

Abstract
IKs, the slow component of the delayed rectifier potassium current, figures prominently in the repolarization of heart cells. The K+ channel gene KvLQT1 is mutated in the heritable long QT (LQT) syndrome. Heterologous coexpression of KvLQT1 and the accessory protein minK yields an IKs-like current. Nevertheless, the links between KvLQT1 and cardiac IKs are largely inferential.
Since the LQT syndrome mutant KvLQT1-G306R suppresses channel activity when coexpressed with wild-type KvLQT1 in a heterologous system, overexpression of this mutant in cardiomyocytes should reduce or eliminate native IKs if KvLQT1 is indeed the major molecular component of this current. To test this idea, we created the adenovirus AdRMGI-KvLQT1-G306R, which overexpresses KvLQT1-G306R channels.
In > 60 % of neonatal mouse myocytes, a sizable IKs could be measured using perforated-patch recordings (8.0 ± 1.6 pA pF−1, n = 13). IKs was increased by forskolin and blocked by clofilium or indapamide but not by E-4031. While cells infected with a reporter virus expressing only green fluorescent protein (GFP) displayed IKs similar to that in uninfected cells, AdRMGI-KvLQT1-G306R-infected cells showed a significantly reduced IKs (2.4 ± 1.1 pA pF−1, n = 10, P < 0.01) when measured 60-72 h after infection. Similar results were observed in adult guinea-pig myocytes (5.9 ± 1.2 pA pF−1, n = 9, for control vs. 0.1 ± 0.1 pA pF−1, n = 5, for AdRMGI-KvLQT1-G306R-infected cells).
We conclude that KvLQT1 is the major molecular component of IKs. Our results further establish a dominant-negative mechanism for the G306R LQT syndrome mutation.
Mutations of the KvLQT1 (or KCNQ1) gene are responsible for many cases of inherited long QT (LQT) syndrome (Wang et al. 1996) including the autosomal dominant Romano-Ward syndrome (RWS; Ward, 1964). The LQT syndrome consists of a prolonged QT interval on the electrocardiogram, syncope, ventricular arrhythmias and sudden death. These clinical findings imply that KvLQT1 is essential for proper cardiac repolarization. When coexpressed with the regulatory protein minK in heterologous systems, KvLQT1 produces currents with biophysical and pharmacological properties resembling the slowly activating component (i.e. IKs) of the delayed-rectifier potassium currents observed in native cardiac cells (Barhanin et al. 1996; Sanguinetti et al. 1996). Despite these compelling lines of evidence, the links between KvLQT1 and cardiac IKs remain largely inferential.
In the present study, we sought to confirm KvLQT1 as a critical molecular component of IKs by overexpressing a KvLQT1 construct carrying the dominant negative Romano-Ward mutation G306R (Wang et al. 1996; Wollnik et al. 1997) in cardiomyocytes via viral gene transfer and looking for changes in native IKs. We found that virally mediated overexpression of this construct results in significantly diminished IKs, confirming the critical role of KvLQT1 in IKs and further establishing a dominant-negative mechanism for the G306R disease mutant.
METHODS
The present investigation conformed to the standards of the US National Institutes of Health regarding the care and use of laboratory animals, and was performed in accordance with the guidelines of the Animal Care and Use Committee of the Johns Hopkins University.
Isolation of ventricular myocytes
Neonatal myocytes were prepared as previously described (Nuss et al. 1994). Briefly, ventricles of neonatal mice (1-2 days old, CD-1, Charles River) were aseptically removed immediately following decapitation. A single litter of mouse pups (≈10) was used for each experiment. Isolated hearts were pooled, minced and digested at room temperature with trypsin. When the digestion was complete, cells were centrifuged, plated at low density (2 × 105 cells ml−1) and incubated at 37 °C in a humidified atmosphere of 95 % O2-5 % CO2. Adult guinea-pigs were anaesthetized with sodium pentobarbital by peritoneal injection. Ventricular myocytes were isolated using Langendorff perfusion and collagenase digestion (Hoppe et al. 1999), and similarly cultured at 37 °C.
