

Abstract
We studied the functions of mitochondria and their hypoxic modulation in the brainstem slices of neonatal mice (postnatal day (P)6-11). The measurements were made in the preBötzinger complex (pBC), a part of the respiratory centre, and in the hypoglossal (XII) nucleus. Using a CCD camera, changes in the redox state were assessed from cell autofluorescence produced by NADH and FAD, while alterations in mitochondrial membrane potential (Δψ) and free Ca2+ concentration ([Ca2+]m) were obtained from fluorescence signals after loading the cells with Rh123 and Rhod-2, respectively.
In the pBC, the cells were functionally identified by correlating the oscillations in [NADH], [FAD], Δψ and [Ca2+]m with the respiratory motor output recorded simultaneously from XII rootlets. In the inspiratory cells, NADH fluorescence showed a brief decrease followed by a slow and long-lasting increase during one oscillation period. The initial decrease in NADH fluorescence was accompanied by an increase in FAD fluorescence and coincided with Δψ depolarization. The slow secondary increase in NADH fluorescence had a time course similar to that of the Rhod-2 signal, indicating the role of Ca2+ uptake by mitochondria in NAD and FADH reduction.
Brief (2-4 min) hypoxia reversibly abolished rhythmic changes in mitochondrial variables and brought them to new steady levels. In parallel, ATP-sensitive K+ (KATP) channels were activated and the respiratory output was depressed. The hypoglossal neurons showed much bigger increases in Δψ and [NADH] during hypoxia than the pBC neurons, which may explain their extreme vulnerability to hypoxia.
We show here that mitochondrial function can be monitored in vitro in neurons constituting the respiratory neural network in slice preparations. Since mitochondrial variables demostrate specific, stereotypic fluctuations during a respiratory cycle, we suggest that mitochondrial function is modulated by spontaneous activity in the respiratory network. Therefore mitochondrial depolarization and Ca2+ uptake can contribute to the biphasic reaction of the respiratory network during hypoxia.
It has long been known that the respiratory rhythm is generated in the brainstem; however, only recently (Smith et al. 1991) has the critical locus been localized to the preBötzinger complex (pBC), a functionally identified subregion of the mammalian ventrolateral medulla oblongata. Neural activity in this region is essential for maintaining rhythmic respiratory activity in brainstem neurons in vivo and in vitro. In vitro brainstem slice preparations from neonatal rodents (Smith et al. 1991) containing the pBC spontaneously generate rhythmic inspiratory activity in cranial hypoglossal (XII) motor nerves whose motoneurons are also present in the slice. Respiratory neurons normally exhibit rhythmic depolarizations with burst discharges, during which periodic changes of cytosolic Ca2+ (Frermann et al. 1999; Koshiya & Smith, 1999) and extracellular K+ and Cl− occur (Richter et al. 1978). Respiratory neurons seem to have a high metabolic demand to maintain ionic homeostasis through active ion transport as revealed by rhythmic changes in PO2 levels (Brockhaus et al. 1993) and the activity of ATP-sensitive K+ (KATP) channels (Haller et al. 1999).
The electrical activity of cells in the respiratory network demonstrates a biphasic reaction during hypoxia, which consists of augmentation and subsequent depression (Cherniack et al. 1970; Richter et al. 1991). Hypoxic augmentation of respiratory activity is due to a direct stimulation of respiratory neurons (Richter et al. 1991) and is possibly caused by membrane depolarization and/or activation of L-type Ca2+ channels (Mironov & Richter, 1998), triggering a concomitant rise in [Ca2+]i and release of glutamate and other transmitters (Richter et al. 1999). As the activity of KATP channels is greatly increased after oxygen withdrawal (Mironov et al. 1998), the respiratory depression might be associated with [ATP]i depletion and activation of other factors during hypoxia that converge on KATP channels (Mironov & Richter, 2000).
Ongoing spontaneous activity in the respiratory network needs continuous control to guarantee the stable periodic activity necessary for eupnoeic breathing. It appears that this network is more resistant to hypoxia than any other system in the brain (Richter et al. 2000), which implies the existence of specific mechanisms that protect the respiratory network against excitotoxicity. The hypoxic changes in the electrical activity of respiratory neurons may be related to changes in energy metabolism, but the functions of mitochondria in neurons within the respiratory network have not yet been documented. As mitochondrial depolarization and changes in [Ca2+]i homeostasis (see Duchen, 1999; Nicholls & Budd, 1999; Bernardi, 1999, for recent reviews) can represent important steps in the hypoxic response of neurons, we aimed to investigate the properties of mitochondria in pBC neurons. The major objectives of this study were: (1) to demonstrate in this slice preparation the applicability of the fluorescence probes that have previously been used to measure mitochondrial variables in isolated cells; (2) to establish whether mitochondrial variables in respiratory neurons oscillate and how these rhythmic changes are correlated with the compound activity of the respiratory network; (3) to monitor hypoxic changes in mitochondrial variables for pBC and hypoglossal motoneurons, which are differently sensitive to oxygen withdrawal. By measuring mitochondrial [NADH], [FAD], potential and [Ca2+], we found that there is indeed a correlation between the rhythmic respiratory output and mitochondrial variables in pBC cells. We also detected significantly greater Δψ changes in hypoglossal motoneurons, which may be responsible for their high vulnerability to damage during hypoxia. Thus, monitoring of mitochondrial variables in brain slice preparations provides an important tool for mapping neuronal activity and can be used in other in vitro preparations to elucidate the role of mitochondria during different physiological and pathophysiological conditions.
