

Abstract
The neuropeptide melanin concentrating hormone (MCH) is synthesised only by neurons of the lateral hypothalamic (LH) area in the CNS. MCH cells project widely throughout the brain. Despite the growing interest in this peptide, in part related to its role in feeding, little has been done to characterise its physiological effects in neurons. Using whole-cell recording with current and voltage clamp, we examined the cellular actions in neurons from the LH.
MCH induced a consistent decrease in the frequency of action potentials and reduced synaptic activity. Most fast synaptic activity in the hypothalamus is mediated by GABA or glutamate. MCH inhibited the synaptic activity of both glutamatergic and GABAergic LH neurons, each tested independently.
MCH reduced the amplitude of glutamate-evoked currents and reduced the amplitude of miniature excitatory currents, indicating an inhibitory modulation of postsynaptic glutamate receptors.
In the presence of tetrodotoxin to block action potentials, MCH caused a depression in the frequency of miniature glutamate-mediated postsynaptic currents, suggesting a presynaptic site of receptor expression.
In voltage clamp experiments, MCH depressed the amplitude of calcium currents, suggesting that a mechanism of inhibition may involve a reduced calcium-dependent release of amino acid transmitter.
Previous reports have suggested that MCH activated potassium channels in non-neuronal cells transfected with the MCH receptor gene. We found no effect of MCH on voltage-dependent potassium channels in LH neurons. Baclofen, a GABAB receptor agonist, activated G-protein gated inwardly rectifying potassium (GIRK)-type channels; in the same neurons, MCH had no effect on GIRK channels. MCH showed no modulation of sodium currents.
Blockade of the Gi/Go protein with pertussis toxin eliminated the actions of MCH.
The inhibitory actions of MCH on both excitatory and inhibitory synaptic events, coupled with opposing excitatory actions of hypocretin, another LH peptide that projects to many of the same loci, suggest a substantial level of complexity in neuropeptide modulation of LH actions.
Melanin concentrating hormone is synthesised in the CNS only by neurons in the lateral hypothalamic (LH) area. MCH efferent axons innervate a wide variety of regions in the brain from the cortex to the spinal cord (Skofitsch et al. 1985; Nahon et al. 1989; Bittencourt et al. 1992). The highest density of MCH axons and boutons is found in the LH (Bittencourt et al. 1992). The receptor for MCH has recently been determined to be the orphan receptor SLC-1 (Chambers et al. 1999; Lembo et al. 1999; Saito et al. 1999). This MCH receptor, similar to the peptide, has a wide distribution throughout the CNS (Lembo et al. 1999), suggesting that MCH may modulate a number of CNS systems.
The LH plays a substantial role in a number of functions including sensorimotor integration, sleep, arousal and regulation of the autonomic nervous system. It is also involved in the regulation of feeding. This has led some to consider the LH a feeding centre (Stellar, 1954; Sawchenko, 1998). LH damage caused by electrolytic lesions (Anand & Brobeck, 1951) or chemical lesions with glutamate agonists (Sticker et al. 1978) depress feeding and body weight regulation, whereas LH stimulation induces feeding. The LH has been called a pleasure centre of the brain, as both rats and humans will administer electrical self-stimulation with electrodes implanted in the LH (Valenstein & Campbell, 1966; Heath, 1972; Moran et al. 1981; Gallistel et al. 1985). Although a large number of neuroactive peptides have been found in the medial hypothalamus, many of which were first discovered there, relatively few peptides have been identified that are synthesised by LH neurons. Recently, two peptides synthesised by LH neurons, MCH and hypocretin (de Lecea et al. 1998), also called orexin (Sakurai et al. 1998), have generated considerable attention due to their potential role in maintaining energy homeostasis. Although MCH and hypocretin are both synthesised by large neurons of the LH and maintain many overlapping projections, the two peptides are not co-localised in single neurons (Broberger et al. 1998; Elias et al. 1998; Peyron et al. 1998).
The potential involvement of MCH in brain function in mammals is of great interest, and probably mirrors to some degree the involvement of LH neurons in a variety of functions. One function that has recently gathered considerable attention is the regulation of food intake. Intracerebral administration of MCH has been reported to evoke feeding in rodents (Qu et al. 1996; Rossi et al. 1999). MCH-deficient mice have reduced body weight and leanness due to reduced feeding and an enhanced metabolism (Shimada et al. 1998). MCH mRNA expression is increased by starvation and c-fos in MCH neurons is upregulated by starvation (Qu et al. 1996) and by leptin administration (Huang et al. 1999). In addition to a role in energy regulation, MCH has also been reported to modify memory retention (Monzon et al. 1999), consistent with the innervation of the hippocampus and cortex (Bittencourt et al. 1992) and the expression of the MCH receptor in the same regions (Chambers et al. 1999; Lembo et al. 1999; Shimomura et al. 1999). MCH expression levels also show sensitivity to oestrogenic steroids (Viale et al. 1999), suggesting some involvement in the reproductive axis. In lower vertebrates such as reptiles, amphibians and fish, MCH modulates pigmentation (Kawauchi et al. 1983; Eberle, 1988). In fish MCH plays a strong neuroendocrine role in the inhibition of corticotropin releasing hormone (Baker et al. 1985; Eberle, 1988).
Both MCH neurons and receptors are found in the LH. The highest density of MCH immunoreactive axons, many of them making synaptic contact with other LH neurons, is found here (Bittencourt et al. 1992). Although MCH appears to play an important role in LH-regulated feeding, and probably in other actions of the LH, the physiological role of MCH has not been examined in neurons at the cellular level. In the present study we used whole-cell recording in current and voltage clamp to study the cellular actions of MCH in neurons from the LH in vitro.