Molecular biology and virus preparation
The human KvLQT1 gene (provided by Dr Mark Keating, University of Utah) was subcloned into the vector pAdEGI (Johns et al. 1999) to make pAdEGI-KvLQT1. Human minK was first amplified from purified genomic DNA by PCR and then subcloned into pAdEGI-KvLQT1 making a fusion gene with minK at the 5′ end and enhanced green fluorescent protein (EGFP, Clontech, Palo Alto, CA, USA) at the 3′ end, creating pAdEMGI-KvLQT1. When expressed in CHO-K1 cells, this tandem construct directed expression of currents similar to those produced by coexpressing minK and KvLQT1 from separate constructs (data not shown). The point mutation G306R was introduced by PCR with overlapping mutagenic primers. The product pAdEMGI-KvLQT1-G306R was sequenced to confirm that the desired mutation was present. The MGI-KvLQT1 and MGI-KvLQT1-G306R sequences were then spliced into pAdRGI, which carries the Rous sarcoma virus long terminal repeat promoter, to create the final constructs pAdRMGI-KvLQT1 and pAdRMGI-KvLQT1-G306R. Adenovirus vectors were generated by Cre-lox recombination of purified ψ5 viral DNA and shuttle vector DNA as previously described (Hardy et al. 1997). The recombinant products were plaque purified, yielding concentrations of the order of 1010 plaque-forming units (PFU) ml−1.
Cardiomyocytes were infected by adding purified viruses to the cells at a final concentration of 10 000 particles ml−1. For transfection of HEK 293 cells (human embryonic kidney cell line), the Lipofectamine Plus transfection kit (Gibco-BRL, Gaithersburg, MD, USA) was used. DNA encoding the channel (0.5 μg per well of 6-well plates) was added to the cells with lipofectamine. Transfected or virally infected cells were incubated at 37 °C.
Electrophysiology
Electrical recordings were performed using the whole-cell patch-clamp or perforated-patch technique (Hamill et al. 1981; Rae et al. 1991) with an integrating amplifier. Pipettes had tip resistances of 1-3 MΩ when filled with an internal solution containing (mm): KCl, 140; ATP (magnesium salt), 4; EGTA, 5; MgCl2, 1; and Hepes, 10; pH 7.4. The external bath solution was composed of (mm): N-methyl-d-glucamine, 140; KCl, 5.4; glucose, 10; MgCl2, 1; CaCl2, 0.1; CdCl2, 0.5; 4-aminopyridine, 5; and Hepes, 10; pH 7.4. Clofilium, indapamide, E-4031, forskolin and isobutylmethylxanthine (IBMX) were added to the bath as noted. Perforated-patch recordings were performed by including 100 μm amphotericin B in the pipette solution (Rae et al. 1991). All recordings were performed at room temperature (≈23 °C) after 12-36 h in culture for uninfected cells and 60-72 h for virally infected or transfected cells. Only successfully infected or transfected cells, as identified by their green fluorescence using epifluorescence microscopy, were selected for experiments. Drug block was assessed from currents elicited at +70 mV (8 s) from a holding potential of -40 mV. Data reported are means ±s.e.m. with P < 0.05 indicating statistical significance.
RESULTS
Properties of IKs in neonatal mouse myocytes
When neonatal mouse ventricular myocytes were depolarized, > 60 % of the cells displayed a time-dependent outward current typical of IKs. Rapid run-down of IKs was observed when using the conventional patch-clamp technique (Fig. 1A and C), as previously reported (Nuss et al. 1994). Little current (< 0.5 pA pF−1) remained after complete run-down of IKs, suggesting that the contribution of the rapid component of the delayed rectifier (IKr) to the total current was minor under our experimental conditions. In contrast, IKs recorded with the perforated-patch technique (Rae et al. 1991) did not run down (Fig. 1B and C). Subsequent data presented here were collected using this technique.
Figure 1. IKs in uninfected neonatal murine ventricular myocytes.
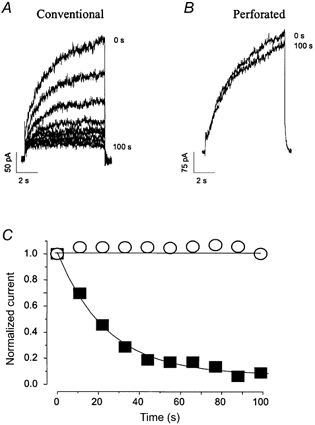
A and B, representative raw current traces of slowly activating time-dependent outward currents (IKs) elicited by long depolarization (8 s) of neonatal mouse myocytes to +70 mV from a holding potential of -40 mV using conventional patch-clamp (A) and perforated-patch (B) techniques. C, current magnitude, measured as the difference in outward current at the beginning and end of the pulse, is plotted against time during these experiments. ▪, conventional; ○, perforated-patch recording. Rapid current run-down was observed when IKs was recorded using the conventional patch-clamp technique but was absent when the perforated-patch technique was employed. Cell capacitance was 42 and 25 pF, respectively, for these cells.
