component (-7 ± 2 pA) (n = 6), which was similarly blocked by nifedipine (0 ± 1 pA; n = 4). Again, there was no block of the sustained component by 1 mm 9-AC or by loading with BAPTA AM (n = 4 per treatment group).
The data above suggest that, in addition to activating a Ca2+-dependent Cl− current, caffeine and CPA also switch on a sustained, voltage-independent, non-selective cation conductance, activated directly by store depletion and inhibited by nifedipine. The fact that the reversal potential of the store-operated current was shifted in a negative direction following removal of extracellular Ca2+ suggests that the underlying channels have a substantial permeability to Ca2+ ions.
Experiments were finally conducted to ensure that the nifedipine-sensitive sustained current was not simply a reflection of a shift in the voltage range of the L-type Ca2+‘window’ current to more negative potentials as a direct consequence of store depletion. Activation and inactivation curves for the L-type Ca2+ current in 2 mm[Ca2+]o were constructed as described previously, but this time for vessels exposed for 1 min to 10 mm caffeine. The Ca2+‘window’ current in the presence and absence of caffeine was compared by multiplying d∞ by f∞ (eqns (1) and (2)) for each respective data set, thereby producing two bell-shaped curves depicting the proportion of current to that at Vpeak against holding voltage. No significant differences were observed between the two curves (P > 0.05; Kolmogorov-Smirnov two-sample test), indicating that store depletion was not acting to alter the voltage range for the L-type Ca2+‘window’ current.
Nifedipine inhibits refilling of caffeine-sensitive Ca2+ stores in cells held at -80 mV
The role of the nifedipine-sensitive cation current in refilling of the Ca2+ stores was examined in the next set of experiments. Vessel segments were voltage clamped at -80 mV. Caffeine (10 mm) was again used to directly deplete the Ca2+ stores and the amount of Ca2+ released assessed by the amplitude of the resulting Ca2+-activated Cl− current. Earlier experiments had shown that 80 s was a sufficient period for the Ca2+ stores to refill fully. Thus, any intervention during the 80 s interlude that reduced the second response to caffeine would provide an indication of attenuated store refilling. When 1 μm nifedipine was present during the 80 s refilling period the caffeine-evoked current was reduced by 38 ± 5 % (Fig. 5Aa; current amplitudes were -631 ± 78 and -393 ± 51 pA, before and during 1 μm nifedipine, respectively; P < 0.001, paired t test; n = 8). After washing out both the caffeine and nifedipine, caffeine was reapplied after another 80 s and the resultant inward current had the same amplitude as that with the first application (Fig. 5Aa; -644 ± 80 pA; P > 0.05, paired t test; n = 8). These experiments were repeated in six isolated single arteriolar smooth muscle cells instead of segments. The caffeine-induced current was reduced by 38 ± 10 % when nifedipine was present during the 80 s refilling period and this effect was fully reversible upon washout. Ethanol alone (0.1 %) had no effect on store refilling in whole vessels (n = 6; P > 0.05; paired t test).
Figure 5. Effect of nifedipine and the removal of extracellular Ca2+ on Ca2+ store refilling after caffeine depletion in cells held at -80 mV.
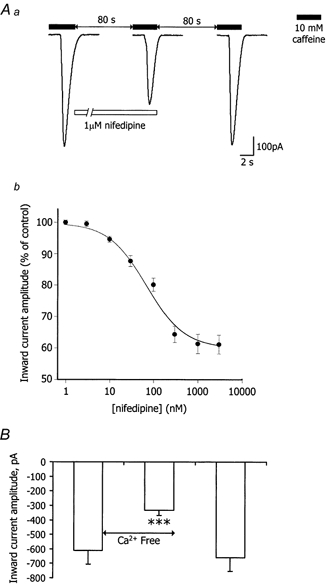
These experiments were repeated in vessel segments with various concentrations of nifedipine between 1 and 3000 nm present during the 80 s refilling period. The second response to caffeine was reduced in a concentration-dependent manner with the effect saturating at 1 μm and the half-maximal effect at 64 nm (Fig. 5Ab).
To assess the total contribution of extracellular Ca2+ to store refilling, vessels were challenged with caffeine and then were bathed in a nominally Ca2+-free solution for the 80 s refilling period. The second response to caffeine was reduced, by 46 ± 6 % (Fig. 5B; P < 0.001, paired t test; n = 8).
