

Abstract
Vascular endothelial growth factor (VEGF) chronically increases microvascular permeability, compliance and vessel diameter. To determine the signalling pathways by which VEGF exerts these effects, we investigated the role of Ca2+ influx and mitogen-activated protein kinase (MAPK) phosphorylation on the increase in hydraulic conductivity (Lp), diameter and compliance in mesenteric microvessels in the anaesthetised frog (Rana species).
The VEGF-mediated chronically increased permeability was attenuated by co-perfusion of VEGF with 5 mm NiCl2, previously shown to inhibit Ca2+ influx. MAPK phosphorylation inhibition by PD98059 did not affect the chronic increase in Lp. To determine whether other agonists which increased Ca2+ influx also chronically increased Lp, the effect of ATP perfusion on chronic Lp was measured. ATP perfusion also chronically increased Lp. The chronic increase in Lp was therefore dependent on an initial transient Ca2+ influx, and not MAPK activation, and was not unique to VEGF stimulation.
Inhibition of Ca2+ influx did not inhibit the increase in microvascular diameter or compliance brought about by VEGF. Both these increases were inhibited by PD98059. The VEGF-mediated increase in compliance and diameter was therefore dependent on MAPK activation, not on Ca2+ influx.
The chronic increase in Lp stimulated by VEGF perfusion 24 h previously was reduced when the vessel was perfused with 5 mm NiCl2. The sustained, high Lp was therefore dependent on Ca2+ influx.
The endothelial cell calcium concentration ([Ca2+]i) of vessels previously perfused with VEGF or ATP, and with a chronically increased Lp, was not significantly increased compared to [Ca2+]i of endothelial cells in vessels before agonist perfusion
These experiments show that VEGF acts through different pathways to stimulate increased permeability and compliance. The data are consistent with the hypothesis that VEGF chronically increases Lp through an acute stimulation of Ca2+ influx, but increases compliance and diameter by acute stimulation of the MAPK signalling pathway. They also suggest that the increase in Lp is dependent on a sustained Ca2+ influx, even though the endothelial [Ca2+]i is not raised.
Vascular endothelial growth factors (VEGFs) are endogenous vascular peptides that result in angiogenesis, vasodilatation and increased microvascular permeability in vivo (Bates et al. 1999). The overall action of the most well characterised and widely distributed member of this family of peptides, known as VEGF or VEGF-A, is therefore to increase delivery of nutrients from blood to tissue. They are critically necessary for development of the cardiovascular system, wound healing and other physiological circumstances requiring new blood vessel growth (Ferrara & Bunting, 1996). Various forms of VEGFs are also over-expressed in many pathological conditions that are associated with new vessel formation, including tumour growth, arthritis and diabetes (Dvorak et al. 1995). Despite the fact that the permeability-enhancing properties of VEGF were first suggested during its initial isolation and characterisation in 1983 (Senger et al. 1983), and despite the importance of this factor in both health and disease, the mechanism by which it exerts its effects are still being elucidated. VEGF is known to stimulate endothelial cells in culture to produce matrix metalloproteinases (MMPs) that break down their surrounding extracellular matrix, to form gaps in the vessel wall, to invade the surrounding tissue, and to undergo cell division (Ferrara & Bunting, 1996). VEGF has also been shown to increase vascular permeability in a biphasic manner - a transient increase lasting no more than a few minutes, and a chronic sustained increase in vascular permeability that is apparent 24 h after stimulation with VEGF (Bates & Curry, 1996). The acute, transient phase has been shown to be dependent on calcium influx (Bates & Curry, 1997) through a calcium store-independent mechanism (Pocock et al. 2000). It has also been shown that the magnitude of the chronic increase in permeability is correlated with the magnitude of the acute transient phase (Bates, 1998). However, it is not known if these two phases are causally related. There are other agonists which can also stimulate an acute increase in permeability (measured as hydraulic conductivity, Lp) through an acute increase in calcium influx. These include ATP, histamine and bradykinin (He et al. 1996). It is not known whether these agents can also result in a chronically increased Lp.
VEGF has also been shown to chronically increase microvascular compliance and diameter in non-dilatory microvessels (capillaries and post-capillary venules) (Bates, 1998). It has been hypothesised that this increase in diameter and compliance may be associated with VEGF-mediated production of MMPs. These proteins break down the surrounding basement membrane and hence increase the compliance of the vessels. In endothelial cells in culture, and in mesenteric tissue in vivo it has been shown that VEGF stimulates activation of mitogen-activated protein kinase (MAPK) by raf activation of MAPK and ERK (extracellular signal-related kinase) kinase (MEK) (D'Angelo et al. 1995). Stimulation of this raf-MEK- MAPK pathway is one mechanism for MMP production in endothelial cells (Ridley et al. 1997). However, whether this is the cause of the functional changes described for vessels exposed to VEGF has not been shown to be the case, and the signalling pathways through which changes in permeability, compliance and diameter are brought about are therefore not known. In order to investigate the signalling pathways for both the chronic increase in permeability and the chronic increase in microvascular compliance and diameter, we have determined the effect of inhibiting two well-characterised signalling pathways, calcium influx and MAPK activation on the VEGF-mediated microvascular permeability, diameter and compliance.
METHODS
Frog preparation
All anaesthetic and surgical procedures were carried out according to local and national guidelines. All experiments were carried out as previously described on male leopard frogs (Rana temporaria or R. pipiens, 20-35 g) supplied by Blades (UK), or JM Hazen (VT, USA). All chemicals were purchased from Sigma Chemical Company unless otherwise specified. The animals were anaesthetised by immersion in 1 mg ml−1 MS222 (3-aminobenzoic acid ethyl ester) in water or injection into the dorsal lymph sac of 1 mg ml−1 MS222 in frog Ringer solution (111 mm NaCl, 2.4 mm KCl, 1 mm MgSO4, 1.1 mm CaCl2, 0.20 mm NaHCO3, 2.63 mm Hepes acid and 2.37 mm Hepes sodium salt). The pH of this solution was 7.40 ± 0.02 at room temperature. The animal was laid supine and the limbs lightly secured. A small incision (8-10 mm) was made in the right lateral skin and muscular body wall and the distal ileum draped over a quartz pillar. The mesenteric microvessels were visualised under a Leica inverted microscope (Leica DMIL). A video camera (Panasonic WVBP32, 8 mm) attached to the top of the microscope allowed binocular visualisation of the entire vessel and simultaneous recording of a 180-270 μm segment. The video was connected through an electronic timer (ForA VT33) to a video cassette recorder (Panasonic AG7350, Panasonic). The anaesthesia was maintained by continuous superfusion of the mesentery with 1-2 mg ml−1 MS222 in Ringer solution. All experiments were done at room temperature (20-22 °C).