Figure 2A shows the current-voltage relationship of IKs. As is characteristic of this current, IKs increased with progressive depolarization. Current density in these cells was 8.0 ± 1.6 pA pF−1(n = 13) at the end of 8 s depolarizing pulses to +70 mV. We next examined the pharmacological effects of indapamide, clofilium, E-4031 and forskolin on IKs (Fig. 2B). Application of 100 μm clofilium, a delayed-rectifier blocker (Arena et al. 1988; Colatsky et al. 1990) inhibited the current to 55.7 ± 5.6 % (n = 5) of the control level. Similarly, application of 1 mm indapamide, a putatively selective blocker of IKs (Turgeon et al. 1994), reduced current to 47.6 ± 11.9 % (n = 5). In contrast, application of 10 μm E-4031, a benzenesulphonamide agent which blocks IKr selectively (Sanguinetti et al. 1990), failed to inhibit the current (96.5 ± 3.2 %, n = 3) consistent with the notion that IKs was the predominant component. Adrenergic agonists such as forskolin and isoproterenol (isoprenaline) are known to upregulate IKs, but not IKr, by elevating the level of intracellular cAMP (Varnum et al. 1993). We thus tested the effect of forskolin (10 μm) on IKs by co-applying it with IBMX (100 μm), a phosphodiesterase inhibitor, which potentiates the cAMP elevation by forskolin. As anticipated, application of forskolin + IBMX enhanced the amplitude of IKs (130 ± 20 %, n = 2).
Figure 2. IKs in neonatal mouse myocytes.
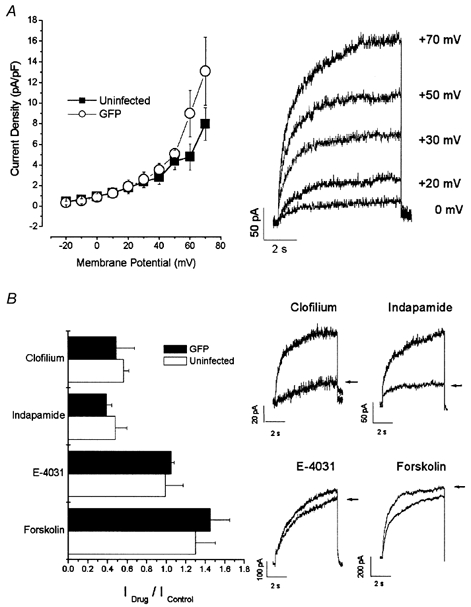
A, current-voltage (I-V) relationship (left panel, ▪) and raw records of a family (right panel) of IKs recorded from uninfected myocytes using the perforated-patch technique. B, left, the effects of 100 μm clofilium, 1 mm indapamide, 10 μm E-4031 and 10 μm forskolin + 100 μm IBMX (Forskolin) on the current amplitude of IKs were normalized to those measured under drug-free conditions and plotted as bar graphs (□). Right, typical IKs currents recorded before and after (arrows) application of these drugs. I-V relationships (left panel of A, ○) and pharmacology (left panel of B, ▪) of IKs recorded from AdRGI-infected cells are also shown.
Viral gene transfer of KvLQT1-G306R to cardiomyocytes
Since the Romano-Ward mutation G306R is known to exert a dominant-negative effect on WT KvLQT1 channel activity in a heterologous expression system (Wollnik et al. 1997), overexpression of a construct carrying this mutation in cardiomyocytes should reduce or eliminate native IKs if KvLQT1 is indeed the major molecular component of this cardiac current. To test this hypothesis, we created the constructs pAdRMGI-KvLQT1 and pAdRMGI-KvLQT1-G306R (see Methods). Figure 3A shows the experiments that we performed to verify the efficacy of our dominant-negative construct. While transfection with 3 times the DNA (by mass) of pAdRMGI-KvLQT1 in HEK 293 cells more than doubled the current magnitude recorded after 72 h, co-transfection of pAdRMGI-KvLQT1 with pAdRMGI-KvLQT1-G306R (DNA ratio of 1:2) reduced current by ≈60 % compared to expression of WT KvLQT1 alone over the same period. In contrast, co-transfection of a plasmid encoding the Kv1.3 channel with pAdRMGI-KvLQT1-G306R (1:2), did not suppress current relative to expression of Kv1.3 alone. These observations confirm both the specificity and the inhibitory effect of pAdRMGI-KvLQT1-G306R.