METHODS
Preparation
Experiments were performed on medullary slices from neonatal mice (P6-11) that contained the functional respiratory network generating spontaneous oscillatory activity. The preparation was obtained following the approach developed for rats by Smith et al. (1991) as applied to mice (Mironov et al. 1998). All animals were housed and cared for and, prior to experiments, killed in accordance with the recommendations of the European Commission (no. L358, ISSN 0378-6978) and the Committee for Animal Research, Göttingen University. Mice (NMRI) of both sexes were anaesthetized with ether and decapitated at the C3-C4 spinal level. A single transverse 700 μm thick slice containing the pBC and XII motor nucleus was cut from the brainstem, transferred to a recording chamber and continuously superfused at 28 °C with artificial cerebrospinal fluid (ACSF) that was saturated with carbogen (95 % O2+ 5 % CO2). ACSF contained (mm): 128 NaCl, 3 KCl, 1.5 CaCl2, 1.0 MgSO4, 21 NaHCO3, 0.5 NaH2PO4 and 30 d-glucose, pH adjusted to 7.4 with NaOH after saturating ACSF with carbogen. All salts were obtained from Sigma (Deisenhofen, Germany). To obtain a respiratory rhythm, the extracellular K+ concentration was elevated to 9 mm. Hypoxia was induced by replacing O2 in the bubbling gas mixture with N2, which brought tissue PO2 level close to zero (Mironov et al. 1998).
The perfusion chamber had a volume of 0.5 ml, and solutions were exchanged at a flow of 40-50 ml min−1. The perfusing solutions were delivered via stainless steel tubes to prevent the loss of dissolved gases and solution exchange was achieved by switching between two reservoirs (it took about 10 s to completely exchange the chamber contents). The respiratory output (Fig. 2) was monitored through a suction electrode from the hypoglossal (XII) nerve rootlets. Cell-attached patch recordings were obtained from superficial cells visualized with infrared differential interference contrast (IR-DIC) illumination as described previously (Mironov et al. 1998, 1999). The KATP channels were identified by their conductance (72 ± 4 pS), gating pattern and sensitivity to hypoxia, diazoxide and suphonylureas.
Figure 2. Simultaneous measurements of [NADH] and mitochondrial potential (as assessed by Rh123 fluorescence), inspiratory cell activity and respiratory motor output from XII nerve rootlets during hypoxia.
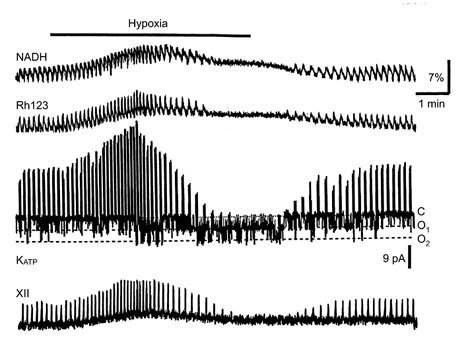
NADH and Rh123 fluorescence (upper two traces) were obtained as described in Methods. The membrane current (third trace) was recorded in cell-attached mode at a command potential of 0 mV (-60 mV in terms of membrane voltage). The patch was bathed by ‘symmetrical’ K+ concentrations (140 mm K+ in the pipette). The downward current deflections correspond to fluctuations in KATP channel activity between the closed (C) and two open (O1 and O2) levels and the upward deflections are inspiratory spikes. Note a close correspondence between the peaks in XII activity, NADH and Rh123 signals and the bursts of action potentials. NADH and Rh123 fluorescence are presented as relative increases (%) above the resting values.
Fluorescence measurements
Slices were viewed under a ×10 objective (Achroplan, NA 0.30), whereas the cells were viewed under a ×63 objective (Achroplan, NA 0.95). The optical recording system included a Zeiss Axioscope equipped with a monochromator light source (Till Photonics, Planegg, Germany). Images from a cooled CCD camera (MicroMax, Princeton Instruments, USA) were digitized (782 pixels ×512 pixels at 12-bit resolution) and collected using WinView software (Visitron System, Puchheim, Germany). MetaMorph software (Princeton Instruments) was used for image analysis.
Changes in Δψ and [Ca2+]m were measured by using the fluorescent dyes Rh123 and Rhod-2, respectively. In some experiments, mitochondria were also stained with MitoTracker Green. All dyes were from Molecular Probes (Leiden, The Netherlands). Apart from Rh123, which is water soluble, fresh solutions were made in dehydrated DMSO before each experiment and the detergent Pluronic (0.02 %) was added to the incubation medium. The slices were incubated at 28 °C in darkness with 10 μg ml−1 Rh123 for 10 min or with 1 μm MitoTracker Green for 30 min and then perfused with fresh ACSF for 60 min before experiments began. Rhod-2 AM (15 μm) was loaded for 30 min and washed for 2 h. The procedures used did not affect the functional properties of the slice after loading, since neither respiratory output nor cell viability was changed.
In optical recordings the cells were illuminated with light at wavelengths of 360 nm (NADH), 450 nm (FAD), 465 nm (MitoTracker Green) or 535 nm (Rh123 and Rhod-2). Selective excitation and emission were achieved by using UV-fluorescein or UV-rhodamine filter sets (AFanalysetechnik, Tübingen, Germany), consisting of dual-band beam splitters and emission filters. In the first case, the incident light at excitation wavelengths of 360 nm and 450 or 465 nm was directed to a dichroic mirror (510 mid-deflection) and the emission was collected using a wide-pass filter (535 ± 25 nm). A dichroic mirror with mid-reflection at 380 and 570 nm and a double transmittance emission filter (515 ± 15 and 600 ± 30 nm, respectively) were used for simultaneous measurement of UV-excited and rhodamine-based fluorescence signals of Rh123 or Rhod-2. To prevent dye bleaching, the light intensity was attenuated to 25 % by using a neutral density filter. Rh123 and Rhod-2 gave reproducible signals, which were in line with the changes in Δψ and [Ca2+]m expected from known mitochondrial properties (see Duchen, 1999; Nicholls & Budd, 1999; Bernardi, 1999, for recent reviews), indicating that they monitor changes in mitochondrial variables, but not in other intracellular organelles. The changes in all fluorescence signals (NADH, FAD, Rh123 and Rhod-2) are expressed as the percentage change in fluorescence over baseline ((ΔF/F)× 100). Means ±s.e.m. were compared using Student's t test for statistical analysis, with P < 0.05 being the criterion for statistical significance.