METHODS
Cell culture
Hypothalamic neuron cultures were prepared from rat embryos as described elsewhere (Gao et al. 1998). Use of rat tissue was approved by the university committee on animal use. Pregnant rats were given an overdose of Nembutal (100 mg kg−1), embryos were collected, and then the pregnant rats were given a second lethal dose of Nembutal (150 mg kg−1). Briefly, lateral hypothalami were dissected out of the brains of Sprague-Dawley rats at embryonic days (E) 15-18 and cut into small pieces (smaller than 1 mm3). The tissue was digested at 37 °C for 15 min in an enzyme solution (containing 20 units ml−1 papain, 0.5 mm EDTA, 1.5 mm CaCl2, and 0.2 mg ml−1l-cysteine) and mechanically triturated in culture medium to obtain dissociated cells. After washing with culture medium (containing 10 % fetal calf serum), LH cells were plated in 35 mm culture dishes at a density of 200 000-300 000 cells per dish and maintained at 37 °C and 5 % CO2. Serum-containing medium was replaced by serum-free medium 1-2 h after plating. The serum-free culture medium contained neurobasal medium (Gibco), 5 % B27 supplement (Gibco), 0.5 mml-glutamine, 100 units ml−1 penicillin-streptomycin and 6 g ml−1 glucose. Neurons were fed twice a week.
Electrophysiological recording
All experiments were performed at room temperature in neurons between 14 and 21 days in vitro. The recording chamber was continuously perfused at a rate of 2 ml min−1 with a bath solution containing (mm): NaCl 150, KCl 2.5, MgCl2 2, CaCl2 2, Hepes 10 and glucose 10; pH 7.3 with NaOH. Whole-cell current and voltage clamp (at -60 mV) were used to observe action potentials, spontaneous, miniature and evoked postsynaptic currents with a L/M EPC-7 amplifier. The patch pipette was made of borosilicate glass (World Precision Instruments) with a Narashige puller (PP-83). The tip resistance of the recording pipettes was 4-6 MΩ after filling with a pipette solution containing (mm): KCl (or KMeSO4) 145, MgCl2 1, Hepes 10, EGTA 1.1, Mg-ATP 2, Na2-GTP 0.5; pH adjusted to 7.3 with KOH. In experiments on calcium currents, the recorded neuron was clamped at -80 mV and then given a voltage step to +20 mV. During these experiments, the bath solution contained (mm): NaCl 100, TEA-Cl 40, KCl 2.5, BaCl2 5, Hepes 10, glucose 10, TTX 1 (μm); pH adjusted to 7.3 with KOH. The pipette solution contained (mm): CsCl 135, MgCl2 1, Hepes 10, BAPTA-Cs 5, Mg-ATP 4, Na2-GTP 0.5; pH adjusted to 7.3 with CsOH. After a gigohm seal and whole-cell access were achieved, the series resistance was between 20 and 40 MΩ and partially compensated by the amplifier. Both input resistance and series resistance were monitored throughout the experiments. Only those recordings with seal resistance higher than 0.8 GΩ and stable series resistance were accepted.
The response of postsynaptic glutamate receptors was tested during voltage clamp by brief pressure application (20 ms duration, 8 p.s.i, 5 s interval between each application) of glutamate (100 μm) in the absence or presence of MCH; MCH was applied continuously for 4 min prior to glutamate application. Under current clamp, negative currents (20 pA, 100 ms) were injected to monitor membrane conductance for control purposes. To study voltage-dependent sodium currents, 40 mm TEA-Cl (an equal amount of NaCl was removed) and 200 μm CdCl2 were added to all extracellular solutions and CsCl was used in the pipette solution. To examine voltage-dependent potassium currents, 1 μm TTX and 200 μm CdCl2 were added to all extracellular solutions.
All data were sampled at 3-10 kHz and filtered at 1-3 kHz with an Apple Macintosh computer using AxoData 1.2.2 (Axon Instruments). Data were analysed with Axograph 3.5 (Axon Instruments) and plotted with Igor Pro software (WaveMetrics, Lake Oswego, OR, USA). Both miniature and spontaneous postsynaptic currents were detected and measured with an algorithm in Axograph 3.5 (Clements & Bekkers, 1997; Gao & van den Pol, 1999). Briefly, a simulated template with the width and time course of a typical synaptic event was moved along the recorded data trace one point at a time. At each position, this template was optimally scaled and offset to fit the data so that an event could be detected. The template function used to approximate the time course of miniature and spontaneous synaptic events had a flat baseline region followed by an exponential rise and decay typical of a synaptic event:
where τrise is the time constant of the rising phase, τdecay is the time constant of the falling phase of the template and t is the time from onset of an idealised synaptic event.
The same criteria were applied during control periods, drug treatment and washout. To eliminate electronic noise, we only used signals > 5 pA. All data were reported as means ±s.e.m. The median and cumulative probability of miniature EPSC (mEPSC) and IPSC amplitudes were analysed based on more than 50 events per condition/cell detected with the above method. In each tested neuron, mEPSCs were obtained by analysing at least 50 detected events from control, MCH and washout groups. The rise time was measured from 10 to 90 % of the maximum amplitude using Axograph software (Axon Instruments). The decay time constant of mEPSC events was determined by fitting the averaged mEPSC events with a monoexponential function with a simplex optimisation procedure in Axograph software. A likelihood estimator was used to generate a goodness of fit with a precision of 1 %. Student's t test and ANOVA were used to compare two and more groups of data, respectively. The Kolmogorov-Smirnov test was used to evaluate the significance of change in the amplitude of miniature postsynaptic currents.