Figure 3. Effects of constructs carrying the dominant-negative mutation G306R.
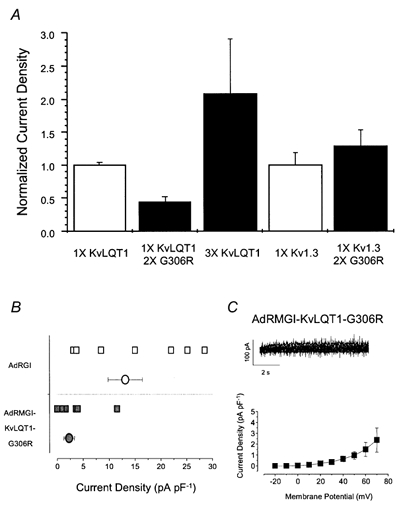
A, bar graphs summarizing the specific inhibitory effects of transfection of pAdRMGI-KvLQT1-G306 on KvLQT1 channels. For each group of channels, current amplitudes were normalized by the mean current of the same channel type (transfected with 1 × DNA) expressed alone. Current through KvLQT1 channels increased when more pAdRMGI-KvLQT1 DNA was used for transfection but was substantially suppressed when it was co-expressed with pAdRMGI-KvLQT1-G306R. In contrast, co-expression of Kv1.3 channels with KvLQT1-G306R did not lead to current suppression compared to expression of Kv1.3 alone. Currents were measured at the end of a 4 s, +70 mV or 1 s, +60 mV pulse from a holding potential of -40 or -100 mV for KvLQT1 and Kv1.3 channels, respectively. B, distribution of raw (squares) and averaged (circles) current densities at +70 mV recorded from AdRGI- (open symbols) and AdRMGI-KvLQT1-G306R-infected (filled symbols) neonatal mouse myocytes. The latter was significantly (P < 0.05) reduced. C, top, typical current traces of IKs recorded during a family of stepping voltages from AdRMGI-KvLQT1-G306R-infected myocytes. Bottom, I-V relationship.
Since myocytes are generally resistant to conventional methods of transfection, viral gene transfer techniques were employed to deliver our genes of interest. From the plasmids pAdRMGI-KvLQT1 and pAdRMGI-KvLQT1-G306R, we generated the adenoviruses AdRMGI-KvLQT1 and AdRMGI-KvLQT1-G306R, respectively (see Methods). As a control, we infected myocytes in parallel with AdRGI, which expresses only GFP. Myocytes infected with AdRGI displayed IKs with a current density (13.1 ± 3.3 pA pF−1 at +70 mV, n = 8, P > 0.05) and pharmacology profile similar to those of uninfected cells (Fig. 2). IKs measured from AdRGI-infected cells was enhanced by 10 μm forskolin with 100 μm IBMX (145 ± 20 %, n = 2), blocked by 100 μm clofilium (48.3 ± 18.8 %, n = 2) or 1 mm indapamide (38.8 ± 5.6 %, n = 3) but was not affected by 10 μm E-4031 (105 ± 3.2 %, n = 2; Fig. 2B). These observations indicate that infection of cells with an adenovirus reporter construct did not alter the biophysical or pharmacological properties of native IKs.
In contrast to AdRGI-infected cells (Fig. 3B, open symbols), IKs of AdRMGI-KvLQT1-G306R-infected cells (filled squares) was markedly suppressed (2.4 ± 1.1 pA pF−1 at +70 mV, n = 10, P < 0.05). Figure 3C shows the current-voltage relationship of IKs measured from AdRMGI-KvLQT1-G306R-infected cells. Application of 10 μm forskolin + 100 μm IBMX substantially increased the small remaining current (data not shown) but further pharmacological studies with blockers were not performed because of the small basal current amplitude.