Rh123 and Rhod-2 have relatively broad excitation spectra, therefore they can potentially be excited with 465 nm light and emit at 535 nm, i.e. at wavelengths that were used to excite and to collect the fluorescence of MitoTracker Green. The extent of the possible bleeding between signals of Rh123 and MitoTracker Green was estimated as follows. Cells were first stained with MitoTracker Green and images were acquired using the UV-fluorescein or UV-rhodamine sets described above. Then the cells were stained with Rh123 and two images were again acquired. In comparison to the initial image, the mean signal at 600 nm (535 nm excitation) increased 8 to 10-fold (mean increase, 9.1 ± 0.5 times; n = 9, n here and below corresponds to the number of cells examined), indicating the major contribution of Rh123 fluorescence when the UV-rhodamine filter set was used. When cells were viewed using the UV-fluorescein filter set, after staining with Rh123, the signal at 535 nm (465 nm excitation) increased by only 0.11 ± 0.03 (n = 9), indicating a small contribution of Rh123 fluorescence to the signal generated by MitoTracker Green. Our data are similar to the results of Monteith & Blaustein (1999), who detected no mitochondrial fluorescence signal at 535 nm (488 nm excitation) in isolated smooth muscle cells loaded with Rhod-2, but a strong signal after applying MitoTracker Green.
The Ca2+ indicator dye Rhod-2 has been used to measure the [Ca2+] in intracellular organelles (Miyata et al. 1991; Minezaki et al. 1994; Babcock & Hille, 1998; Monteith & Blaustein, 1999). The delocalized positive charge of Rhod-2 and long incubation time after dye loading promote dye sequestration into mitochondria. Rhod-2 is a non-ratiometric dye, and calibration in terms of Ca2+ concentration is difficult and imprecise. Therefore, we have adopted the convention of most authors and refer to relative increases in mitochondrial Rhod-2 fluorescence in terms of increases in [Ca2+]m. After 10-15 min of application of ACSF containing 0.2 mm Mn2+, the diffuse staining in the cytoplasm disappeared, but the bright spots remained; therefore they were attributed to the dye localized within mitochondria. All measurements with Rhod-2 were made after the cytoplasmic signal was quenched with Mn2+. The use of Mn2+ has potential complications as it may influence mitochondrial calcium handling. The reported Mn2+ effects on the Ca2+ uniporter range from blockade to permeation (see Bernardi, 1999, for a recent review). When the Rhod-2 signals and their changes during hypoxia were recorded before and after Mn2+ quenching, the signals in the second trial had amplitudes smaller by 21 ± 3 % (n = 7), but the wave-form of the Rhod-2 oscillations in inspiratory neurons was unchanged, indicating no modification of [Ca2+]m kinetics by intracellular Mn2+. The hypoxic changes in Rhod-2 fluorescence measured before and after Mn2+ quenching were also similar. The absence of Mn2+ effects on mitochondrial calcium handling observed in this study is in line with data recorded from myocytes: the threshold for mitochondrial Ca2+ uptake was similar to that without Mn2+ quench (Zhou et al. 1998) and 20 μm Mn2+ only slightly inhibited Ca2+ uptake (Miyata et al. 1991).
RESULTS
Staining and identification of respiratory cells
The staining of slices with specific mitochondrial probes was uneven such that only some cells became visible (Fig. 1A). We believe that the majority of fluorescent cells were of neuronal origin, because they exhibited spikes when patched in the cell-attached mode and synaptic currents in the whole-cell mode (n = 21). The frequency of inspiratory bursts increased after elevation of [K+]o from 9 to 12 mm or after addition of 100 μm glutamate (authors' unpublished observations). In parallel, [Ca2+]m (as measured by Rhod-2 fluorescence) and Δψ (as measured by Rh123 fluorescence) slowly increased. Elevated [K+]o and glutamate also increased [Ca2+]i (as measured with Calcium green-2), suggesting that increases in [Ca2+]i underlay the changes in [Ca2+]m and Δψ.
Figure 1. Mitochondria-related signals in respiratory neurons.
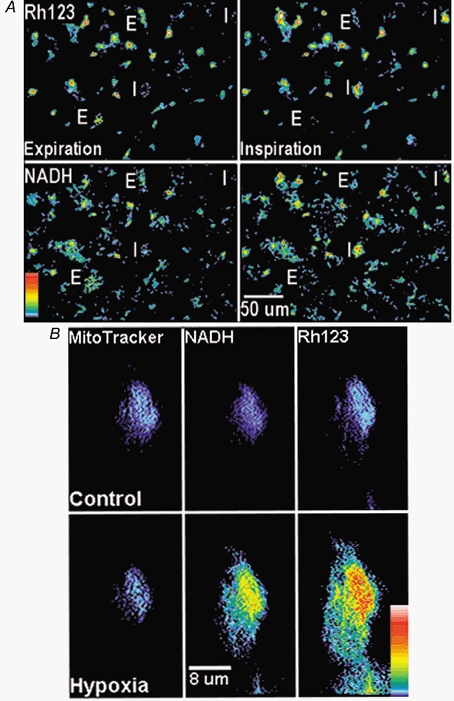
A, images of Rh123 (top) and NADH fluorescence (bottom) at low magnification obtained 2 s before the inspiratory burst began (Expiration, left) and 2 s after its completion (Inspiration, right). Two inspiratory and two expiratory neurons (marked I and E, respectively) were identified as demonstrating increases and decreases, respectively, in both Rh123 and NADH fluorescence signals caused by inspiratory motor output as recorded from XII nerve rootlets. The original 12-bit images were filtered using a 2 × 2 median space filter with MetaMorph software in order to remove scarce bright spots, which sometimes appeared after staining on the slice surface. B, double staining with MitoTracker Green and Rh123. After obtaining control images of MitoTracker Green, NADH and Rh123, the fluorescence being selectively excited and collected as described in Methods, hypoxia was applied for 3 min and the images were acquired again. The acquisition time was 2 s for each image. The cell was identified as inspiratory by correlating changes in mitochondrial variables with the respiratory output. In the images presented in the top and bottom panels, the background (defined as mean fluorescence level measured outside the slice) was subtracted. Vertical bar indicates relative fluorescence scale ΔF/F (white indicates highest fluorescence changes of 10 %).