MCH was obtained from Phoenix Pharmaceuticals, Inc. (Belmont, CA, USA) The lyophilised peptide was reconstituted in DMSO to a concentration of 1 mm and stored at -70 °C. The stock solution was diluted 1000-fold to the working concentration of 1 μm just prior to use. DMSO (0.1 %) was used in control buffers and did not contribute to the actions of MCH. 2-Amino-5-phosphono-pentanoic acid (AP5), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), and bicuculline methobromide (BIC) were obtained from Sigma-RBI (Research Biochemicals International, MO, USA). Tetrodotoxin (TTX) was obtained from Alomone Labs, Ltd (Jerusalem, Israel). Pertussis toxin (PTX) was obtained from Sigma (St Louis, MO, USA).
RESULTS
To study the actions of MCH at a cellular level, we used synaptically coupled LH neurons in tissue culture. Cultured neurons have an advantage for studying some mechanistic questions relating to peptide actions; the peptide can be delivered rapidly to the cell surface, and complete washout can be achieved rapidly. Although brain slices preserve the local connections, some peptides take several minutes just to reach the recorded cell, and consequently the peptide concentrations can ramp up slowly, rather than arriving rapidly. Furthermore, washout in slices may be substantially retarded, taking up to an hour, particularly for peptides that may stick non-specifically to cell surfaces. Another advantage of simple systems is that they are more amenable to quantal analysis of miniature postsynaptic currents, used to investigate pre- and postsynaptic actions of neuromodulators (Bekkers & Stevens, 1995).
In our initial experiments, flow pipette application of MCH (1 μm) strongly depressed action potentials in cultured LH neurons within 2 min (n = 5), as shown in an example from a typical cell in Fig. 1. Prior to peptide application, the mean frequency of action potentials was 8 ± 4 spikes min−1. MCH reduced the spike frequency to 0.4 ± 0.2 spikes min−1. MCH also hyperpolarised the membrane potential from a pre-MCH level of -52.7 ± 2.9 mV to -59.8 ± 5.0 mV (n = 5) in the presence of MCH. The MCH-mediated changes in spike frequency and membrane potential were statistically significant (P < 0.05, t test).
Figure 1. MCH inhibits action potentials in LH neurons.

The trace shown here was recorded under current clamp. Resting membrane potential is -60.4 mV. MCH (1 μm) induced a dramatic depression of spontaneous action potentials, which was reversible after MCH washout. The mean membrane potential was slightly hyperpolarised in the presence of MCH.
As a further control, we compared the actions of MCH with hypocretin, another LH peptide that has been linked to feeding. Application of hypocretin (1 μm) had the opposite effect to MCH. Hypocretin stimulated an increase in general synaptic activity, as previously reported (de Lecea et al. 1998; van den Pol et al. 1998). Even single LH neurons that showed a decrease in synaptic activity in response to MCH showed an increase in response to hypocretin; the actions of the two peptides could be mediated by different presynaptic cells in synaptic contact with the recorded cell. The opposing actions of MCH and hypocretin are further evaluated below in the specific context of excitatory and inhibitory synaptic actions.
MCH does not change membrane potential and conductance
The mechanism underlying the inhibitory effect of MCH on action potentials and the resting membrane potential might be a direct action on membrane properties or an indirect effect on neurotransmission. Previous reports in Xenopus oocytes transfected with the MCH receptors indicate that MCH can activate G-protein gated inwardly rectifying potassium (GIRK) currents (Bächner et al. 1999). If MCH activated GIRK currents in our study, a hyperpolarisation in membrane potential would be expected (Hille, 1992). To address this question, the effect of MCH on resting membrane potential and conductance was tested in the presence of AP5, CNQX, BIC and TTX. Blocking glutamate and GABA receptors effectively eliminated synaptic activity. No effect of MCH on the resting membrane potential was detected (Fig. 2A). The average resting membrane was -58.3 ± 0.7 mV (n = 15) prior to MCH application. The mean resting membrane potential was -57.7 ± 1.5 mV during the application of MCH (from 40 s to 1.5 min) and -57.7 ± 1.4 mV after the withdrawal of MCH (Fig. 2B, n = 15). No significant difference was found (P = 0.91, ANOVA test).
Figure 2. MCH does not modulate membrane potential and conductance directly.

A, the trace presented here was recorded under current clamp in the presence of AP5 (100 μm), CNQX (10 μm) BIC (30 μm) and TTX (1 μm). Hyperpolarising current (20 pA, 100 ms duration, 1 Hz) was injected at regular intervals to test membrane conductance (inset). MCH exerted no effect on membrane potential or conductance. Resting membrane potential was -63.5 mV. B and C, bar graphs showing resting membrane potential and conductance before, during and after MCH application in all 15 neurons. MCH had no effect. D, the trace was from one of seven typical neurons, whose resting membrane potential and conductance was not changed by MCH. The experiments were also performed in the presence of of AP5 (100 μm), CNQX (10 μm), BIC (30 μm) and TTX (1 μm). Baclofen application (4 μm) exerted a substantial decrease in membrane potential, and a modest increase in conductance, shown by the smaller amplitude hyperpolarisation upon current injection. Initial resting membrane potential was -63.4 mV. E and F, bar graphs show baclofen evokes a hyperpolarisation of the resting membrane potential and a slight increase in conductance during application (n = 7).
As a control to demonstrate that G-protein activated K+ channels do exist in these cells, the effect of baclofen, an agonist of GABAB receptors, was tested in seven neurons. Experiments were done in the presence of AP5, CNQX, BIC and TTX to block synaptic activation of glutamate and GABA receptors (Fig. 2D). None of these cells showed any hyperpolarisation in response to MCH. A 40 s application of baclofen (4 μm) induced a rapid and dramatic hyperpolarisation of the neuronal membrane in six of seven neurons. The membrane potential was hyperpolarised from -56.8 ± 1.9 to -64.1 ± 2.7 mV in the presence of baclofen and returned to -56.9 ± 1.9 mV after washout of baclofen (Fig. 2E, n = 7); this was a statistically significant difference (P < 0.05, ANOVA test). At the same time, membrane conductance increased slightly from 1.72 ± 0.16 to 1.90 ± 0.19 nS in the presence of baclofen and returned to 1.75 ± 0.16 nS after the removal of baclofen (Fig. 2F).