Measurement of Lp
The Lp of isolated perfused mesenteric microvessels was measured using the Landis micro-occlusion method previously described (Michel et al. 1974), which has been extensively discussed in the literature (Curry et al. 1983) and adapted to measure rapid as well as chronic changes in Lp (Bates & Curry, 1996). Baseline Lp was defined as the conductivity during perfusion with 1 % bovine serum albumin (BSA) in frog Ringer solution, adjusted to pH 7.4 with 0.115 mm NaOH. Microvessels were selected that had flowing blood, no white cells adhering to the wall, and a length of at least 800 μm with no side branches. Microvessels chosen for Lp measurement were either true capillaries (divergent flow at one end and convergent at the other), or first order venules (convergent flow from two true capillaries at one end and convergent flow at the other), and had a diameter of 12-30 μm. The vessel was cannulated with a bevelled glass micropipette filled with 1 % BSA in frog Ringer solution and rat red blood cells as flow markers. The rat red cells were collected by direct cardiac puncture of halothane-anaesthetised rats (5 % halothane by inhalation), and washed three times in frog Ringer solution before use. Rats were killed by cervical dislocation while still anaesthetised. The micropipette was connected to a water manometer. The vessel was occluded with a glass rod for 3-7 s, and then allowed to flow freely for at least 7 s before another occlusion was made. The flow of red cells was recorded during the occlusion on the videotape.
Calculation of Lp
The transcapillary water flow per unit area of vessel wall (Jv/S) was calculated from the initial velocity of the red cells (dl/dt) after occlusion, the vessel radius (r) and the length between the marker cell and the point of occlusion (l), all of which were measured off-line from the videotape:
| (1) |
The hydraulic conductivity (Lp) was calculated from the Starling equation:
| (2) |
where ΔP is the hydrostatic pressure difference and σΔπ is the effective oncotic pressure difference between the capillary and the interstitium. For 1 % BSA the capillary pressure was set at 30 cmH2O so ΔP - σΔπ was 26.4 cmH2O (Bates, 1998), assuming tissue pressure was negligible, and tissue oncotic pressure was equivalent to that in the superfusate (zero).
Measurements of Lp during perfusion with VEGF or ATP and antagonists
Lp was measured during perfusion with frog Ringer solution containing 1 % BSA and, where specified, antagonist (e.g. NiCl2, SK&F 96365 (1-{β-[3-(4-methoxyphenyl)propyl]-4-methoxyphenethyl}-1H-imidazole, HCl) or PD98059 (Calbiochem, UK)). The pipette was then filled with 1 nm VEGF (Genentech, CA, USA) or 100 μm ATP, rat red cells, 1 % BSA and, where specified, the antagonist, using a refilling system based on that described by Neal (1998) and Hillman et al. (2001). The vessel was occluded for 3-5 s within 30 s of re-filling. The occlusion was released and Lp measured approximately every 10 s over the next 3-5 min. The vessel was perfused for 10 min with either VEGF or ATP, before the pipette was removed. It is not known how long the agents remain in the interstitium.
Measurement of compliance
The compliance of the vessel wall was measured by determining the distance moved by a marker red cell during a reduction in pressure from 30 to 20 cmH2O as previously described (Smaje et al. 1980). While the vessel was being perfused with 1 % BSA the vessel was occluded with the pressure at 30 cmH2O. After approximately 5 s, the perfusion line was switched to a manometer set at 20 cmH2O by turning a three-way stopcock. The pressure was switched back about 3 s later and the process repeated twice. Compliance (C) is defined as the change in radius (Δr) per unit change in pressure (ΔP):
| (3) |
The change in radius was calculated assuming that the fluid in the vessel was incompressible, and that fluid filtration during the time of the pressure drop (4-8 ms) was negligible:
| (4) |
where l is the length of the column between the block site and the red cell and Δx is the distance moved by the red cell during the pressure step. The subscripts 0 and 1 denote values at the higher and lower pressures, respectively (Bates, 1998).
Measurement of Lp and compliance on day 2
Experiments to measure Lp chronically were carried out as described previously in detail (Bates & Curry, 1996). After the final occlusion, a map was made of the mesenteric vasculature and the gut replaced in the body cavity of the animal, the skin and body wall were sutured, and the frog was allowed to recover. Twenty-four hours later (day 2) the frog was anaesthetised as above and the same vessel was cannulated. The Lp and compliance were measured as above for baseline solutions (1 % BSA). Where specified the vessel was then reperfused with solution containing 1 % BSA in frog Ringer solution containing 5 mm NiCl2. The superfusate was also switched to one containing 5 mm NiCl2. We have previously shown that laparotomy, exposure of the vessel, cannulation, permeability and compliance measurement, replacement of the gut, surgery, recovery, re-anaesthetisation, and a second cannulation result in an unchanged permeability and compliance (Bates & Curry, 1996; Bates, 1998). To ensure that the inhibitors PD98059 and nickel also did not affect the permeability, experiments were performed as outlined above, but without perfusing with VEGF. At the end of the experiment frogs were killed by destruction of the cranium (crushing), before recovery to consciousness.