As a further confirmation of the identification of KvLQT1 as a critical component of IKs, we also infected isolated adult guinea-pig ventricular myocytes with our viruses. Similar results were obtained. IKs density was reduced from 5.9 ± 1.2 pA pF−1(n = 9) in control uninfected cells to 0.1 ± 0.1 pA pF−1(n = 5) in AdRMGI- KvLQT1-G306R-infected cells (P < 0.05) when measured at +60 mV (Fig. 4A and B). Tail current measurements from uninfected cells further indicate that the current recorded was fairly potassium selective (Fig. 4C); although the value of Vrev (-68.7 ± 3.1 mV, n = 6) was somewhat more positive than the predicted K+ equilibrium potential (EK, -86.9 mV under our conditions), our findings are consistent with prior reports of Vrev for IKs in native cardiomyocytes (Sanguinetti & Jurkiewicz, 1990). Tail currents were not studied in neonatal cells owing to the relatively small IKs amplitude in these cells, resulting from their small size (typically 5-40 pF), despite the sizable IKs current density.
Figure 4. IKs in isolated adult guinea-pig ventricular myocytes.
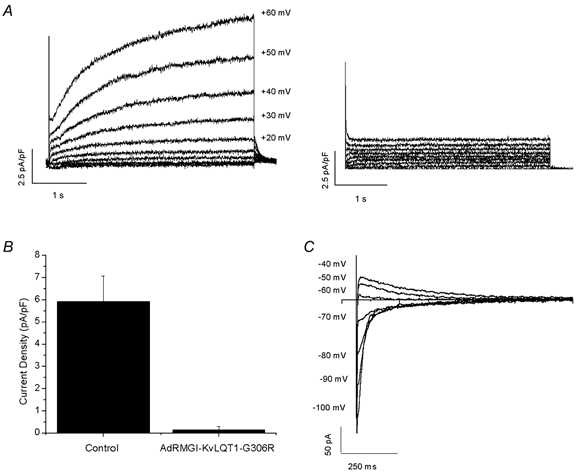
A, representative raw current traces of IKs recorded from control uninfected (left) and AdRMGI-KvLQT1-G306R-infected (right) myocytes. B, bar graph summarizing the current densities measured at the end of a 4 s depolarizing pulse to +60 mV. C, typical tail currents of IKs recorded from a control uninfected myocyte during a family of stepping voltages after a 4 s prepulse to +70 mV.
DISCUSSION
Overexpression of dominant-negative constructs in native cells has been a useful tool to dissect the contributions of various ion channel genes to excitability and different native ionic currents (Johns et al. 1997; Barry et al. 1998). In the present study, we employed this strategy to confirm the identity of KvLQT1 as the major molecular component of native cardiac IKs by introducing a dominant-negative construct (G306R) into cardiomyocytes to cripple IKs. The specific inhibitory effect of the construct on KvLQT1 channels was confirmed by its lack of effect on other channels. Though the exact mechanism of such a dominant-negative effect on channel activity is still unknown, it is generally assumed that coassembly of WT and mutant subunits results in non-functional heteromeric KvLQT1 channels. However, since such stoichiometric combinations are stochastic, some functional homotetramers that are made up of only WT subunits also exist, leading to incomplete elimination of IKs as observed in AdRMGI-KvLQT1-G306R-infected cells. Alternatively, incomplete elimination may result from competition between expression of the suppressive gene products and turnover of pre-existing functional channel proteins. Regardless of the underlying mechanism, our work demonstrates for the first time that ‘poison pill’ KvLQT1 constructs suppress IKs in native myocytes. Such suppression could not be due to dilution of minK since AdRMGI-KvLQT1-G306R coexpresses its own minK (see Methods). Our observation is pathophysiologically relevant to the clinical manifestations of LQT syndrome, since diminished IKs would lead to prolongation of the action potential and the electrocardiographic QT interval, and subsequently increase the susceptibility of afflicted individuals to malignant arrhythmias. These data support the idea that KvLQT1 is the major molecular component of IKs and further establish a dominant-negative mechanism for this Romano-Ward mutant. This dominant-negative approach, applied here in cardiomyocytes via gene transfer, is generally useful for dissecting the molecular identity of various native ionic currents.