In addition to the lack of effect on membrane potential, MCH had no effect on membrane conductance (P = 0.95, ANOVA test). Baseline membrane conductance was 1.42 ± 0.15 nS prior to peptide application, 1.37 ± 0.14 nS in the presence of MCH and 1.36 ± 0.14 nS after the washout of MCH (Fig. 2C, n = 15).
Thus, our data suggest that MCH has no direct effect on the resting membrane potential or membrane conductance of LH neurons.
MCH inhibits synaptic transmission
After failing to find any direct inhibitory effect of MCH on the resting membrane potential or membrane conductance, we next tested the hypothesis that MCH-mediated inhibition was due to an enhancement of GABA activity, or to a reduction in glutamate actions, or to a reduction in both. Almost all the fast synaptic activity in the hypothalamus appears to be mediated by glutamate or GABA release (van den Pol et al. 1990; Kim & Dudek, 1992; van den Pol & Trombley, 1993). We therefore selectively blocked GABAA or ionotropic glutamate receptors to study the actions of MCH on each of the transmitters independently. LH neurons 14-21 days in vitro (DIV) were used. After a 10 min recording of the control baseline of spontaneous inhibitory postsynaptic currents (sIPSCs), MCH (1 μm) was applied through a flow pipe. MCH induced a dramatic reduction in the frequency of sIPSCs in 11 neurons tested in the presence of AP5 and CNQX. A striking reduction in the frequency of sIPSCs occurred within 2 min after the application of MCH, as shown in Fig. 3C. The sIPSC frequency was reduced from a control level of 103 ± 41 min−1 to 57 ± 18 min−1 and returned to 82 ± 19 min−1 after MCH washout (Fig. 3A). When normalised to control, MCH reduced the frequency of sIPSCs by 33.8 ± 5.8 % (n = 11), a very significant decrease (Fig. 3D; P < 0.01, ANOVA test).
Figure 3. MCH and hypocretin: opposing effects on the frequency of spontaneous inhibitory postsynaptic currents (sIPSCs).

A and B, raw traces recorded in the presence of glutamate receptor antagonists CNQX (10 μm) and AP5 (50 μm) under voltage clamp. A, MCH (1 μm) reduced the frequency of spontaneous IPSCs. B, hypocretin (1 μm) increased frequency of IPSCs. C, normalised data from five neurons were plotted to show the time course of MCH application. The frequency of sIPSCs was normalised to the mean of control sIPSC frequency. During application of MCH (1 μm), a depression of GABAergic synaptic transmission developed. D, bar graph showing the relative effect of MCH and hypocretin on the frequency of sIPSCs.
As a control for the specificity of the actions of MCH, we compared it to the actions of hypocretin. In contrast to the inhibitory actions of MCH, hypocretin (1 μm) caused a dramatic increase in the frequency of sIPSCs in the same group of LH neurons. The frequency of sIPSCs was increased from a baseline of 105 ± 38 min−1 to 189 ± 64 min−1 during hypocretin administration, and returned to 84 ± 12 min−1(n = 5) after hypocretin washout (Fig. 3B). The increase in the frequency of sIPSCs by hypocretin (97.6 ± 34.2 %, n = 5) was statistically significant (Fig. 3D; P < 0.05, ANOVA test).
In the presence of the GABAA receptor antagonist bicuculline, the effect of MCH on glutamatergic synaptic transmission was tested. After a 10 min control period, MCH was given through a flow pipe to the recorded neuron. As shown in Fig. 4A, MCH dramatically reduced the frequency of spontaneous excitatory postsynaptic currents (sEPSCs). As indicated in Fig. 4C, the reduction in the frequency of sEPSCs began within 1 min after the initial application of MCH. The frequency of sEPSCs was reduced from a baseline of 383 ± 82 min−1 to 217 ± 89 min−1 and returned to 368 ± 212 min−1 after peptide washout in six tested neurons. The reduction in the frequency of sEPSCs was 66.0 ± 5.3 %, and very significant statistically (Fig. 4D; P < 0.01, ANOVA test). In contrast, hypocretin caused a significant increase in the frequency of sEPSCs by 36.8 ± 14.0 % (Fig. 4B and D; P < 0.05, ANOVA test, n = 6). Together our data suggest that MCH significantly inhibits both GABA and glutamate synaptic transmission; in contrast, hypocretin enhances synaptic transmission of both amino acid transmitters in LH neurons. There was no significant change in the holding current during the application of MCH when recording either EPSCs or IPSCs.
Figure 4. MCH and hypocretin: opposing effects on the frequency of spontaneous excitatory postsynaptic currents (sEPSCs).

A and B, raw traces recorded in the presence of GABAA receptor antagonist bicuculline (30 μm) under voltage clamp. A, MCH (1 μm) reduced the frequency of spontaneous EPSCs. B, hypocretin (1 μm) increased the frequency of sEPSCs. C, normalised data from two neurons were plotted to indicate the time course of MCH treatment. D, comparison of the effect of MCH and hypocretin on the frequency of sEPSCs demonstrates the opposing actions of the two LH peptides.
MCH decreased both glutamate- and GABA-mediated synaptic transmission. The MCH-mediated inhibition of glutamate transmission (66 % decrease) was substantially more pronounced than the inhibition of GABA transmission (34 % decrease) (P < 0.01, t test).