Chronic effect of VEGF on endothelial intracellular calcium concentration
Intracellular calcium concentration ([Ca2+]i) was measured in frog mesenteric microvessels as described by Bates & Curry (1997). This system has previously been used to measure nanomolar changes in [Ca2+]i in endothelial cells of frog mesenteric microvessels in vivo (Bates & Curry, 1997; Pocock et al. 2000). In brief, after Lp was measured on both days as described above, the pipette was removed, the abdominal cavity opened up and the mesentery spread over a glass coverslip. The vessel was visualized under an epifluorescence microscope (Leitz Diavert) and measurements made using a photomultiplier tube (Leitz MPV), an excitation filter changer (Kinetek, New York), and a 100 W mercury lamp. The vessel was re-cannulated and perfused with control solution. Fluorescence intensity (If), collected by a dry Fluotar lens (× 20, NA 0.75), was measured at excitation wavelengths of 340 ± 5 and 380 ± 5 nm and emission at 500 ± 35 nm using a 0.25 s exposure to give an initial background If for each vessel to estimate loading. The vessel was then loaded with 5 μm Fura-2 AM and 1 % BSA for 45-60 min in the dark. Once the fluorescence intensity had reached 5-10 times background, and loading was even, the vessel was perfused for 10 min with 1 % BSA to give a baseline [Ca2+]i reading. The vessel was then perfused with 5 mm Mn2+ and 10 μm ionomycin to quench the fluorescence from the Ca2+-sensitive form of Fura-2.
Intracellular calcium concentration was calculated from the equation:
| (5) |
(He et al. 1990; Poenie, 1990), where R is the normalised ratio of If calculated as R =Rexp/Rmin, where Rexp= (If340 - B340)/(If380 - B380), If340 is the fluorescence intensity with excitation at 340 nm, If380 is the fluorescence intensity with excitation at 380 nm, B340 and B380 are the background fluorescence intensities at excitations of 340 and 380 nm, respectively (measured as the If after Mn2+ quenching), and Rmin is the in vitro ratio for zero [Ca2+]. Rmax is the in vitro ratio at saturating calcium, and K (the product of the effective dissociation constant for Fura-2 and the in vitro fluorescence intensity ratio of zero to saturating calcium) was estimated from the in vitro calibration curve for Fura-2.
Effect of VEGF on MAPK activation and inhibition of MAPK in the frog
Frog lung was collected from animals pithed by cervical dislocation and destruction of the brain. Parasites were removed from the lung if present, and the lung tissue diced up coarsely. The tissue was then gently homogenised in 100 μl of extraction buffer (50 mm Tris, 10 mm EDTA) containing protease inhibitors: 10 μg ml−1 leupeptin, 20 μm E64 (trans-epoxysuccinyl-l-leucylamido (4-guanidine)-butane), 2 μg ml−1 aprotinin, 1 μm pepstatin A, 50 mm sodium fluoride (NaF), 2.5 mm sodium orthovanadate (Na3VO4), 62.5 mmβ-glycerophosphate and 1 mm phenyl methyl sulphonyl fluoride (PMSF, added immediately prior to use). The tissue was incubated with 100 μm ATP or 1 nm VEGF or 30 μm PD98059, or VEGF and PD98059 for up to 30 min, and then snap frozen in liquid nitrogen to stop the reaction. Homogenates were spun in a benchtop centrifuge for 10 min at 10 000 g to separate the protein and cell debris and 10 μl aliquots were removed to determine protein concentration by the Bradford method (BioRad). For each sample, 200 μg of protein was used for sodium dodecyl sulphate-1 % polyacrylamide gel electrophoresis (SDS-PAGE). The gel was electroblotted onto a nitrocellulose membrane at 30 mV, overnight. The membrane was washed in 1 × PBS-0.1 % Tween then incubated in 10 % Marvel in PBS-Tween for 2 h, and probed with a phospho-p44/p42-MAPK antibody at 1:1000 in 5 % Marvel for 2 h. The membrane was washed in PBS-Tween, incubated with horseradish peroxidase-conjugated anti-rabbit secondary antibody (Amersham Life Science, 1: 2000) and the previous wash step repeated. Protein was detected with enhanced chemiluminescence Western blotting detection reagent (Amersham Life Science) and exposed to photosensitive film for 30-300 s and developed. Any bands present were measured against coloured markers (Sigma) also loaded onto the gel. The developed film was scanned (AGFA 130C) into Adobe Photoshop on a G3 Macintosh, and the NIH Image program was used to calculate optical densities.
Statistics
Means ±s.e.m. are shown unless stated. Differences between Lp, compliance and diameter of vessels on successive days were compared using Student's paired t tests. Differences between groups of vessels were compared using Alternate Welch unpaired t tests. The chronic Lp measured during the experiments to determine the effect of sustained Ca2+ influx were not normally distributed and therefore median ± interquartile range (IQR) values are shown and non-parametric Wilcoxon paired tests were used. A P value of < 0.05 was accepted as significant.
RESULTS
Effect of VEGF on MAPK activation and inhibition of MAPK in the frog
VEGF has been shown to result in activation of raf which phosphorylates MEK, which in turn phosphorylates MAPK, in mesenteric microvessels of mice (Mukhopadhyay et al. 1998). PD98059 is an inhibitor of MEK and therefore blocks MAPK phosphorylation. However, this has not been shown in the frog Rana. Therefore in order to check that VEGF did stimulate MAPK activation in frog tissue and that this could be inhibited by the MEK specific inhibitor PD98059, MAPK activation was assayed in homogenised frog lung tissue using an antibody to phosphorylated MAPK.
Incubation of frog lung tissue in 1 nm VEGF for 10 and 30 min significantly increased MAPK phosphorylation. ATP (100 μm) also weakly increased MAPK phosphorylation over 5-20 min (see Fig. 1A). The VEGF-mediated increase in phosphorylation was inhibited by incubation of lung tissue with 30 μm PD98059 for 10 min before addition of VEGF (Fig. 1B). These data are consistent with experiments carried out in Xenopus showing that PD98059 is an effective inhibitor of MAPK phosphorylation in amphibia (Chau & Shibuya, 1999).
Figure 1. VEGF-stimulated MAPK activation in frog lung tissue is inhibited by PD98059.
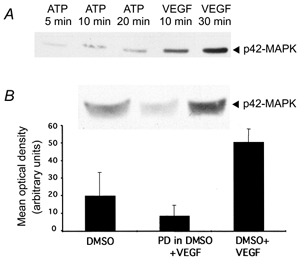
A, effect of incubation of lung tissue with 100 μm ATP for 5, 10 or 20 min or 1 nm VEGF for 10 or 30 min. B, effect of 20 min incubation of lung tissue with PD98059 on VEGF-induced MAPK activation. Lungs were incubated with PD98059 (PD) or vehicle (DMSO) for 20 min and then either exposed to VEGF or saline for 20 min (n = 3). An example of one of the Western blots is shown above the graph.