To exclude any direct blockade of the Ca2+-activated Cl− channels by nifedipine or any interference with Ca2+ release, the Ca2+-free protocol described above was repeated, first in the absence and then in the presence of 5 μm nifedipine (80 s exposure) during refilling. Nifedipine had no direct effect on the action of caffeine (current amplitudes for the second response to caffeine were -231 ± 33 and -245 ± 27 pA, in the absence and presence of 5 μm nifedipine, respectively; P > 0.05; paired t test; n = 5).
Nifedipine reduces endothelin (Et-1)-induced Ca2+ influx in cells held at -80 mV
The experiments above suggest the presence of a store-filling Ca2+ influx pathway that is not the classical voltage-dependent L-type Ca2+ channel, but is nevertheless blocked by similar concentrations of nifedipine. The next series of experiments were designed to assess whether this pathway could also be stimulated by agonist depletion of IP3-sensitive Ca2+ stores. In vascular smooth muscle, endothelin-1 (Et-1) is a potent vasoconstrictor, which induces Ca2+ release from intracellular stores via formation of IP3 (Neylon, 1999).
All these experiments were conducted on single isolated cells voltage clamped at -80 mV. Figure 6Aa shows a record of [Ca2+]i in a choroidal cell. Application of Et-1 (10 nm) produced a biphasic increase in [Ca2+]i: an initial transient rise (344 ± 38 nm in amplitude) and a subsequent sustained phase (198 ± 28 nm above basal levels) (n = 7). In most vessels, [Ca2+]i peaked within 10-15 s and declined to a steady plateau level after 1-2 min. This elevated [Ca2+]i persisted whether or not Et-1 was washed out (for > 20 min). A transient Ca2+-activated Cl− current was evoked by the initial transient rise in [Ca2+]i. The cells contracted into spheres with Et-1 and failed to relax on washout.
In nominally Ca2+-free solution, [Ca2+]i was reduced from 84 ± 5 to 65 ± 4 nm (n = 6) in cells clamped at -80 mV. Et-1 also caused a transient increase in [Ca2+]i in Ca2+-free medium (230 ± 25 nm; n = 6), but this subsided back to the resting level observed in Ca2+-free solution alone, i.e. there was no plateau phase of elevated [Ca2+]i (Fig. 6b). This suggests that in normal bathing solution (2 mm Ca2+) the plateau phase of [Ca2+]i came from the surrounding medium.
Nifedipine (1 μm) by itself had no effect on resting [Ca2+]i (from 87 ± 3 to 88 ± 3 nm with nifedipine; n = 7). During the plateau phase of Et-1 action, nifedipine produced a progressive fall in [Ca2+]i from 198 ± 28 to 135 ± 22 nm (Fig. 6Ab). Higher concentrations of nifedipine (i.e. up to 5 μm) caused no further inhibition of plateau [Ca2+]i (133 ± 20 nm; n = 7). On washing out the nifedipine, [Ca2+]i recovered to 198 ± 32 nm (Fig. 6A). Ethanol (0.1 %) alone did not affect the Et-1-stimulated Ca2+ influx (n = 6).
DISCUSSION
In choroidal arteriolar smooth muscle cells, depolarisation reduced the rate of store refilling. Refilling times depended more on changes in the electrochemical driving force for Ca2+ entry than on increased Ca2+ influx through voltage-operated L-type Ca2+ channels. These results were surprising since voltage-dependent Ca2+ channels have very high Ca2+ permeation rates (one million Ca2+ ions per second at -50 mV; Gollash & Nelson, 1997), and hence very few channels would need to be open to satisfy store refilling. In other types of smooth muscle, store-refilling rates are well maintained over a wide voltage range (Casteels & Droogmans, 1981; McCarron et al. 2000). In colonic myocytes, for instance, Ca2+ influx through channels which lack voltage gating provides for store refilling at negative membrane potentials (-70 mV), while at positive potentials (+40 mV) voltage-gated channels are largely responsible (McCarron et al. 2000).