The action of MCH on miniature postsynaptic currents
The inhibitory effect of MCH on the spontaneous IPSCs and EPSCs led to a second question. Is the inhibitory action of MCH related to depression of presynaptic release machinery or postsynaptic neurotransmitter receptors? The investigation of miniature postsynaptic currents (mPSCs) has been commonly used to address questions related to pre- or postsynaptic modulator actions, often associated with calcium-independent release mechanisms (Bekkers & Stevens, 1995; Chen & van den Pol, 1997; Kondo & Marty, 1997). Miniature currents occur in the presence of tetrodotoxin (TTX) which blocks Na+-dependent action potentials and functionally separates the axon terminal from somatic activity. In this study both miniature inhibitory postsynaptic currents (mIPSCs) and miniature excitatory postsynaptic currents (mEPSCs) were studied.
In the presence of TTX (1 μm), AP5 (50 μm) and CNQX (10 μm), the frequency of bicuculline-blockable GABA mIPSCs was changed from 88 ± 39 min−1 to 75 ± 32 min−1 during the application of MCH and returned to 88 ± 37 min−1 in seven neurons tested (Fig. 5A). The reduction in the frequency of mIPSCs (87.0 ± 7.0 % of control) was not significant as suggested by ANOVA test (Fig. 5B; P = 0.26). In addition to the frequency of mIPSCs, two parameters (median and cumulative probability of detected synaptic events) related to the amplitude of mIPSCs were examined here. The median of detected events was only slightly reduced, from 12.7 ± 0.8 pA to 11.9 ± 0.7 pA, in the presence of MCH and returned to 13.4 ± 2.0 pA after the washout of MCH. The difference between the median of control and MCH-treated events (6.2 ± 2.5 %, n = 7) was small and statistically insignificant (Fig. 5C). The Kolmogorov-Smirnov test also suggested that the cumulative probability of detected events did not change during the treatment of MCH (Fig. 5D; P = 0.13). These data suggest that MCH did not evoke a substantial change in the release machinery or in the postsynaptic receptor at GABAergic synapses.
Figure 5. MCH and miniature IPSCs.

A, raw traces recorded in the presence of TTX (1 μm), CNQX (10 μm) and AP5 (50 μm) under voltage clamp (-60 mV). B, normalised frequency of mIPSCs from all tested neurons was plotted. ANOVA test suggested insignificant change. C, mean normalised medians of detected synaptic events from all tested neurons were plotted. D, cumulative distribution of detected events before, during and after MCH application was plotted. Although a small MCH-induced depression in mIPSC frequency was noted, a Kolmogorov-Smirnov test revealed no statistically significant change during the application of MCH.
At glutamate synapses, the action of MCH was investigated by examining the change in mEPSCs. In the presence of TTX (1 μm) and bicuculline (30 μm), MCH induced a dramatic decrease in the frequency of glutamate-mediated mEPSCs (blockable by 100 μm AP5, 10 μm CNQX, not shown), as shown in Fig. 6A. The reduction in the frequency of mEPSCs occurred within 2 min of the initial application of MCH (Fig. 6B). The frequency was reduced from a control level of 157 ± 66 min−1 (100 %) to 74.7 ± 7.8 % of control in the presence of MCH and returned to 87.9 ± 9.1 % of control after the washout of MCH in eight tested neurons. As normalised to percentage of control, the reduction in the frequency of mEPSCs (25.3 %, n = 8) was significant (P < 0.05, ANOVA test).
Figure 6. MCH-mediated depression of frequency and amplitude of mEPSCs.

A, raw traces presented here were recorded in the presence of TTX (1 μm) and BIC (30 μm) under voltage clamp (-60 mV). B, normalised frequency of mEPSCs from two neurons was plotted to show the time course of MCH actions. C, normalised frequency of mEPSCs from all eight neurons was plotted in this bar graph. D, a representative cumulative distribution from five of eight neurons showing that the cumulative probability of detected events was changed during the application of MCH. E, a representative cumulative distribution from three of eight neurons showing that the cumulative probability of detected events was not changed during the application of MCH, although the frequency was reduced by 15 %. F, mean normalised median of mEPSC amplitudes before, during and after MCH treatment was plotted.
The postsynaptic actions at glutamatergic synapses were also altered by MCH. The median of detected miniature events from eight neurons was decreased from 12.4 ± 1.4 pA to 9.15 ± 1.0 pA by MCH (tested after 4 min in MCH) and returned to 12.2 ± 1.4 pA after washout of MCH. The reduction in amplitude of the excitatory events (21.9 ± 9.2 %, n = 8) was significant as indicated by an ANOVA test (Fig. 6F). Furthermore, the change (left shift) in the cumulative probability curve was also significant as indicated by a Kolmogorov-Smirnov test (Fig. 6D). A change in the frequency of miniature postsynaptic currents (mPSCs), as measured here, has previously been suggested to indicate a presynaptic site of action (Bekkers & Stevens, 1995; Carroll et al. 1999). Based on previous studies that detected similar reductions in amplitude and frequency of mEPSCs during neuromodulation, the reduction in amplitude was determined to account for only a minor apparent reduction in frequency due to the loss of small events at the threshold of detection (Carroll et al. 1999). In the present study, in some cells the frequency of mEPSCs was reduced by MCH to 85 % of control frequency with no decrease in EPSC amplitude (Fig. 6E; three of eight neurons), further supporting the view that in some cells MCH could act primarily at a presynaptic site, with no postsynaptic component.
We also examined the effect of MCH on the kinetics of mEPSC events (rise time and decay time constants), but found little effect. The rise time was 1.14 ± 0.02 ms prior to, 1.18 ± 0.03 ms during and 1.15 ± 0.02 ms after the application of MCH. The monoexponential time constants for averaged mEPSC events prior to, during and after the application of MCH were 3.43 ± 0.20 ms, 3.58 ± 0.15 ms and 3.66 ± 0.20 ms, respectively (n = 8 cells with 50 events per cell).