The chronic increase in Lp is dependent on the acute calcium influx
To determine whether the chronic increase in permeability was dependent on the acute, transient calcium influx stimulated by VEGF, we perfused six vessels with 5 mm NiCl2, which has previously been shown to block VEGF-mediated calcium influx into endothelial cells in vivo (Pocock et al. 2000). We then perfused these vessels with VEGF and measured Lp. The vessels were then replaced in the animal and 24 h later Lp was once again measured. In contrast to the effect of VEGF described previously (Bates & Curry, 1996), perfusion of the vessel with 5 mm NiCl2 before and during VEGF perfusion significantly attenuated both the acute and chronically increased permeability (Fig. 2). Permeability (measured as Lp) during VEGF perfusion did not significantly increase in the presence of NiCl2 ((4.1 ± 2.0) × 10−7 cm s−1 cmH2O−1 with Ni2+ to (4.9 ± 2.0) × 10−7 cm s−1 cmH2O−1 with VEGF and Ni2+). The following day there was a small, but not significant, increase in permeability (to (9.4 ± 2.7) × 10−7 cm s−1 cmH2O−1, P > 0.05, paired t test), which was a significantly lower increase than that measured in vessels not exposed to nickel (P < 0.01, unpaired t test). Perfusion of five additional vessels with 5 mm NiCl2 on the first day, but without exposure to VEGF, caused a very small, but statistically significant increase in Lp from (1.9 ± 0.7) to (4.2 ± 0.7) × 10−7 cm s−1 cmH2O−1(P = 0.03). In order to ensure that the chronic increase in Lp was dependent on calcium influx as opposed to any other effect of NiCl2, the experiments were repeated in three vessels using the calcium channel blocker SK&F 96365. Perfusion of vessels with 100 μm SK&F 96365 immediately before VEGF and during VEGF perfusion did not result in an increase in Lp the following day. Lp was (4.8 ± 2.3) × 10−7 cm s−1 cmH2O−1 before perfusion with VEGF, and (3.8 ± 2.5) × 10−7 cm s−1 cmH2O−1 (P > 0.05) the following day.
Figure 2. Chronic effect of acute inhibition of Ca2+ influx or MEK on VEGF-mediated increased Lp.
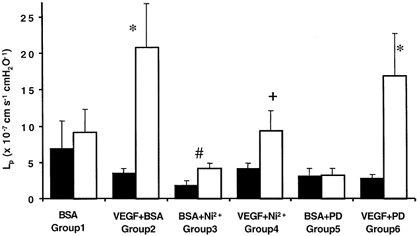
Permeability was measured on day 1 (▪) during perfusion with 1 % BSA alone or 1 % BSA and inhibitor (5 mm Ni2+ or 30 μm PD98059). Vessels were then perfused with BSA (group 1, n = 6), 1 nm VEGF and BSA (group 2, n = 28), VEGF, BSA and the inhibitor (group 4, n = 6 and group 6, n = 8), or with just BSA and the inhibitor (group 3, n = 5 and group 5, n = 5). The following day the Lp was measured on the same vessel (□) during perfusion with 1 % BSA alone. *P < 0.01 compared to day 1, #P = 0.03 compared to day 1, + P < 0.01 compared to vessels exposed to VEGF in the absence of nickel (unpaired t test).
The chronic increase in Lp is not dependent on MAPK activation
Since both VEGF and ATP are known to increase both calcium and MAPK activation in endothelial cells we determined whether the increase in permeability could be attenuated by inhibition of MAPK phosphorylation. Lp was measured in eight vessels with 1 % BSA and 30 μm PD98059, which inhibits MAPK activation (see Fig. 1). The vessels were then perfused with 1 nm VEGF and PD98059 and Lp was measured. The mesentery was then replaced in the animal and the animal was allowed to recover. Twenty-four hours later the Lp was once again measured. The Lp was 5.9-fold greater on day 2 than on day 1, an increase from (2.9 ± 0.5) to (16.9 ± 4.0) × 10−7 cm s−1 cmH2O−1 (P < 0.01; see Fig. 2). This was not significantly different from the increase in Lp seen without inhibition of MAPK phosphorylation. Perfusion of five additional vessels with 30 μm PD98059 on the first day, but without exposure to VEGF, did not result in a change in Lp ((3.2 ± 1.1) × 10−7 cm s−1 cmH2O−1 on day 1, (3.3 ± 1.0) × 10−7 cm s−1 cmH2O−1 on day 2, P > 0.05).
The chronic increase in diameter is dependent on MAPK activation, not calcium influx
To determine whether the previously described chronic increase in diameter is stimulated by calcium influx or MAPK activation, we measured the increase in diameter brought about by VEGF in the presence of Ni2+ or PD98059, at 30 cmH2O. Figure 3 shows the diameter of vessels before and 24 h after perfusion with 1 nm VEGF alone, 1 nm VEGF in the presence of 5 mm Ni2+ and 1 nm VEGF in the presence of 30 μm PD98059. VEGF alone increased the diameter of 28 vessels from 23 ± 1.1 to 31 ± 2.1 μm (P < 0.001), as previously described (Bates & Curry, 1996). Perfusion of vessels with 1 nm VEGF in the presence of 5 mm Ni2+ also significantly increased the diameter of six vessels from 23 ± 5.8 to 31 ± 8.1 μm (P < 0.05), an increase that was not significantly different from that with VEGF alone. Perfusion of seven vessels with 30 μm PD98059 attenuated the increase in diameter brought about by VEGF, so that there was no significant increase in diameter (24 ± 4.6 μm without PD98059; 26 ± 7.3 μm with PD98059). Interestingly, in the 10 vessels perfused with 100 μm ATP, the diameter increased from 15.3 ± 3.1 μm on day 1 to 20.6 ± 3.6 μm (P < 0.01) on day 2. Furthermore, the increase in diameter was also seen when vessels were perfused with 100 μm SK&F 96365 and VEGF. Exposure to VEGF during SK&F 96365 application resulted in an increase in diameter from 24.7 ± 1.9 to 28.7 ± 2.8 μm in three vessels studied. The diameter of five vessels perfused with 30 μm PD98059 (26.2 ± 2.9 μm on day 1 to 28.4 ± 4.0 μm on day 2) or 5 mm Ni2+ (25.4 ± 1.6 μm on day 1 to 28.6 ± 1.0 μm on day 2) without VEGF did not significantly alter.