Acknowledgments
This work was supported by National Institutes of Health grant P50 HL-52307 (to E.M.) and American Heart Association Scientist Development Grant 9730261N (to H.B.N.). R.A.L. is the recipient of a fellowship award from the Heart and Stroke Foundation of Canada. U.C.H. is supported by Deutsche Forschungsgemeinschaft. D.C.J. is supported by a Research Career Development Award of the CARE Foundation. We thank Sara A. Johns for supplying genomic DNA and Eisai Co. (Tokyo, Japan) for kindly providing E-4031. E.M. holds the Michel Mirowski, MD Professorship of Cardiology of the Johns Hopkins University.
R. A. Li and J. Miake contributed equally to this work.
References
- Arena JP, Kass RS. Block of heart potassium channels by clofilium and its tertiary analogs: relationship between drug structure and type of channel blocked. Molecular Pharmacology. 1988;34:60–66. [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. KvLQT1 and lsK (minK) proteins associate to form the IKs cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Barry DM, Xu H, Schuessler RB, Nerbonne JM. Functional knockout of the transient outward current, long-QT syndrome, and cardiac remodeling in mice expressing a dominant-negative Kv4 alpha subunit. Circulation Research. 1998;83:560–567. doi: 10.1161/01.res.83.5.560. [DOI] [PubMed] [Google Scholar]
- Colatsky TJ, Follmer CH, Starmer CF. Channel specificity in antiarrhythmic drug action. Mechanism of potassium channel block and its role in suppressing and aggravating cardiac arrhythmias. Circulation. 1990;82:2235–2242. doi: 10.1161/01.cir.82.6.2235. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hardy S, Kitamura M, Harris-Stansil T, Dai Y, Phipps ML. Construction of adenovirus vectors through Cre-lox recombination. Journal of Virology. 1997;71:1842–1849. doi: 10.1128/jvi.71.3.1842-1849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe UC, Johns DC, Marbán E, O'rourke B. Manipulation of cellular excitability by cell fusion: effects of rapid introduction of transient outward K+ current on the guinea pig action potential. Circulation Research. 1999;84:964–972. doi: 10.1161/01.res.84.8.964. [DOI] [PubMed] [Google Scholar]
- Johns DC, Marx R, Mains RE, O'rourke B, Marbán E. Inducible genetic suppression of neuronal excitability. Journal of Neuroscience. 1999;19:1691–1697. doi: 10.1523/JNEUROSCI.19-05-01691.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns DC, Nuss HB, Marbán E. Suppression of neuronal and cardiac transient outward currents by viral gene transfer of dominant-negative Kv4. 2 constructs. Journal of Biological Chemistry. 1997;272:31598–31603. doi: 10.1074/jbc.272.50.31598. [DOI] [PubMed] [Google Scholar]
- Nuss HB, Marban E. Electrophysiological properties of neonatal mouse cardiac myocytes in primary culture. Journal of Physiology. 1994;479:265–279. doi: 10.1113/jphysiol.1994.sp020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. Journal of Neuroscience Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. Journal of General Physiology. 1990;96:195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgeon J, Daleau P, Bennett PB, Wiggins SS, Selby L, Roden DM. Block of IKs, the slow component of the delayed rectifier K+ current, by the diuretic agent indapamide in guinea pig myocytes. Circulation Research. 1994;75:879–886. doi: 10.1161/01.res.75.5.879. [DOI] [PubMed] [Google Scholar]
- Varnum MD, Busch AE, Bond CT, Maylie J, Adelman JP. The min K channel underlies the cardiac potassium current IKs and mediates species-specific responses to protein kinase C. Proceedings of the National Academy of Sciences of the USA. 1993;90:11528–11532. doi: 10.1073/pnas.90.24.11528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, Vanraay TJ, Shen J, Timothy KW, Vincent GM, De jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KvLQT1 mutations cause cardiac arrhythmias. Nature Genetics. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- Wollnik B, Schroeder BC, Kubisch C, Esperer HD, Wieacker P, Jentsch TJ. Pathophysiological mechanisms of dominant and recessive KvLQT1 K+ channel mutations found in inherited cardiac arrhythmias. Human Molecular Genetics. 1997;6:1943–1949. doi: 10.1093/hmg/6.11.1943. [DOI] [PubMed] [Google Scholar]