Postsynaptic inhibitory modulation of glutamate receptor
In addition to a presynaptic action, MCH could depress EPSCs by reducing the amplitude of the postsynaptic response. To determine if MCH might depress the actions of glutamate at postsynaptic receptors, glutamate-evoked responses were studied. In five of six cells tested, the response of the glutamate receptor to its agonist (100 μm glutamate) was decreased by up to 25 % by the application of MCH; one cell showed no decrease. In these experiments, glutamate was applied in 10 baseline trials, and then 4 min after incubation in MCH another 10 trials were done, followed by washout and recovery 5 min later. The mean glutamate response prior to MCH was 97.8 ± 28.0 pA; this was depressed to 92.1 ± 3.6 % (n = 6) in the presence of MCH, and recovered to 99.4 ± 7.4 % (n = 6; Fig. 7). The MCH-induced depression of the evoked inward current was statistically significant at the P < 0.055 level (1-tail t test, n = 6). Multiple trials (n = 10 in each condition) in single cells allowed us to make a statistical comparison of the actions of MCH in each of the six cells. Five of the six neurons showed a reliable MCH-mediated depression of glutamate, with P values ranging from < 0.05 to < 0.01, a significant decrease in each of those five neurons. This result is consistent with the view that MCH can reduce the amplitude of the neuronal response to glutamate.
Figure 7. MCH-mediated depression of glutamate-evoked response.
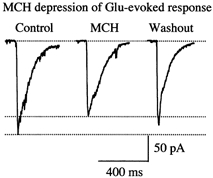
Raw data recorded under voltage clamp (-60 mV) showing an MCH-mediated depression of glutamate-evoked current. l-Glutamate (100 μm) was applied via a flow pipe and pressure (8 p.s.i., 20 ms duration) to the recorded neurons before, during and after the application of MCH.
MCH inhibits voltage-dependent calcium current but not sodium or potassium currents
Our data suggest that MCH inhibits synaptic transmission. One mechanism of inhibition of transmitter release may be a reduction in inward voltage-dependent calcium currents necessary for transmitter exocytosis (Mintz et al. 1995; Turner & Dunlap, 1995; Wu & Saggau, 1994, 1997). In 14-21 DIV LH neurons, BaCl2 was substituted for CaCl2 in the bath solution to increase the conductance at the calcium channel. IBa was activated with a voltage step from -80 to +20 mV for 350 ms under voltage clamp. After recording a control baseline, MCH was applied to the recorded neuron. As shown in Fig. 8A, the peak value of IBa was rapidly and dramatically decreased from 160.0 ± 25.5 to 73.2 ± 18.9 pA within the first minute of MCH application in nine tested neurons. In six of nine neurons maintained in the presence of MCH for an extended time, 5 min, the amplitude of IBa began to return spontaneously to near the control level, suggesting desensitisation. In the other three neurons, the peak value of IBa only partially recovered or remained at a decreased level. After washout of MCH, the IBa recovered to 113.6 ± 20.8 pA in all nine neurons. The reduction in IBa (51.6 ± 7.8 %, n = 9) was very significant as determined by ANOVA (P < 0.01). These data support the conclusion that MCH inhibits voltage-dependent calcium channels.
Figure 8. MCH-mediated depression of voltage-dependent calcium channels.

Left traces in A, whole-cell current of calcium channel in BaCl2 bath solution. Traces show the pre-MCH control (1), inhibition induced by MCH (2), a partial return to control levels after 4 min of MCH application (3) and recovery after MCH washout (4). Right panel in A, mean peak IBa before (□), during ( ) and after (▪) MCH application from all nine neurons is shown. B, whole-cell current of voltage-dependent sodium channels. Experiments were done in the presence of 40 mm TEA and 200 μm CdCl2 in all extracellular solutions and CsCl in the pipette solution. Left traces represent sodium currents prior to (1), during (2) and after (3) MCH application. Right, mean peak INa before (□), during (
) and after (▪) MCH application from all nine neurons is shown. B, whole-cell current of voltage-dependent sodium channels. Experiments were done in the presence of 40 mm TEA and 200 μm CdCl2 in all extracellular solutions and CsCl in the pipette solution. Left traces represent sodium currents prior to (1), during (2) and after (3) MCH application. Right, mean peak INa before (□), during ( ) and after (▪) MCH application from all seven neurons is shown. C, whole-cell current recordings of voltage dependent potassium channels. Experiments were done in the presence of TTX (1 μm) and CdCl2 (200 μm) in all extracellular solutions. Left traces represent potassium currents prior to (1), during (2) and after (3) MCH application. Right, mean peak IK before (□), during (
) and after (▪) MCH application from all seven neurons is shown. C, whole-cell current recordings of voltage dependent potassium channels. Experiments were done in the presence of TTX (1 μm) and CdCl2 (200 μm) in all extracellular solutions. Left traces represent potassium currents prior to (1), during (2) and after (3) MCH application. Right, mean peak IK before (□), during ( ) and after (▪) MCH application from all seven neurons is shown.
) and after (▪) MCH application from all seven neurons is shown.
In a previous report on non-neuronal cells cotransfected with the MCH receptor, SLC-1, an effect of MCH on potassium currents was noted (Bächner et al. 1999). In experiments described above, we found no effect of MCH on membrane potential. To determine if MCH might modulate other voltage-dependent potassium or sodium currents in LH neurons, IK was activated by a voltage step from -80 mV to +20 mV in the presence of TTX (1 μm) and CdCl2 (200 μm). The mean peak IK was not affected by the presence of MCH (Fig. 8C). Control IK was 967.1 ± 74.3 pA prior to MCH application, and 974.5 ± 76.2 pA during MCH application (n = 7). Similarly, voltage-dependent sodium channels did not change during the application of MCH (Fig. 8B). The mean control INa was 2.15 ± 0.54 nA prior to MCH application and 2.03 ± 0.54 nA during MCH application (n = 7). These data suggest that MCH does not modulate sodium or potassium channels involved in regulating membrane potential in response to depolarisation in these LH cells.