Figure 3. Chronic effect of inhibition of Ca2+ influx or MEK inhibition on vessel diameter.
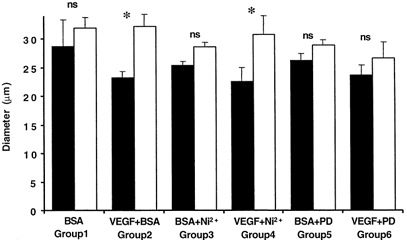
Diameter was measured on day 1 (▪) at 30 cmH2O during perfusion with 1 % BSA and inhibitor (5 mm Ni2+ or 30 μm PD98059). Vessels were then perfused with BSA alone (group 1, n = 6), 1 nm VEGF and BSA (group 2, n = 28), VEGF, BSA and the inhibitor (group 4, n = 6 and group 6, n = 7), or with BSA and the inhibitor (group 3, n = 5 and group 5, n = 5). The following day the diameter was measured on the same vessel at the same pressure (□). * P < 0.01 compared to day 1.
The chronic increase in compliance is dependent on MAPK activation, not calcium influx
We have previously shown that VEGF increases the compliance of vessels and that this may contribute to some, but not all, of the increase in diameter. To determine whether this increase in compliance was also dependent on MAPK activation or calcium influx, we measured the compliance changes before and 24 h after perfusion with 1 nm VEGF alone, 1 nm VEGF in the presence of 5 mm Ni2+ and 1 nm VEGF in the presence of 30 μm PD98059 (see Fig. 4). Perfusion of seven vessels with VEGF significantly increased the compliance of the vessels from 28 ± 5 to 39 ± 7.1 nm cmH2O−1 (P < 0.01) 24 h later. Perfusion of six vessels with 1 nm VEGF in the presence of 5 mm Ni2+ also increased the compliance, from 12 ± 1.9 to 34 ± 7.8 nm cmH2O−1 (P < 0.01) after 24 h. In contrast, perfusion of seven vessels with 1 nm VEGF in the presence of 30 μm PD98059 resulted in no change in compliance (from 23 ± 1.0 to 19 ± 1.9 nm cmH2O−1, P > 0.05). The increase in compliance was also seen when vessels were perfused with 100 μm SK&F 96365 and VEGF. Exposure to VEGF during SK&F 96365 resulted in a significant increase in compliance (from 15.8 ± 2.2 to 23.8 ± 2.3 nm cmH2O−1, P < 0.05) in the three vessels studied. The compliance of five vessels perfused with 30 μm PD98059 (18.4 ± 4.2 nm cmH2O−1 on day 1, 21.7 ± 6.2 nm cmH2O−1 on day 2) or 5 mm nickel (16.6 ± 3.9 nm cmH2O−1 on day 1, 15.6 ± 2.3 nm cmH2O−1 on day 2) without VEGF did not significantly alter. These data indicate that VEGF increases compliance through MAPK activation rather than calcium influx.
Figure 4. Chronic effect of inhibition of Ca2+ influx or MEK inhibition on vessel compliance.
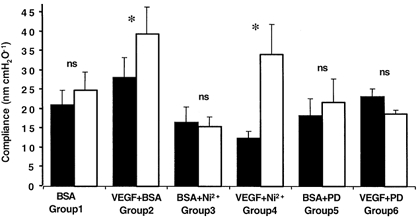
Compliance was measured on day 1 (▪) during perfusion with 1 % BSA alone, or 1 % BSA and inhibitor (5 mm Ni2+ or 30 μm PD98059). Vessels were then perfused with BSA (group 1, n = 6), 1 nm VEGF and BSA (group 2, n = 28), VEGF, BSA and the inhibitor (group 4, n = 6 and group 6, n = 7), or with just BSA and the inhibitor (group 3, n = 5 and group 5, n = 5). The following day the compliance was measured on the same vessel (□). *P < 0.01 compared to day 1.
The chronic increase in Lp is dependent on a sustained calcium influx
To determine whether the chronic, sustained increase in Lp was due to a sustained increase in calcium influx into endothelial cells, in addition to the transient increase seen on day 1, six vessels were perfused with VEGF and the chronic increase in Lp measured on day 2, during perfusion with 1 % BSA in normal Ringer solution. The Ringer solution in the perfusate and superfusate was then switched to one containing 5 mm Ni2+ and Lp was measured. Perfusion of these six vessels with VEGF resulted in a significant increase in Lp from a baseline on day 1 of (3.35 ± 0.08) × 10−7 cm s−1 cmH2O−1 to a chronic sustained increase of (59 ± 45) × 10−7 cm s−1 cmH2O−1 (P < 0.05, Wilcoxon paired test). Subsequent simultaneous perfusion and superfusion of 5 mm Ni2+ resulted in a rapid and significant reduction in Lp to (6.4 ± 6.3) × 10−7 cm s−1 cmH2O−1 within a few minutes (P < 0.05 compared to before Ni2+ perfusion), which was not significantly different from the baseline on the previous day. Figure 5 shows the effect of 5 mm Ni2+ perfusion on the ratio of the Lp during perfusion with Ni2+ compared to that measured before perfusion. It can be seen that the Lp was rapidly reduced to less than 20 % of its value before calcium influx inhibition within 5 min. In three further vessels we investigated the effect of an alternative inhibitor of calcium influx, SK&F 96365. Hydraulic conductivity was measured in three vessels before and during perfusion with 1 nm VEGF. The mesentery was then replaced in the animal and the following day baseline Lp was measured again. The vessel was then perfused with 100 μm SK&F 96365 and Lp was determined. The mean Lp measurements of these three vessels relative to that measured before perfusion with SK&F 96365 are shown in Fig. 5B. In all three vessels SK&F 96365 reduced the Lp on day 2 towards that on day 1. VEGF chronically increased Lp from (3.8 ± 0.2) to (72.9 ± 13.5) × 10−7 cm s−1 cmH2O−1. Perfusion of these vessels with 100 μm SK&F 96365 reduced the Lp to (3.4 ± 0.2) × 10−7 cm s−1 cmH2O−1. Inhibition of calcium influx can therefore reduce the chronically increased permeability of vessels exposed to VEGF.