The role of L-type Ca2+ channels in store refilling was further assessed by applying depolarising voltage pulses from -60 to -20 mV during a 30 s refilling period following store depletion. Despite a substantial elevation in global cell Ca2+ via L-type Ca2+ channel activation, this procedure failed to contribute to refilling. This reinforces the view that unlike the situation in other types of smooth muscle, L-type Ca2+ channels do not contribute to store filling in choroidal arteriolar smooth muscle. It is difficult to give a precise reason for this difference, but one factor that might be important is the lower density of L-type Ca2+ channels in these cells. When normalised to capacitance, the L-type current in choroidal arteriolar smooth muscle is ˜0.3 pA pF−1, while in larger smooth muscle cells it varies between 10 and 30 pA pF−1 (McDonald et al. 1994). Variation in the spatial architecture of the choroidal arteriolar smooth muscle cells might also be relevant. For instance, perhaps the L-type Ca2+ channels are not co-localised to the stores, as they are in other forms of smooth muscle (Carrington et al. 1995; Gollash et al. 1998).
L-type Ca2+ channels do not contribute to the replenishing of the Ca2+ stores in choroidal arteriolar smooth muscle, yet store refilling was nifedipine sensitive. Our data suggested that nifedipine was acting by blocking a store-operated, non-selective cation current. Both caffeine and CPA activated a small, sustained inward current during prolonged exposure in vessels held at -80 mV. The current was not inhibited by 9-AC or by loading with BAPTA AM, indicating that it was not a chloride conductance or Ca2+ activated. The current was sensitive to a low concentration of nifedipine, but did not result from a negative shift in the voltage range of the L-type Ca2+ ‘window’ current following store depletion. The current-voltage relationship for the nifedipine-sensitive current reversed at around +30 mV suggesting that it was probably due to the activation of a non-selective cation conductance. The reversal potential shifted to a less positive potential following removal of extracellular Ca2+, indicating that the underlying channels have considerable permeability to Ca2+ ions. Store-operated cation currents have been detected in a variety of cell types (for review see Parekh & Penner, 1997), including smooth muscle (Gibson et al. 1998). In common with other cell types, the store-operated current in choroidal arteriolar smooth muscle shows some inward rectification at negative voltages. The inward rectification in store-operated channels has been attributed to Mg2+ ions preferentially blocking outward current (Kershbaum & Cahalan, 1998). In our experiments, rectification became more prominent in nominally Ca2+-free solution. Ca2+ ions preferentially block inward current through store-operated channels (Kershbaum & Cahalan, 1998) and removal of this inhibition would be expected to amplify the rectification at negative voltages. In some respects, the store-operated cation current in choroidal arteriolar smooth muscle has properties similar to those described in other cell types. However, the high susceptibility of the current to nifedipine suggests that there are fundamental differences in the structure of the store-operated channels in this tissue.
Store refilling at -80 mV in choroidal arteriolar smooth muscle was inhibited by nifedipine with an EC50 of 64 nm. This modest concentration is comparable to that effective on L-type Ca2+ channels in many studies on whole cells and tissues. However, a direct comparison of the potency of nifedipine at L-type Ca2+ channels is complicated by the high voltage dependency of its action (Lopez et al. 1989; Zheng et al. 1992), variation in binding between the open and inactivation states of the channel (McDonald et al. 1994), the type of α1 subunit (Morel et al. 1998) and splice variants (Soldatov et al. 1995; Welling et al. 1997; Zuhlke et al. 1998). Nifedipine at 1 μm produced a maximal inhibition of the store-operated pathway in choroidal arteriolar smooth muscle. This concentration is commonly used to accomplish complete blockade of L-type Ca2+ channels and to test for any functional involvement of these channels in cellular processes. The present data indicate that the only satisfactory way of distinguishing between Ca2+ entering through the different nifedipine-sensitive pathways is by assessing voltage dependence and kinetic parameters.
Ca2+ influx stimulated by Et-1 was only partially inhibited by 1 μm nifedipine. Thus, in choroidal arteriolar smooth muscle, Et-1 appears to activate two Ca2+ entry pathways, one nifedipine sensitive and the other nifedipine resistant. Since the cells were held at -80 mV, it seems likely that the nifedipine-sensitive component with Et-1 represents activation of the store-operated channels described above. The additional Ca2+ entry pathway is possibly receptor operated, although further investigation is required to verify this. Multiple Ca2+ influx pathways also exist in other vascular smooth muscle cells (Iwamuro et al. 1999). With caffeine-induced depletion of stores, the depression in refilling produced by nifedipine was only slightly less than that observed with Ca2+-free solution. This implies that, in contrast to Et-1, most of the Ca2+ influx was through the nifedipine-sensitive pathway. Presumably, the remainder of the store filling was due to re-uptake of discharged Ca2+ from the cytosol.