Actions of MCH are dependent on Gi/Go G-proteins
In was recently determined that MCH is the natural ligand for the orphan receptor SLC-1 (Chambers et al. 1999; Saito et al. 1999; Shimomura et al. 1999). There is evidence that the MCH receptor may couple to the Gi/Go-mediated signal transduction pathway. MCH has been shown to reduce forskolin-stimulated rises in cAMP in SLC-1-transfected frog oocytes and HEK and CHO cells (Bächner et al. 1999; Chambers et al. 1999; Saito et al. 1999). To determine the involvement of Gi/Go G-proteins in the action of MCH in neurons, 14-21 DIV LH neurons were pretreated with pertussis toxin (PTX, 300 ng ml−1) for 48 h before MCH was tested. As shown in Fig. 9, the action of MCH was completely abolished in PTX-pretreated cultures. MCH evoked no inhibition of GABAergic synaptic transmission after PTX treatment, with the frequency of sIPSCs being 18 ± 6 min−1 before MCH treatment, 20 ± 9 min−1 during MCH treatment and 16 ± 6 min−1 after washout of MCH. The change in the frequency of sIPSCs was statistically insignificant (n = 5). Similarly, after PTX treatment, MCH caused no decrease in glutamatergic synaptic transmission, with the frequency of sEPSCs being 32 ± 4 min−1 before MCH treatment, 36 ± 4 min−1 during MCH treatment and 39 ± 2 min−1 after washout of MCH, a change that was not statistically significant (n = 5). Thus, our results suggest that the inhibition of the Gi/Go pathway abolished the actions of MCH in LH neurons.
Figure 9. MCH actions are dependent on pertussis toxin-sensitive Gi/Go pathway.
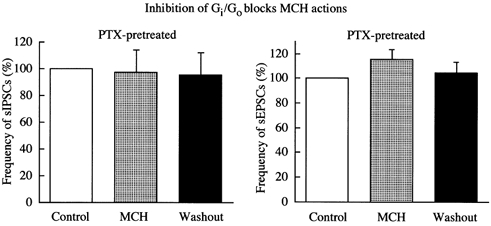
LH neurons were treated with 300 ng ml−1 pertussis toxin (PTX) for 48 h before MCH was tested. The inhibitory effects of MCH on sEPSCs and sIPSCs were abolished by pertussis toxin.
DISCUSSION
Our study appears to be the first to examine the cellular actions of MCH in mammalian neurons. We find that MCH exerts a potent inhibition on synaptic activity of LH neurons. MCH reduced and sometimes completely abolished spontaneous action potentials in LH neurons. Glutamate, and to a lesser degree, GABA synaptic transmission was inhibited by MCH. Since MCH did not appear to exert a direct effect on membrane potential or conductance, the membrane hyperpolarisation and depression of action potential frequency may result from the reduction of excitatory input to the postsynaptic neurons, parallel to the hyperpolarisation evoked by ionotropic glutamate receptor antagonists.
Mechanisms of MCH-mediated inhibition
Our work showing that pertussis toxin blocks the actions of the native MCH receptor in neurons is consistent with previous experiments done in transfected non-neuronal cells showing that MCH acts via a G protein, particularly Gi/Go, after binding to its cloned receptor, SLC-1 (Chambers et al. 1999; Bächner et al. 1999; Lembo et al. 1999). Although MCH does not appear to have been studied with whole-cell recording in mammalian neurons before, in non-neuronal cells transfected with SLC-1, MCH was demonstrated to modulate the activity of several ions. MCH activated G protein-coupled inwardly rectifying potassium channels and modulated calcium-dependent chloride currents in frog oocytes (Bächner et al. 1999). MCH also modulated calcium mobilisation from intracellular stores via a PTX-sensitive Gi/Go protein pathway in HEK 293 cells (Lembo et al. 1999; Chambers et al. 1999). In neurons, we found that MCH modulated voltage-dependent calcium channels (VDCCs), as shown by the MCH-mediated depression of whole-cell calcium currents. We were unable to detect MCH-mediated modulation of voltage-dependent Na+ and K+ channels. Although we found evidence that GABAB agonists activated GIRK channels, we found no MCH-mediated activation of GIRK channels in the same LH cells showing a GABAB response. We cannot exclude the possibility that MCH might modulate the behaviour of other K+ channel types not directly tested by the stimulus protocols in the present set of experiments. These data underline the importance of studying neuropeptides on neurons, in addition to transfected non-neuronal cells, to investigate possible mechanisms of peptide actions in the brain.
MCH appears to act through a PTX-sensitive Gi/Go protein, leading to a reduced amplitude of calcium currents. Modulation of VDCCs by G-proteins (e.g. Gi/Go protein) has been reported with other neurotransmitter receptors, such as α-adrenoceptors (Schofield, 1991), somatostatin receptors (Shapiro & Hille, 1993) and GABAB receptors (Mintz & Bean, 1993). In studies of MCH inhibition of calcium currents, we found desensitisation of this effect in six of nine neurons; a similar desensitisation was reported for opioid receptor agonists which also activate a Gi/Go protein (Kaneko et al. 1997; 1999) Somatic calcium may play an important role as a second messenger in a number of cellular systems, including gene expression, enzyme activation, action potential shape and nuclear signalling.