Figure 1A shows images of Rh123 and NADH fluorescence at low magnification recorded before and after the inspiratory burst discharges. On the basis of changes in fluorescence generated by mitochondrial probes, cells were defined as silent (not marked in Fig. 1A), inspiratory (I) or expiratory (E), as the signals measured in I and E cells increased and decreased, respectively, during the inspiration phase of XII motoneuron activity. Such correlations can be expected from the patterns of electrical activity of these cell types (cf. Bianchi et al. 1995; Richter et al. 2000).
The intracellular distributions of Rh123 and Rhod-2 appeared inhomogeneous and correlated well with the patterns of NADH fluorescence (Fig. 1B). When mitochondria were double stained using either Rh123 (Fig. 1B) or Rhod-2 (data not shown) and then MitoTracker Green, all intracellular fluorescence patterns overlapped. When Rh123 (N = 3, N here and below corresponds to the number of slices examined) or MitoTracker Green (N = 4) was applied alone, the corresponding cell images also resembled the pattern of NADH fluorescence. Digital imaging of mitochondria is complicated by their submicrometre dimensions, but they are often clustered and form intracellular networks (Dedov & Roufogalis, 1999). We were not able to resolve individual mitochondria, but the strong correlation between the intracellular distribution of NADH, Rh123 and MitoTracker Green indicates their common target. The localization of MitoTracker Green and Rhod-2 trapped by mitochondria is also similar in isolated smooth muscle cells (Monteith & Blaustein, 1999). Figure 1B shows that after 3 min of hypoxia, the Rh123 fluorescence became much brighter while the distribution of the MitoTracker Green signal remained unchanged. The images of MitoTracker Green, when it was applied alone, did not change during 3 min of hypoxia (N = 4, data not shown). As the reference signal (MitoTracker Green) was constant during all treatments used in this study, this justifies the use of non-ratiometric dyes for monitoring mitochondrial variables.
Oscillations in mitochondrial variables
Figure 2 shows that rhythmic variations in NADH and Rh123 fluorescence signals in inspiratory cells correlated both with the respiratory motor output and with cell discharges monitored in cell-attached mode. Figures 3–5 show the changes during one oscillation cycle in greater detail. It can be seen that a typical NADH response consisted of a brief decrease in fluorescence followed by a slow and long-lasting increase that subsided before the next inspiratory burst began. The initial decrease in [NADH] occurred simultaneously with an increase in [FAD] Fig. 3) and coincided with Δψ depolarization (Fig. 4). The slow secondary increase in [NADH] (and corresponding [FAD] decrease, Fig. 3) had a time course similar to that of the [Ca2+]m increase as measured with Rhod-2 fluorescence (Fig. 5), indicating that Ca2+ entry into the mitochondria is a prerequisite for NAD and FAD reduction.
Figure 3. Modulation of autofluorescence and the respiratory (XII) motor output during hypoxia.
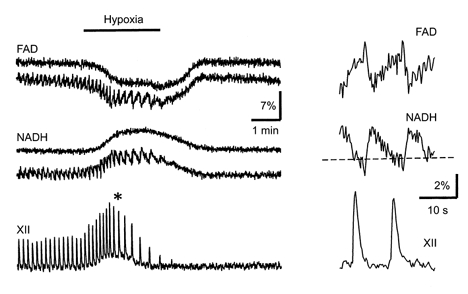
The fluorescence signals of NADH and FAD at excitation wavelengths of 360 and 450 nm, respectively, were recorded as described in Methods. The data obtained for two representative pBC cells (one with and one without periodic NADH and FAD signal changes) are shown. The measurements at the time marked by an asterisk on the left are presented on an expanded time scale on the right. The dashed baselines on the right in this figure and in Figs 4 and 5 show the steady values of NADH fluorescence immediately before the next inspiratory burst began. They were obtained by averaging from 4 to 6 individual oscillations. Note also the different calibration for relative fluorescence changes in the two panels.
Figure 5. Mitochondrial [Ca2+] and [NADH] as measured in pBC cells during hypoxia.
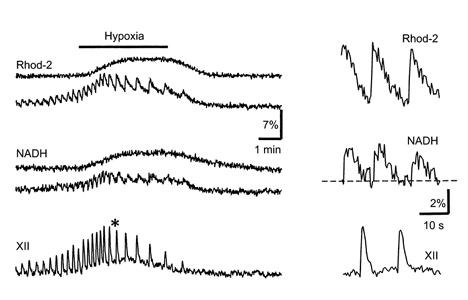
The cells were stained with Rhod-2 to monitor [Ca2+]m changes and the fluorescence signals were collected as described in Methods. The results for two representative cells are shown, one with and one without [Ca2+]m and NADH signal oscillations, which correlated with respiratory (XII) output. The data at the time marked by an asterisk on the left are presented on an expanded time scale on the right.
Figure 4. Changes in Δψ and [NADH] during hypoxia.
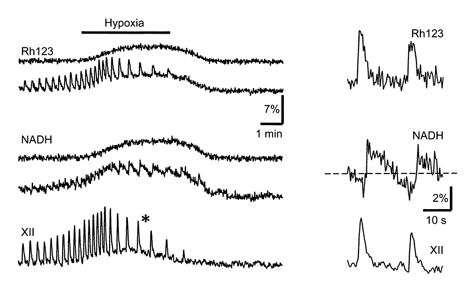
The pBC cells were stained with Rh123 and the fluorescence signals were collected at 515 and 600 nm using excitation at 360 and 535 nm, corresponding to the absorption maxima of Rh123 and NADH, respectively (see also Methods). The results for two different cells are presented, one with and one without Rh123 and NADH signal oscillations, which correlated with respiratory (XII) motor output. The measurements at the time marked by an asterisk on the left are shown on an expanded time scale on the right.