Figure 5. Effect of inhibition of Ca2+ influx on chronically increased vascular permeability.
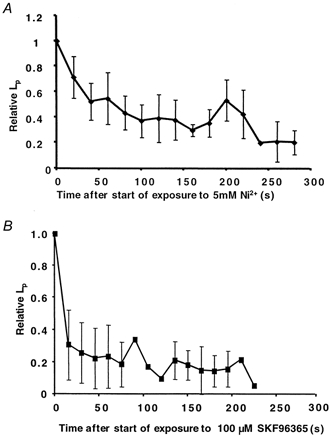
Twenty-four hours after exposure to 1 nm VEGF, Lp was measured on the same vessel during perfusion with 1 % BSA in normal Ringer solution, and then the Ringer solution in the perfusate and superfusate was switched to one containing either 5 mm Ni2+(A; n = 6) or 100 μm SK&F 96365 (B; n = 3). Lp measurements are expressed as the mean (±s.e.m.) fraction of the permeability on day 2 before Ni2+ or SK&F 96365 perfusion.
In order to determine whether VEGF increased endothelial [Ca2+]i we measured [Ca2+]i in five vessels (see Fig. 6) in which we had already measured Lp before, during and 24 h after VEGF perfusion. In addition we measured the [Ca2+]i in four vessels which had been perfused with 100 μm ATP on the previous day. In the five vessels studied, VEGF caused a median (± IQR) (6.0 ± 2.0)-fold transient increase in Lp from (3.5 ± 0.5) to (35 ± 6.0) × 10−7 cm s−1 cmH2O−1. The following day the median Lp was (16 ± 77) × 10−7 cm s−1 cmH2O−1, (8.7 ± 2.1)-fold higher than the baseline on day 1. Despite this increase in permeability on day 2, which occurred in all the vessels studied, the mean endothelial [Ca2+]i of the five vessels perfused with VEGF (111 ± 34 nm) was not significantly different from the normal baseline [Ca2+]i measured in 16 vessels perfused with 1 % BSA (76 ± 10 nm, P > 0.05).
Figure 6. Effect of VEGF and ATP perfusion on chronically increased permeability and endothelial [Ca2+]i.
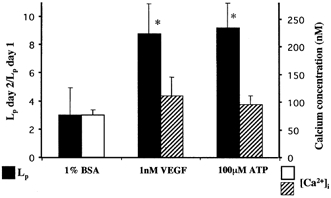
VEGF (n = 5) and ATP (n = 4) both increase Lp 24 h after perfusion compared to 1 % BSA (▪, left-hand axis, n = 11). However the endothelial [Ca2+]i (right-hand axis) was no greater 24 h after ATP (n = 4) or VEGF (n = 5) perfusion ( ) than it was in vessels in which calcium was measured without agonist perfusion (□, n = 16). *P < 0.05 compared to 1 % BSA.
) than it was in vessels in which calcium was measured without agonist perfusion (□, n = 16). *P < 0.05 compared to 1 % BSA.
ATP, which acutely increases [Ca2+]i, also chronically increases Lp
If the chronic increase in Lp is dependent on an acute increase in permeability brought about by calcium influx, then agonists other than VEGF, such as ATP, which also acutely increases [Ca2+]i and Lp, would be expected to chronically increase Lp. We have tested this hypothesis by determining the effect of acute ATP perfusion on the sustained chronic vascular permeability. Perfusion of 10 vessels with 100 μm ATP for 10 min on day 1 resulted in an acute transient significant increase in Lp from (5.7 ± 2.1) to (56.2 ± 22.4) × 10−7 cm s−1 cmH2O−1 that returned to baseline. The following day the baseline Lp was significantly increased compared to the previous day ((33.3 ± 6.8) × 10−7 cm s−1 cmH2O−1, a (9.2 ± 1.8)-fold increase, P < 0.001; see Fig. 6). This was similar to the effect of VEGF, which we have previously shown (Bates, 1998) to chronically increase Lp (8.7 ± 2.1)-fold (Fig. 6). The endothelial [Ca2+]i was not raised 1 day after perfusion with 100 μm ATP either (95 ± 15 nm, n = 4, Fig. 6). We have previously shown that perfusion of vessels with BSA alone does not result in a significant increase in Lp (P > 0.05; Bates & Curry, 1996, and Fig. 2). Therefore increased permeability by perfusion with either VEGF or ATP was not accompanied by a sustained increase in endothelial [Ca2+]i.
DISCUSSION
VEGF chronically increases permeability through calcium influx
We have previously shown that a 10 min perfusion with VEGF results in a chronic increase in microvascular permeability 24 h later (Bates, 1998), that VEGF stimulates increased endothelial [Ca2+]i in microvessels, which lasts only 3-5 min (Bates & Curry, 1997), and that this increase in Ca2+ is dependent on Ca2+ influx (Pocock et al. 2000). The evidence we have presented here shows that 5 mm NiCl2, which is known to block the acute calcium influx, attenuates the VEGF-mediated chronically increased microvascular permeability. In addition, another calcium channel blocker, SK&F 96365, also blocks the chronic increase in Lp. Although these findings were suggested by the positive correlation between the acute and the chronic permeability increases (Bates, 1998), this is the first demonstration of the signalling pathway through which VEGF may result in chronically increased microvascular permeability.