These experiments used only rabbit choroidal microvascular smooth muscle cells. We have also conducted a preliminary investigation using rat retinal vessels (Curtis & Scholfield, 2000c) where the entire sustained Ca2+ rise with Et-1 is nifedipine sensitive and not mediated by L-type Ca2+ channels. Many workers use nifedipine to prevent Ca2+ movements through L-type Ca2+ channels that might otherwise interfere with alternative store-filling pathways. In doing so, this influx pathway would be overlooked and could be more widespread.
Some authors have questioned the specificity of nifedipine because in several systems it produces effects unrelated to L-type Ca2+ channels. Nifedipine has been reported to inhibit enzymes, e.g. cytochrome P450 (Katoh et al. 2000), as well as other channels including the T-type Ca2+ channel (for review see McDonald et al. 1994). However, these non-specific effects have been achieved using supra-micromolar concentrations, well above those producing effects in the present study. The fact that the effective concentration range of nifedipine for the store-operated channel (10-1000 nm) was in good agreement with that for L-type Ca2+ channels suggests that nifedipine did not inhibit the current through one of these other mechanisms. Thus, the store-operated channel in choroidal arteriolar smooth muscle might resemble the L-type Ca2+ channel in structure, but without the voltage sensor. This theory is made more credible by the fact that one of the TRP channels (the putative channels for store-operated Ca2+ entry) shares sequence homology with the α1 subunit of the L-type Ca2+ channel (Phillips et al. 1992), but lacks the charged residues in the S4 segment, the part of the α1 subunit believed to sense voltage changes (Catterall, 1994).
The present results demonstrate that Ca2+ influx through L-type Ca2+ channels fails to contribute to store refilling in choroidal microvascular smooth muscle. Moreover, they provide strong evidence for a novel store-operated Ca2+ influx pathway that is not voltage dependent, but is nevertheless blocked by nifedipine. If this store-filling current proves to be widespread in microvascular smooth muscle, in the future, pharmacological targeting of these channels to leave L-type Ca2+ channels in other cell types unaffected may provide more selective drug therapies for cardiovascular disease.
Acknowledgments
We thank the Wellcome Trust for financial support.
References
- Beach JM, McGahren ED, Xia J, Duling BR. Ratiometric measurement of endothelial depolarization in arterioles with a potential-sensitive dye. American Journal of Physiology. 1996;39:H2216–2227. doi: 10.1152/ajpheart.1996.270.6.H2216. [DOI] [PubMed] [Google Scholar]
- Birnbaumer L, Zhu X, Jiang M, Boulay G, Peyton M, Vannier B, Brown D, Platano D, Sadeghi H, Stefani E, Birnbaumer M. On the molecular basis and regulation of cellular capacitative calcium entry: roles for Trp proteins. Proceedings of the National Academy of Sciences of the USA. 1996;93:15195–15202. doi: 10.1073/pnas.93.26.15195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolsover S, Ashworth R, Archer F. Activator of calcium influx proves a slippery customer. Journal of Physiology. 1999;517:2. doi: 10.1111/j.1469-7793.1999.0002z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad LM, Cannon TR, Taylor CW. A non-capacitative pathway activated by arachidonic acid is the major Ca2+ entry mechanism in rat A7r5 smooth muscle cells stimulated with low concentrations of vasopressin. Journal of Physiology. 1999;517:121–134. doi: 10.1111/j.1469-7793.1999.0121z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington WA, Lynch RM, Moore ED, Isenberg G, Fogarty KE, Fay FS. Superresolution three-dimensional images of fluorescence in cells with minimal light exposure. Science. 1995;268:1483–1487. doi: 10.1126/science.7770772. [DOI] [PubMed] [Google Scholar]