Electrical activity in the respiratory network demonstrates a biphasic response during hypoxia, consisting of initial augmentation and subsequent depression (Cherniack et al. 1970; Richter et al. 1991). Figure 2 shows that the depression developed in parallel with the activation of KATP channels. On average, the open probability (Popen) slowly increased from 0.05 ± 0.02 to 0.18 ± 0.04 after 4 min of hypoxia (n = 12). The mitochondrial depolarization preceded the activation of KATP channels, suggesting that the Δψ decrease caused [ATP]i depletion, leading to a diminished production of ATP (McCormack et al.1990). Figures 3–5 present measurements of different pairs of mitochondrial variables each made in two representative cells, one with and one without rhythmic activity. It can be seen that slow increases in the NADH fluorescence signal and decreases in the FAD fluorescence signal during hypoxia developed simultaneously (Fig. 3) and corresponded well with the time courses of Δψ and [Ca2+]m changes (Figs 4 and 5). Applications of cyanide and hypoxia had similar effects on NADH and FAD signals, and either treatment occluded the action of the other (n = 9, data not shown), suggesting a mitochondrial origin of these signals (Duchen & Biscoe, 1992). The changes in mitochondrial variables were similar during two subsequent hypoxic episodes performed in ACSF containing 3 and 9 mm[K+]o(n = 4, data not shown), i.e. when the rhythmic activity was absent and present, respectively.
Differences in hypoxic responses in brainstem neurons
The responses of hypoglossal (XII) motoneurons during hypoxia (Fig. 6B) were typically larger than those recorded in pBC cells. The relative increases in Rh123 fluorescence during hypoxia were 86 ± 7 % for XII cells and 36 ± 5 % for pBC cells. The Rh123 signals were calibrated by using the uncoupler FCCP (1 μm). This protonophore brings the Rh123 signal to a maximum and the NADH signal to a minimum, which is useful to normalize the hypoxic changes in Δψ and [NADH] (Duchen & Biscoe, 1992). The mean values of the ratio of the change in [NADH] during hypoxia to that in the presence of FCCP (Δ[NADH]hypoxia/Δ[NADH]FCCP) for inspiratory and non-respiratory pBC cells were 0.19 ± 0.04 (n = 6) and 0.18 ± 0.03 (n = 8), respectively. In contrast, in hypoglossal cells this ratio was 0.44 ± 0.09 (n = 11). The values for pBC cells are smaller than those measured in chromaffin cells (2.70; Duchen, 1992), rat sensory neurons (1.08; Duchen, 1992), hepatocytes (0.72; Nieminen et al. 1997), type I cells of the carotid body (2.70; Duchen & Biscoe, 1992) and cardiomyocytes (0.50; Brandes & Bers, 1996, who also estimated the ratio [NADH]/[NAD+]= 0.63 at rest). This may be due to differences between PO2 levels in isolated cells and those in slices, because the latter are typically smaller than those in the perfusing solution and decay steeply with distance from the slice surface (Brockhaus et al. 1993).
Figure 6. Different responses of mitochondrial variables in pBC (A) and hypoglossal (XII, B) cells to hypoxia.
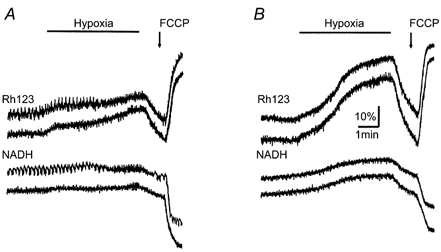
NADH and Rh123 signals were acquired as described in Methods. FCCP (1 μm) was applied at the end of the experiment to estimate Δψ and the relative [NADH] changes during hypoxia as described in Results. Note the much larger mitochondrial depolarization during hypoxia in XII cells.
FCCP was toxic in this preparation and therefore it was applied to a given slice only once. Wash-out of the protonophore with fresh ACSF did not restore rhythmic activity and the cells were barely viable. The deleterous effects of FCCP can be attributed to extensive [ATP]i depletion and concomitant cell damage (Nicolls & Budd, 1999), since during electrophysiological recordings gigaseals were suddenly lost several minutes after FCCP addition to the bath. Popen values for KATP channels measured shortly before this occurred were 0.29 ± 0.03 vs. 0.09 ± 0.02 in control (n = 9).
Inspiratory bursts, whose frequency increases during hypoxic augmentation, should lead to enhancement of [ATP]i consumption due to activation of different ion pumps, operating to restore intracellular ion homeostasis. An increase in the [ADP]i/[ATP]i ratio can contribute to Δψ depolarization due to increases in the ATP-producing activity of F1F0 ATP synthase, as this will increase H+ influx into mitochondria. To test this mechanism, we appled oligomycin (5 μg ml−1) to block ATP synthase activity. The effectiveness of bath-applied oligomycin was evident from the slight, irreversible decrease in Rh123 signal by 5.3 ± 0.9 % (n = 4), indicating weak Δψ hyperpolarization. However, the rhythmic changes in Δψ, [Ca2+]m and autofluorescence persisted, indicating that the oscillations were probably determined by Ca2+ uptake caused by rhythmic [Ca2+]i transients due to network respiratory activity. The steady changes in these variables produced by hypoxia had the same amplitude as in control (n = 4, data not shown). The long-term effects of oligomycin resembled those of FCCP: KATP channel activity increased and the motoneuron output was irreversibly lost. Both effects probably indicate a significant [ATP]i depletion due to inhibition of mitochondrial ATP production.