The only studies investigating the signalling pathways by which VEGF increases the permeability of the vascular wall, as opposed to the combined effect of increases in permeability, capillary surface area and vascular pressure (Bates et al. 1999), have focused on the role of nitric oxide (NO) production and phospholipase C (PLC) and protein kinase C activation in the VEGF-mediated permeability increase (Wu et al. 1999). These have all been shown to be involved in the acute increase in permeability, and it is therefore possible that the calcium influx is PLC mediated. Since NO and PKC activation are calcium dependent, the data presented here are consistent with the hypothesis that the VEGF-mediated chronic increase in permeability may involve NO synthase and PKC activation. VEGF has also been shown to activate MAPK both in vitro (D'Angelo et al. 1995) and in vivo (Mukhopadhyay et al. 1998), and the VEGF-induced increase in endothelial cell monolayer permeability was dependent on MAPK activation (Kevil et al. 1998). Although we have recently shown that inhibition of MAPK does not affect the acute increase in Lp brought about by VEGF perfusion (Bates, 2000), there have been no in vivo studies showing that MAPK activation is necessary for chronically increased vascular permeability. The experiments described here showed that inhibition of MAPK activation did not affect the chronic increase in permeability either. They are therefore consistent with the hypothesis that VEGF chronically, as well as acutely, increases permeability through a calcium influx-dependent pathway
VEGF increases microvascular compliance and diameter through MAPK activation
We have previously shown that VEGF increases microvascular compliance and diameter 24 h after perfusion with VEGF (Bates, 1998). The underlying reasons for these increases are not known. However, it has been shown that the compliance of microvessels is set by the strength of the basement membrane (Smaje et al. 1980) and surrounding extracellular matrix (Swayne et al. 1989). This matrix and membrane consist of type IV collagen, laminin, fibronectin and other matrix proteins (Nerlich & Schleicher, 1991). VEGF is known to stimulate production of matrix metalloproteinases by endothelial cells (Unemori et al. 1992), which helps the cells migrate and invade the surrounding tissue. Furthermore, MMP production has been shown in endothelial cells in vitro to be dependent on MAPK activation (Ridley et al. 1997). It has been proposed that the compliance of a vessel will increase if MMP production results in basement membrane or matrix degradation, and since MMP production is dependent on MAPK activation, this may be the pathway through which VEGF increases compliance. It is interesting to note, however, that since the increase in compliance and diameter was not blocked by Ni2+ or SK&F 96365, and the increase in compliance is dependent on MAPK activation but not Ca2+ influx, then these data suggest that MAPK activation was not dependent on calcium influx. It has previously been shown that VEGF-mediated MAPK activation is through an atypical pathway, involving PKC activation (Doanes et al. 1999; Takahashi et al. 1999). VEGF activates PKC through PLC-mediated diacylglycerol (DAG) production, independently of Ca2+ influx, and this therefore provides a mechanism by which these two functional endpoints, increased permeability and increased compliance, may signal differently.
The VEGF-mediated chronic, sustained increase in Lp is dependent on sustained Ca2+ influx, not increased [Ca2+]i
We have previously shown that chronically increased Lp is reduced in the presence of NOS inhibitors (Bates, 1998). Since NOS is a Ca2+-dependent enzyme, we determined whether the chronic increase in permeability was dependent on sustained increased Ca2+ influx. The results presented support this hypothesis. The sustained, chronic increase in Lp appears to be dependent on sustained Ca2+ influx, since the increase in Lp brought about by VEGF exposure 24 h earlier is significantly reduced by either Ni2+ or SK&F 96365 perfusion. We used a concentration of 5 mm nickel since this has previously been shown to inhibit Ca2+ entry into endothelial cells in vivo. Although previous data on human umbilical vein endothelial cells in culture have shown that similar Ni2+ concentrations (2 mm) can result in an increase in store release in endothelial cells in culture, we found no evidence for this effect of nickel in endothelial cells in vivo. Nevertheless, interpretation of results using inhibitors such as this must be made with some caution.
Since Lp was reduced by inhibiting calcium influx, we hypothesised that the endothelial [Ca2+]i would be significantly higher than in control vessels. However, this was not the case, since the cytoplasmic [Ca2+] appeared not to be significantly higher than control. The reasons for this discrepancy between calcium influx and [Ca2+]i are not clear. It is possible that the Lp is set by the rate of influx and not by the concentration inside the endothelial cells, or that the increase in Lp is dependent on repetitive calcium spiking of short duration (Jacob, 1990). It is interesting to note that the increase in Lp after Ringer solution perfusion is also dependent on sustained calcium influx without a sustained increase in [Ca2+]i (He & Curry, 1993).
In summary, we have shown that VEGF chronically increases microvascular permeability through an acute calcium transient, but that the increase in microvascular compliance and diameter, early indications of the angiogenic response, are mediated by a MAPK pathway that is physiologically distinct. In addition we have shown that the sustained increase in Lp is dependent on sustained calcium influx, in the absence of a sustained significant rise in [Ca2+]i.
Acknowledgments
The authors would like to thank the British Heart Foundation (FS98023 and PG97198) and the Wellcome Trust (50742) for their support. F.E.C is supported by NIH R37 grant HL28607.