- Casteels R, Droogmans GJ. Exchange characteristics of the noradrenaline-sensitive calcium store in vascular smooth muscle cells or rabbit ear artery. Journal of Physiology. 1981;317:263–279. doi: 10.1113/jphysiol.1981.sp013824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. Molecular properties of a superfamily of plasma-membrane cation channels. Current Opinion in Cell Biology. 1994;6:607–615. doi: 10.1016/0955-0674(94)90083-3. [DOI] [PubMed] [Google Scholar]
- Curtis TM, Scholfield CN. Nifedipine-sensitive Ca2+ influx independent of L-type Ca2+ channels in microvascular smooth muscle of the rabbit choroid. Journal of Physiology. 2000a;523.P:154–155P. [Google Scholar]
- Curtis TM, Scholfield CN. Transient Ca2+-activated Cl− currents with endothelin in isolated arteriolar smooth muscle cells of the choroid. Investigative Ophthalmology and Visual Science. 2000b;41:2279–2285. [PubMed] [Google Scholar]
- Curtis TM, Scholfield CN. Endothelin stimulates a nifedipine sensitive store-filling Ca2+ influx but inhibits L-type Ca2+ channels in microvascular smooth muscle of rat retina in vitro. British Journal of Pharmacology. 2000c;129:20P. [Google Scholar]
- Gagov HS, Duridanova DB, Boev KK, Daniel EE. L-type calcium channels may fill directly the IP3-sensitive calcium store. General Physiology and Biophysics. 1994;13:75–84. [PubMed] [Google Scholar]
- Ganitkevich VYa, Isenberg G. Caffeine-induced release and reuptake of Ca2+ by Ca2+ stores in myocytes from guinea-pig urinary bladder. Journal of Physiology. 1992;458:99–117. doi: 10.1113/jphysiol.1992.sp019408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson A, McFadzean I, Wallace P, Wayman CP. Capacitative Ca2+ entry and the regulation of smooth muscle tone. Trends in Pharmacological Sciences. 1998;19:266–269. doi: 10.1016/s0165-6147(98)01222-x. [DOI] [PubMed] [Google Scholar]
- Gillo B, Ma YS, Marks AR. Calcium influx in induced differentiation of murine erythroleukemia cells. Blood. 1993;81:783–792. [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gollasch M, Nelson MT. Voltage-dependent Ca2+ channels in arterial smooth muscle cells. Kidney and Blood Pressure Research. 1997;20:355–371. doi: 10.1159/000174250. [DOI] [PubMed] [Google Scholar]
- Gollasch M, Wellman GC, Knot HJ, Jaggar JH, Damon DH, Bonev AD, Nelson MT. Ontogeny of local sarcoplasmic reticulum Ca2+ signals in cerebral arteries: Ca2+ sparks as elementary physiological events. Circulation Research. 1998;83:1104–1114. doi: 10.1161/01.res.83.11.1104. [DOI] [PubMed] [Google Scholar]
- Gomez-Hernandez JM, Stuhmer W, Parekh AB. Calcium dependence and distribution of calcium-activated chloride channels in Xenopus oocytes. Journal of Physiology. 1997;502:569–574. doi: 10.1111/j.1469-7793.1997.569bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst GDS, Edwards FR, Gould DJ, Sandow SL, Hill CE. Electrical properties of iridial arterioles of the rat. American Journal of Physiology. 1997;42:H2465–2472. doi: 10.1152/ajpheart.1997.273.5.H2465. [DOI] [PubMed] [Google Scholar]
- Hopf FW, Reddy P, Hong J, Steinhardt RA. A capacitative calcium current in cultured skeletal muscle cells is mediated by the calcium-specific leak channel and inhibited by dihydropyridine compounds. Journal of Biological Chemistry. 1996;271:22358–22367. doi: 10.1074/jbc.271.37.22358. [DOI] [PubMed] [Google Scholar]
- Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. Journal of General Physiology. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamuro Y, Miwa S, Zhang XF, Minowa T, Enoki T, Okamoto Y, Hasegawa H, Furutani H, Okazawa M, Ishikawa M, Hashimoto N, Masaki T. Activation of three types of voltage-independent Ca2+ channel in A7r5 cells by endothelin-1 as revealed by a novel Ca2+ channel blocker LOE 908. British Journal of Pharmacology. 1999;126:1107–1114. doi: 10.1038/sj.bjp.0702416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. American Journal of Physiology - Cell Physiology. 2000;278:C235–256. doi: 10.1152/ajpcell.2000.278.2.C235. [DOI] [PubMed] [Google Scholar]