DISCUSSION
The present view on mitochondria is that their multiple influences on cell functions are determined not only by ATP synthesis, but also by their role in maintaining and regulating [Ca2+]i homeostasis (see Duchen, 1999; Nicholls & Budd, 1999; Bernardi, 1999, for recent reviews). Mitochondria accumulate Ca2+ when [Ca2+]i rises only slightly above resting levels, and stimulus-induced [Ca2+]i elevations as small as 300 nm increase [Ca2+]m (Colegrove et al. 2000). Thus, mitochondrial Ca2+ transport can be activated in situ faster and at much lower [Ca2+]i than previously thought (McCormack et al. 1990). In neurons, Ca2+ uptake by mitochondria is usually followed by rapid re-release, which shapes the time course of [Ca2+]i transients during synaptic activity (Babcock & Hille, 1998). Prolongation of elevated [Ca2+]i levels was suggested to underlie many events that accompany synaptic plasticity (Zucker, 1999). Ca2+ entry into mitochondria can activate rate-limiting steps in aerobic metabolism, thus coupling energy demands to ATP production (McCormack et al. 1990). In cardiac cells, for example, rapid increases in [Ca2+]m and activation of Ca2+-sensitive mitochondrial dehydrogenases are synchronized to the rising phase of the [Ca2+]i spikes, indicating the possibility of regulating the Krebs cycle through Ca2+ uptake by mitochondria during physiological stimulation (Szalai et al. 2000).
The aim in the present investigation was to determine whether coupling exists between bursting cell activity and mitochondrial variables in the respiratory network. The dyes used in the present study were non-ratiometric, but possible changes in cell volume, which may occur during longer periods of hypoxia (Haller et al. 1998), seemed not to influence fluorescence measurements during brief hypoxic episodes for the following reasons: (a) the slow signals changed in either direction: NADH and Rh123 fluorescence increased during hypoxia, whereas that of FAD decreased; conversely, after FCCP addition, Rh123 fluorescence increased while that of NADH decreased; (b) the fluorescence of MitoTracker Green was practically unchanged after 3 min of hypoxia. As in chemoreceptor cells (Duchen & Biscoe, 1992), in this preparation the responses to hypoxia and cyanide were mutually exclusive, indicating the mitochondrial localization of enzyme(s) that mediate the hypoxia-induced increase in [NADH]. This suggestion is also suported by changes in FAD fluorescence, which were exactly opposite to the changes in NADH fluorescence.
Oscillations in mitochondrial variables
In the present study, rhythmic changes in Δψ, [Ca2+]m, [NADH] and [FAD] were observed in inspiratory neurons. The oscillatory responses of [NADH] and [FAD] were biphasic, thus resembling depolarization-induced transients in sensory neurons (Duchen, 1992), chemoreceptor cells (Duchen & Biscoe, 1992) and cardiomyocytes (Brandes & Bers, 1996). These studies showed that brief depolarization of the cell membrane raised [Ca2+]i, depolarized Δψ and produced biphasic changes (fast decrease and slow increase) in the NADH autofluorescence. The FAD signal showed a brief initial increase (an increased oxidation), followed by a prolonged decrease, as expected from an increased reduction of the mitochondrial coenzymes. In sensory neurons (Duchen, 1992), the changes in Δψ (measured with Rh123) were dependent on extracellular Ca2+ and reflected those of the Ca2+ current.
On the other hand, Brandes & Bers (1996) attributed depolarization-induced [NADH] changes in cardiomyocytes to a falling [ATP]i/[ADP]i ratio. Previously we observed cyclic modulation of KATP channel activity in respiratory neurons (Haller et al. 1999), which was ascribed to depletion of submembrane [ATP]i following each inspiratory burst. On the other hand, Frermann et al. (1999) and Koshiya & Smith (1999) detected rhythmic [Ca2+]i activities in inspiratory neurons that were in phase with rhythmic membrane depolarizations and bursts of action potentials. Thus, two factors, [ATP]i and [Ca2+]i, may potentially be responsible for the cyclic variations in mitochondrial function observed in the present study.
Ca2+ uptake by mitochondria plays a key role in regulating metabolism and bursting electrical activity in pancreatic β-cells. Its influence was analysed by Magnus & Keizer (1998), who assumed that the cycling of mitochondrial Ca2+ is induced by Ca2+ influx into the cytosol during the active phase, leading to a transient rise in the activity of the Ca2+ uniporter and Na+-Ca2+ exchange, and causes Δψ depolarization. Although small, this depolarization is sufficient to diminish oxygen consumption and thus the driving force for ATP synthesis. [ATP]i depletion is probably responsible for the oscillatory behaviour in mitochondrial variables observed in cardiomyocytes (O'Rourke et al. 1994), pancreatic β-cells (Pralong et al. 1994) and hepatocytes (Robb-Gaspers et al. 1998). For example, during metabolic oscillations in heart cells (O'Rourke et al. 1994), a decrease in mitochondrial [NADH] precedes an increase in KATP conductance and ADP release from a caged precursor by flash photolysis can induce such oscillations.
However, the absence of an effect of oligomycin, a blocker of ATP synthetase, on mitochondrial [NADH] transients in sensory (Duchen, 1992) and respiratory neurons (this study) indicates that [ATP]i fluctuations may be less important than elevations of [Ca2+]i, leading to Δψ depolarization and [Ca2+]m increase. Indeed, the initial decrease in [NADH] and the increase in [FAD] during oscillations (Fig. 3) occurred simultaneously with mitochondrial depolarization (Fig. 4), whereas the late increase in [NADH] and the decrease in [FAD] coincided with the delayed rise in [Ca2+]m (Fig. 5). As it is difficult to reliably remove [Ca2+]o in slice preparations, the Ca2+ dependence of the autofluorescence was not tested, but the changes in Δψ, [NADH], [FAD] and [Ca2+]m are all consistent with a direct relationship, suggesting that the rise in [Ca2+]i depolarizes mitochondria.
Ongoing activity of respiratory neurons periodically increases oxygen consumption (Brockhaus et al. 1993) and elevates [Ca2+]m (this study). Since mitochondrial depolarization precedes both [Ca2+]m and [NADH] increases, the simplest scenario would involve Ca2+ uptake via an electrogenic Ca2+ uniporter. The dominant mechanism probably corresponds to the rapid uptake (Gunter et al. 1998). In sensory neurons (Duchen, 1992), the blockade of the Ca2+ uniporter with Ruthenium Red prevented the changes in autofluorescence.