References
- Bates DO. The chronic effect of vascular endothelial growth factor on individually perfused frog mesenteric microvessels. Journal of Physiology. 1998;513:225–233. doi: 10.1111/j.1469-7793.1998.225by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates DO. VEGF uses different signalling pathways for increasing capillary permeability and vascular compliance in vivo. Circulation. 2000;102:248. [Google Scholar]
- Bates DO, Curry FE. Vascular endothelial growth factor increases hydraulic conductivity of isolated perfused microvessels. American Journal of Physiology. 1996;271:H2520–2528. doi: 10.1152/ajpheart.1996.271.6.H2520. [DOI] [PubMed] [Google Scholar]
- Bates DO, Curry FE. Vascular endothelial growth factor increases microvascular permeability via a Ca2+-dependent pathway. American Journal of Physiology. 1997;273:H687–694. doi: 10.1152/ajpheart.1997.273.2.H687. [DOI] [PubMed] [Google Scholar]
- Bates DO, Lodwick D, Williams B. Vascular endothelial growth factor and microvascular permeability. Microcirculation. 1999;6:83–96. [PubMed] [Google Scholar]
- Chau AS, Shibuya EK. Inactivation of p42 mitogen-activated protein kinase is required for exit from M-phase after cyclin destruction. Journal of Biological Chemistry. 1999;274:32085–32090. doi: 10.1074/jbc.274.45.32085. [DOI] [PubMed] [Google Scholar]
- Curry FE, Huxley VH, Sarelius IH. Techniques in microcirculation: measurement of permeability, pressure and flow. In: Linden RJ, editor. Cardiovascular Physiology. Techniques in the Life Sciences. New York: Elsevier; 1983. pp. 1–34. [Google Scholar]
- D'angelo G, Struman I, Martial J, Weiner RI. Activation of mitogen-activated protein kinases by vascular endothelial growth factor and basic fibroblast growth factor in capillary endothelial cells is inhibited by the antiangiogenic factor 16-kDa N-terminal fragment of prolactin. Proceedings of the National Academy of Sciences of the USA. 1995;92:6374–6378. doi: 10.1073/pnas.92.14.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doanes AM, Hegland DD, Sethi R, Kovesdi I, Bruder JT, Finkel T. VEGF stimulates MAPK through a pathway that is unique for receptor tyrosine kinases. Biochemical and Biophysical Research Communications. 1999;255:545–548. doi: 10.1006/bbrc.1999.0227. [DOI] [PubMed] [Google Scholar]
- Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. American Journal of Pathology. 1995;146:1029–1039. [PMC free article] [PubMed] [Google Scholar]
- Ferrara N, Bunting S. Vascular endothelial growth factor, a specific regulator of angiogenesis. Current Opinion in Nephrology and Hypertension. 1996;5:35–44. doi: 10.1097/00041552-199601000-00008. [DOI] [PubMed] [Google Scholar]
- He P, Curry FE. Albumin modulation of capillary permeability: role of endothelial cell [Ca2+]i. American Journal of Physiology. 1993;265:H74–H82. doi: 10.1152/ajpheart.1993.265.1.H74. [DOI] [PubMed] [Google Scholar]
- He P, Pagakis SN, Curry FE. Measurement of cytoplasmic calcium in single microvessels with increased permeability. American Journal of Physiology. 1990;258:H1366–1374. doi: 10.1152/ajpheart.1990.258.5.H1366. [DOI] [PubMed] [Google Scholar]
- He P, Zhang X, Curry FE. Ca2+ entry through conductive pathway modulates receptor-mediated increase in microvessel permeability. American Journal of Physiology. 1996;271:H2377–2387. doi: 10.1152/ajpheart.1996.271.6.H2377. [DOI] [PubMed] [Google Scholar]
- Hillman NJ, Whittles CE, Pocock TM, Williams B, Bates DO. Differential effects on microvascular hydraulic conductivity (Lp) of vascular endothelial growth factor C (VEGF-C) and placental growth factor-1 (PlGF-1) Journal of Vascular Research. 2001 doi: 10.1159/000051044. in the Press. [DOI] [PubMed] [Google Scholar]
- Jacob R. Calcium oscillations in electrically non-excitable cells. Biochimica et Biophysica Acta. 1990;1052:427–438. doi: 10.1016/0167-4889(90)90152-4. [DOI] [PubMed] [Google Scholar]
- Kevil CG, Payne DK, Mire E, Alexander JS. Vascular permeability factor/vascular endothelial cell growth factor-mediated permeability occurs through disorganization of endothelial junctional proteins. Journal of Biological Chemistry. 1998;273:15099–15103. doi: 10.1074/jbc.273.24.15099. [DOI] [PubMed] [Google Scholar]
- Michel CC, Mason JC, Curry FE, Tooke JE, Hunter PJ. A development of the Landis technique for measuring the filtration coefficient of individual capillaries in the frog mesentery. Quarterly Journal of Experimental Physiology and Cognitive Medical Science. 1974;59:283–309. doi: 10.1113/expphysiol.1974.sp002275. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D, Nagy JA, Manseau EJ, Dvorak HF. Vascular permeability factor/vascular endothelial growth factor-mediated signaling in mouse mesentery vascular endothelium. Cancer Research. 1998;58:1278–1284. [PubMed] [Google Scholar]
- Neal C. A method for changing the contents of a micropipette in situ. Journal of Physiology. 1998;513:P4. [Google Scholar]
- Nerlich AG, Schleicher E. Identification of lymph and blood capillaries by immunohistochemical staining for various basement membrane components. Histochemistry. 1991;96:449–453. doi: 10.1007/BF00316003. [DOI] [PubMed] [Google Scholar]
- Pocock TM, Williams B, Curry FE, Bates DO. VEGF and ATP act by different mechanisms to increase microvascular permeability and endothelial [Ca2+]i. American Journal of Physiology. 2000;279:H1625–1634. doi: 10.1152/ajpheart.2000.279.4.H1625. [DOI] [PubMed] [Google Scholar]
- Poenie M. Alteration of intracellular Fura-2 fluorescence by viscosity: A simple correction. Cell Calcium. 1990;11:85–91. doi: 10.1016/0143-4160(90)90062-y. [DOI] [PubMed] [Google Scholar]
- Ridley SH, Sarsfield SJ, Lee JC, Bigg HF, Cawston TE, Taylor DJ, Dewitt DL, Saklatvala J. Actions of IL-1 are selectively controlled by p38 mitogen-activated protein kinase: regulation of prostaglandin H synthase-2, metalloproteinases, and IL-6 at different levels. Journal of Immunology. 1997;158:3165–3173. [PubMed] [Google Scholar]
- Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–985. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- Smaje LH, Fraser PA, Clough G. The distensibility of single capillaries and venules in the cat mesentery. Microvascular Research. 1980;20:358–370. doi: 10.1016/0026-2862(80)90064-3. [DOI] [PubMed] [Google Scholar]
- Swayne GT, Smaje LH, Bergel DH. Distensibility of single capillaries and venules in the rat and frog mesentery. International Journal of Microcirculation: Clinical and Experimental. 1989;8:25–42. [PubMed] [Google Scholar]
- Takahashi T, Ueno H, Shibuya M. VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene. 1999;18:2221–2230. doi: 10.1038/sj.onc.1202527. [DOI] [PubMed] [Google Scholar]
- Unemori EN, Ferrara N, Bauer EA, Amento EP. Vascular endothelial growth factor induced interstitial collagenase expression in human endothelial cells. Journal of Cellular Physiology. 1992;153:557–562. doi: 10.1002/jcp.1041530317. [DOI] [PubMed] [Google Scholar]
- Wu HM, Yuan Y, Zawieja DC, Tinsley J, Granger HJ. Role of phospholipase C, protein kinase C, and calcium in VEGF-induced venular hyperpermeability. American Journal of Physiology. 1999;276:H535–542. doi: 10.1152/ajpheart.1999.276.2.H535. [DOI] [PubMed] [Google Scholar]