- Kamishima T, McCarron JG. Regulation of the cytosolic Ca2+ concentration by Ca2+ stores in single smooth muscle cells from rat cerebral arteries. Journal of Physiology. 1997;501:497–508. doi: 10.1111/j.1469-7793.1997.497bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh M, Nakajima M, Shimada N, Yamazaki H, Yokoi T. Inhibition of human cytochrome P450 enzymes by 1,4-dihydropyridine calcium antagonists: prediction of in vivo drug-drug interactions. European Journal of Clinical Pharmacology. 2000;55:843–852. doi: 10.1007/s002280050706. [DOI] [PubMed] [Google Scholar]
- Kerschbaum HH, Cahalan MD. Monovalent permeability, rectification, and ionic block of store-operated calcium channels in Jurkat T lymphocytes. Journal of General Physiology. 1998;111:521–537. doi: 10.1085/jgp.111.4.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutetskaia ZI, Lebedev OE, Krutetskaia NI, Petrova TV. Organic and inorganic blockers of potential-dependent Ca2+ channels inhibit store-dependent entry of Ca2+ into rat peritoneal macrophages. Tsitologiia. 1997;39:1131–1141. [PubMed] [Google Scholar]
- Large WA, Wang Q. Characteristics and physiological role of the Ca2+-activated Cl− conductance in smooth muscle. American Journal of Physiology. 1996;40:C435–454. doi: 10.1152/ajpcell.1996.271.2.C435. [DOI] [PubMed] [Google Scholar]
- Lenz T, Kleineke JW. Hormone-induced rise in cytosolic Ca2+ in axolotl hepatocytes: properties of the Ca2+ influx channel. American Journal of Physiology. 1997;273:C1526–1532. doi: 10.1152/ajpcell.1997.273.5.C1526. [DOI] [PubMed] [Google Scholar]
- Lewis RS. Store-operated calcium channels. Advances in Second Messenger Phosphoprotein Research. 1999;33:279–307. doi: 10.1016/s1040-7952(99)80014-7. [DOI] [PubMed] [Google Scholar]
- Lopez MG, Moro MA, Castillo CF, Artalejo CR, Garcia AG. Variable, voltage-dependent, blocking effects of nitrendipine, verapamil, diltiazem, cinnarizine and cadmium on adrenomedullary secretion. British Journal of Pharmacology. 1989;96:725–731. doi: 10.1111/j.1476-5381.1989.tb11874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma HT, Patterson RL, Van Rossum DB, Birnbaumer L, Mikoshiba K, Gill DL. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 2000;287:1647–1651. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- McCarron JG, Flynn ER, Bradley KN, Muir TC. Two Ca2+ entry pathways mediate InsP3-sensitive store refilling in guinea-pig colonic smooth muscle. Journal of Physiology. 2000;525:113–124. doi: 10.1111/j.1469-7793.2000.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiological Reviews. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- Matsuoka T, Nishizaki T, Nomura T. The voltage-dependent non-selective cation channel sensitive to the L-type calcium channel blocker efonidipine regulates Ca2+ influx in brain vascular smooth muscle cells. Biochemical and Biophysical Research Communications. 1997;240:484–487. doi: 10.1006/bbrc.1997.7624. [DOI] [PubMed] [Google Scholar]
- Missiaen L, De Smedt H, Droogmans G, Himpens B, Casteels R. Calcium ion homeostasis in smooth muscle. Pharmacology and Therapeutics. 1992;56:191–231. doi: 10.1016/0163-7258(92)90017-t. [DOI] [PubMed] [Google Scholar]
- Montell C. TRP trapped in fly signaling web. Current Opinion in Neurobiology. 1998;8:389–397. doi: 10.1016/s0959-4388(98)80066-4. [DOI] [PubMed] [Google Scholar]