The effects of hypoxia in the respiratory centre
It was described previously (Mironov et al. 1998, 1999) that the respiratory motor output shows no appreciable deterioration after 10-20 sequential hypoxic episodes. This observation and the comparison of [NADH] changes during hypoxia (see Results) with those measured in cells isolated from different tissues (Duchen, 1992; Duchen & Biscoe, 1992; Brandes & Bers, 1996; Nieminen et al. 1997) indicate that the brainstem in vitro (and possibly in vivo) may operate at moderate O2 levels (see also Brockhaus et al. 1993). This feature may determine the relative robustness of pBC neurons (and correspondingly of the whole respiratory network) to hypoxia. The pBC cells with and without respiratory activity responded to hypoxia similarly, but in comparison to other CNS neurons (cf. Nicholls & Budd, 1999) they seem to be less vulnerable. However, this is not a property of the brainstem in general, since XII motoneurons underwent much larger Δψ changes during hypoxia, demonstrating almost complete mitochondrial depolarization. Thus, these findings may explain the low tolerance of hypoglossal motoneurons to oxygen withdrawal, which was previously observed in electrophysiological studies in vivo (Pierrefiche et al. 1997).
The relative insensitivity of the respiratory network to hypoxia may also be influenced by another temporal profile of neurotransmitter release, as compared to other CNS areas. Hypoxia and/or ischaemia usually trigger the release of glutamate, which is accompanied by a massive Ca2+ influx (Duchen, 1999; Nicholls & Budd, 1999), leading to a large Δψ depolarization and Ca2+ overload of mitochondria. Hypoxic glutamate release in the cat brainstem in vivo is only transient (Richter et al. 1999), and the excitotoxic action of glutamate via NMDA and AMPA/kainate ionotropic receptors, which are both important in the rhythmogenesis (Bianchi et al. 1995), may well be compensated by the inhibitory action of other neurotransmitters (adenosine, 5-HT and GABA). These neurotransmitters are also released during hypoxia (Richter et al. 1999) and may act as protective neuromodulators via corresponding metabotropic receptors (Mironov et al. 1999; Mironov & Richter, 2000). Additional evidence for the absence of massive Ca2+ overload in brainstem cells during hypoxia is that a mitochondrial permeability transition (see Bernardi, 1999, for a recent review) does not participate in the respiratory hypoxic response as indicated by the absence of the effects of cyclosporine A in this preparation (authors' unpublished data).
Acknowledgments
The study was supported by SFB 406 and INTAS grant 99-01915. The authors thank Nicole Hartelt for excellent technical assistance and Peter Lalley for critical reading of the manuscript.
References
- Babcock DF, Hille B. Mitochondrial oversight of cellular Ca2+ signaling. Current Opinion in Neurobiology. 1998;8:398–404. doi: 10.1016/s0959-4388(98)80067-6. [DOI] [PubMed] [Google Scholar]
- Bernardi P. Mitochondrial transport of cations: Channels, exchangers and permeability transition. Physiological Reviews. 1999;79:1127–1150. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Bianchi AL, Denavit-Saubie M, Champagnat J. Central control of breathing in mammals: neuronal circuitry, membrane properties, and neurotransmitters. Physiological Reviews. 1995;75:1–45. doi: 10.1152/physrev.1995.75.1.1. [DOI] [PubMed] [Google Scholar]
- Brandes R, Bers DM. Increased work in cardiac trabeculae causes decreased mitochondrial NADH fluorescence followed by slow recovery. Biophysical Journal. 1996;71:1024–1035. doi: 10.1016/S0006-3495(96)79303-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhaus J, Ballanyi K, Smith JC, Richter DW. Microenvironment of respiratory neurons in the in vitro brainstem-spinal cord of neonatal rats. Journal of Physiology. 1993;462:421–445. doi: 10.1113/jphysiol.1993.sp019562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherniack NS, Edelman NH, Lahiri S. Hypoxia and hypercapnia as respiratory stimulants and depressants. Respiration Physiology. 1970;11:113–126. doi: 10.1016/0034-5687(70)90107-6. [DOI] [PubMed] [Google Scholar]
- Colegrove SL, Albrecht MA, Friel DD. Quantitative analysis of mitochondrial Ca2+ uptake and release pathways in sympathetic neurons. Reconstruction of recovery after depolarization-evoked [Ca2+]i elevations. Journal of General Physiology. 2000;115:371–388. doi: 10.1085/jgp.115.3.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedov VN, Roufogalis BD. Organisation of mitochondria in living sensory neurons. FEBS Letters. 1999;456:171–174. doi: 10.1016/s0014-5793(99)00951-5. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Ca2+-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochemical Journal. 1992;283:41–50. doi: 10.1042/bj2830041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. Journal of Physiology. 1999;516:1–17. doi: 10.1111/j.1469-7793.1999.001aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Biscoe TJ. Mitochondrial function in type I cells isolated from rabbit arterial chemoreceptors. Journal of Physiology. 1992;450:13–31. doi: 10.1113/jphysiol.1992.sp019114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frermann D, Keller BU, Richter DW. [Ca2+]i oscillations in rhythmically active respiratory neurons in the brainstem of the mouse. Journal of Physiology. 1999;515:119–131. doi: 10.1111/j.1469-7793.1999.119ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, Buntinas L, Sparagna GC, Gunter KK. Ca2+ transport mechanisms of mitochondria and Ca2+ uptake from physiological-type Ca2+ transients. Biochimica et Biophysica Acta. 1998;1366:5–15. doi: 10.1016/s0005-2728(98)00117-0. [DOI] [PubMed] [Google Scholar]
- Haller M, Mironov SL, Karschin A, Richter DW. KATP channels in respiratory neurons of newborn mice. Pflügers Archiv. 1999;437S:R68. [Google Scholar]
- Haller M, Mironov SL, Richter DW. Changes in the IR transmittance of the respiratory center as induced by hypoxia. Pflügers Archiv. 1998;434S:R34. [Google Scholar]
- Koshiya N, Smith JC. Neuronal pacemaker for breathing visualized in vitro. Nature. 1999;400:360–363. doi: 10.1038/22540. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian mitochondrial metabolism. Physiological Reviews. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- Magnus G, Keizer J. Model of beta-cell mitochondrial calcium handling and electrical activity. II. Mitochondrial variables. American Journal of Physiology. 1998;274:C1174–1184. doi: 10.1152/ajpcell.1998.274.4.C1174. [DOI] [PubMed] [Google Scholar]
- Minezaki KK, Suleiman MS, Chapman RA. Changes in mitochondrial function induced in isolated guinea-pig ventricular myocytes by calcium overload. Journal of Physiology. 1994;476:459–471. doi: 10.1113/jphysiol.1994.sp020147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov SL, Langohr K, Haller M, Richter DW. Hypoxia activates ATP-dependent potassium channels in inspiratory neurons of neonatal mice. Journal of Physiology. 1998;509:755–766. doi: 10.1111/j.1469-7793.1998.755bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov SL, Langohr K, Richter DW. A1 adenosine receptors modulate respiratory activity of the neonatal mouse via the cAMP-mediated signalling pathway. Journal of Neurophysiology. 1999;81:241–255. doi: 10.1152/jn.1999.81.1.247. [DOI] [PubMed] [Google Scholar]
- Mironov SL, Richter DW. L-type Ca2+ channels in inspiratory neurones and their modulation by hypoxia. Journal of Physiology. 1998;512:75–87. doi: 10.1111/j.1469-7793.1998.075bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov SL, Richter DW. Intracellular signalling pathways modulate KATP channels in inspiratory brainstem neurons and their hypoxic activation: Involvement of metabotropic receptors, G-proteins and cytoskeleton. Brain Research. 2000;853:60–67. doi: 10.1016/s0006-8993(99)02234-9. [DOI] [PubMed] [Google Scholar]
- Miyata HH, Silverman HS, Sollott SJ, Lakatta EG, Stern MD, Hansford RG. Measurement of mitochondrial free Ca2+ concentration in living single ventricular cardiac myocytes. American Journal of Physiology. 1991;261:H1123–1139. doi: 10.1152/ajpheart.1991.261.4.H1123. [DOI] [PubMed] [Google Scholar]
- Monteith GR, Blaustein MP. Heterogeneity of mitochondrial matrix free Ca2+: resolution of Ca2+ dynamics in individual mitochondria in situ. American Journal of Physiology. 1999;276:C1193–1204. doi: 10.1152/ajpcell.1999.276.5.C1193. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiological Reviews. 1999;80:315–345. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Nieminen AL, Byrne AM, Herman B, Lemasters JJ. Mitochondrial permeability transition in hepatocytes induced by t-BOOH: NAD(P)H and reactive oxygen species. American Journal of Physiology. 1997;272:C1286–1294. doi: 10.1152/ajpcell.1997.272.4.C1286. [DOI] [PubMed] [Google Scholar]
- O'Rourke B, Ramza BM, Marban E. Oscillations of membrane current and excitability driven by metabolic oscillations in heart cells. Science. 1994;265:962–966. doi: 10.1126/science.8052856. [DOI] [PubMed] [Google Scholar]
- Pierrefiche O, Bischoff AM, Richter DW, Spyer KM. Hypoxic response of hypoglossal motoneurons in the in vivo cat. Journal of Physiology. 1997;505:785–795. doi: 10.1111/j.1469-7793.1997.785ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pralong WF, Spat A, Wollheim CB. Dynamic pacing of cell metabolism by intracellular Ca2+ transients. Journal of Biological Chemistry. 1994;269:27310–27314. [PubMed] [Google Scholar]
- Richter DW, Bischoff AM, Anders K, Bellingham M, Windhorst U. Response of the medullary respiratory network of the cat to hypoxia. Journal of Physiology. 1991;443:231–256. doi: 10.1113/jphysiol.1991.sp018832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter DW, Camerer H, Sonnhof U. Changes in extracellular potassium during the spontaneous activity of medullary respiratory neurones. Pflügers Archiv. 1978;376:139–149. doi: 10.1007/BF00581577. [DOI] [PubMed] [Google Scholar]
- Richter DW, Mironov SL, Büsselberg D, Bischof AM, Lalley PM, Wilken B. Respiratory rhythm generation: Plasticity of a neuronal network. Neuroscientist. 2000;6:188–205. [Google Scholar]
- Richter DW, Schmidt-Garcon P, Pierrefiche O, Bischof AM, Lalley PM. Neurotransmitters and neuromodulators controlling the hypoxic respiratory response in anaesthetized cats. Journal of Physiology. 1999;514:567–578. doi: 10.1111/j.1469-7793.1999.567ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb-Gaspers LD, Rutter GA, Burnett P, Hajnoczky G, Denton RM, Thomas AP. Coupling between cytosolic and mitochondrial oscillations: role in the regulation of hepatic metabolism. Biochimica et Biophysica Acta. 1998;1366:17–32. doi: 10.1016/s0005-2728(98)00118-2. [DOI] [PubMed] [Google Scholar]
- Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL. Pre-Bötzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science. 1991;254:726–729. doi: 10.1126/science.1683005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalai G, Csordas G, Hantash BM, Thomas AP, Hajnoczky G. Ca signal transmission between ryanodine receptors and mitochondria. Journal of Biological Chemistry. 2000;275:15305–15013. doi: 10.1074/jbc.275.20.15305. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Matlib MA, Bers DM. Cytosolic and mitochondrial Ca2+ signals in patch clamped mammalian ventricular myocytes. Journal of Physiology. 1998;507:379–403. doi: 10.1111/j.1469-7793.1998.379bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Ca2+ and activity-dependent synaptic plasticity. Current Opinion in Neurobiology. 1999;9:305–313. doi: 10.1016/s0959-4388(99)80045-2. [DOI] [PubMed] [Google Scholar]