- Morel N, Buryi V, Feron O, Gomez JP, Christen MO, Godfraind T. The action of calcium channel blockers on recombinant L-type calcium channel α1-subunits. British Journal of Pharmacology. 1998;125:1005–1012. doi: 10.1038/sj.bjp.0702162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neylon CB. Vascular biology of endothelin signal transduction. Clinical and Experimental Pharmacology and Physiology. 1999;26:149–153. doi: 10.1046/j.1440-1681.1999.03013.x. [DOI] [PubMed] [Google Scholar]
- Pacaud P, Loirand G, Gregoire G, Mironneau C, Mironneau J. Calcium-dependence of the calcium-activated chloride current in smooth muscle cells of rat portal vein. Pflügers Archiv. 1992;421:125–130. doi: 10.1007/BF00374818. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Store depletion and calcium influx. Physiological Reviews. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Phillips AM, Bull A, Kelly LE. Identification of a Drosophila gene encoding a calmodulin-binding protein with homology to the trp phototransduction gene. Neuron. 1992;8:631–642. doi: 10.1016/0896-6273(92)90085-r. [DOI] [PubMed] [Google Scholar]
- Scholfield CN, Curtis TM. Heterogeneity in cytosolic calcium regulation among different microvascular smooth muscle cells of the rat retina. Microvascular Research. 2000;59:233–242. doi: 10.1006/mvre.1999.2227. [DOI] [PubMed] [Google Scholar]
- Shmigol AV, Eisner DA, Wray S. Properties of voltage-activated [Ca2+]i transients in single smooth muscle cells isolated from pregnant rat uterus. Journal of Physiology. 1998;511:803–811. doi: 10.1111/j.1469-7793.1998.803bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal RR, Rohlf FJ. Biometry: The Principles and Practice of Statistics in Biological Research. 3. New York: W. H. Freeman & Co; 1995. [Google Scholar]
- Soldatov NM, Bouron A, Reuter H. Different voltage-dependent inhibition by dihydropyridines of human Ca2+ channel splice variants. Journal of Biological Chemistry. 1995;270:10540–10543. doi: 10.1074/jbc.270.18.10540. [DOI] [PubMed] [Google Scholar]
- Van Zwieten PA, Pfaffendorf M. Pharmacology of the dihydropyridine calcium antagonists: relationship between lipophilicity and pharmacodynamic responses. Journal of Hypertension. 1993;11(suppl. 6):S3–S11. [PubMed] [Google Scholar]
- Wayman CP, McFadzean I, Gibson A, Tucker JF. Two distinct membrane currents activated by cyclopiazonic acid-induced calcium store depletion in single smooth muscle cells of the mouse anococcygeus. British Journal of Pharmacology. 1996;117:566–572. doi: 10.1111/j.1476-5381.1996.tb15228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welling A, Ludwig A, Zimmer S, Klugbauer N, Flockerzi V, Hofmann F. Alternatively spliced IS6 segments of the α1C gene determine the tissue-specific dihydropyridine sensitivity of cardiac and vascular smooth muscle L-type Ca2+ channels. Circulation Research. 1997;81:526–532. doi: 10.1161/01.res.81.4.526. [DOI] [PubMed] [Google Scholar]
- Willmott NJ, Choudhury Q, Flower RJ. Functional importance of the dihydropyridine-sensitive, yet voltage-insensitive store-operated Ca2+ influx of U937 cells. FEBS Letters. 1996;394:159–164. doi: 10.1016/0014-5793(96)00939-8. [DOI] [PubMed] [Google Scholar]
- Wu SN, Jan CR, Li HF. Characteristics of store-operated Ca2+-permeable current in monocytic U937 cells. Chinese Journal of Physiology. 1997;40:115–120. [PubMed] [Google Scholar]
- Zheng W, Stoltefuss J, Goldmann S, Triggle DJ. Pharmacologic and radioligand binding studies of 1,4-dihydropyridines in rat cardiac and vascular preparations: stereoselectivity and voltage dependence of antagonist and activator interactions. Molecular Pharmacology. 1992;41:535–541. [PubMed] [Google Scholar]
- Zuhlke RD, Bouron A, Soldatov NM, Reuter H. Ca2+ channel sensitivity towards the blocker isradipine is affected by alternative splicing of the human α1C subunit gene. FEBS Letters. 1998;427:220–224. doi: 10.1016/s0014-5793(98)00425-6. [DOI] [PubMed] [Google Scholar]