Presynaptic terminal
The reduction in the frequency of spontaneous EPSCs and IPSCs induced by MCH suggests a decrease in glutamatergic and GABAergic synaptic transmission. Modulation of calcium influx via VDCCs at presynaptic terminals is a powerful mechanism for controlling neurotransmitter release (Lipscombe et al. 1989; Zeilhofer et al. 1996; Wu & Saggau, 1997; Chen & van den Pol, 1998). Although direct evidence demonstrating the modulation of VDCCs by MCH at presynaptic terminals is still needed, our data showing that MCH depresses whole-cell calcium currents at the cell body suggests such a mechanism might also operate at the axon terminal to mediate the decrease in action potential-dependent transmitter release.
MCH may also have an effect on action potential-independent transmitter release (calcium-independent in most cases) at presynaptic sites, as suggested by the reduction in mEPSC frequency. The reduction in the frequency of mEPSCs suggests that MCH reduced the release probability or number of release sites at presynaptic glutamate terminals. Previous work has demonstrated a presynaptic Gi/Go protein modulation of the frequency of miniature PSCs (Boehm & Betz, 1997). Although a reduction in amplitude of mEPSCs may reduce the detection of the events, studies parallel to our own (Carroll et al. 1999) with similar data concluded that both pre- and postsynaptic actions were involved.
Actions on glutamatergic transmission
In most LH cells tested, we found that MCH caused a postsynaptic depression in the amplitude of glutamate-evoked inward currents and glutamate-mediated mEPSCs. This finding is consistent with previous work on AMPA/kainate types of ionotropic glutamate receptors showing that increased phosphorylation of these receptors enhanced glutamate actions (Raymond et al. 1993; Lee et al. 1998). As a PTX-sensitive Gi/Go G-protein may mediate the inhibitory actions of MCH on LH neurons, activation of this G-protein may reduce the phosphorylation state of glutamate receptors, leading to a depressed inward current upon receptor activation. Although not tested in the present work, other mechanisms such as receptor internalisation or receptor binding affinity could also be modulated by MCH.
Thus, MCH may inhibit glutamate activity by three mechanisms; calcium-dependent transmitter release as indicated by the reduction in calcium currents, calcium-independent release as suggested by the reduction in mEPSC frequency in the absence of spikes, and a postsynaptic reduction in inward currents at the glutamate receptor.
Similarities and differences between the actions of MCH and those of NPY and hypocretin
Hypocretin is another peptide synthesised in LH neurons. The administration of hypocretin may enhance feeding (Sakurai et al. 1998), although the increase in feeding may not be robust (Ida et al. 1998; Lubkin et al. 1998; Edwards et al. 1999). Nonetheless, taken together the body of evidence suggests that hypocretin may play a role in feeding or modulation of energy regulation. In this light it is of interest that the two peptides, MCH and hypocretin, each synthesised by neurons in the same region of the LH and with considerable but not complete overlap in their axon projections (Bittencourt et al. 1992; Peyron et al. 1998; van den Pol, 1999), appear to have opposing actions at the cellular level. The hypocretin-mediated enhancement of synaptic activity found here in LH neurons is similar to that we reported previously in medial hypothalamic neurons (de Lecea, 1998; van den Pol et al. 1998). Hypocretin also plays a role in sleep regulation. The absence of the hypocretin receptors in dogs and the absence of hypocretin in knockout mice cause narcolepsy (Lin et al. 1999; Chemelli et al. 1999; Siegel et al. 1999). More recent experiments indicate that narcoleptic humans lack hypocretin in their cerebrospinal fluid (Nishino et al. 2000), and that hypocretin neurons, but not MCH neurons, are missing or dramatically reduced in number in the hypothalamus of narcoleptics (Peyron et al. 2000; Thannickal et al. 2000), perhaps due to the selective degeneration of hypocretin neurons postnatally (van den Pol, 2000). It is highly probable that MCH also plays a role in a number of functions in addition to its putative role in feeding. This is supported by the wide distribution of MCH fibres, and the selective innervation of brain regions that regulate the autonomic nervous system and play a role in general arousal and activation of many brain systems (Bittencourt et al. 1992; Sawchenko, 1998). This is consistent with the findings that neurons in the region of the LH that contain MCH cells receive extensive converging inputs from a wide variety of sensory and motor regions of the brain (Rolls, 1984).
The actions of MCH at the cellular level can be compared with another peptide that evokes a robust increase in feeding, neuropeptide Y (NPY). NPY may be synthesised in the medial hypothalamus, or elsewhere. NPY-containing axons make synaptic contact with LH cells (Horvath et al. 1999). Both MCH and NPY (Chen & van den Pol, 1996; Obrietan & van den Pol, 1996; van den Pol et al. 1996; Rhim et al. 1997) inhibit synaptic activity in the hypothalamus, both of glutamate- and GABA-releasing neurons. Both appear to act through a PTX-sensitive Gi/Go G-protein. Thus if both MCH and NPY are released onto the same cell, similar cellular mechanisms of action may occur, and similar depressive effects on synaptic activity might be expected.
In Results, we indicated some of the benefits of studying modulatory neuropeptides in synaptically coupled cultured cells in vitro, particularly the fast access of the peptide to cell surfaces, which facilitates an understanding of the speed of peptide actions, and the rapidity of peptide washout, and the utility of simpler systems for quantal analysis (Bekkers & Stevens, 1995). In the course of studying other hypothalamic neuromodulators, particularly NPY, we found parallel inhibitory physiological actions in cultured hypothalamic neurons and in slices of the same areas (Obrietan & van den Pol, 1995; van den Pol et al. 1996). Similarly, in experiments examining the modulatory activity of hypocretin, we found similar excitatory actions in cultures and in slices of the hypothalamus. Studies of the GABAB receptor also demonstrate parallel actions in hypothalamic slices (Kim & Dudek, 1992) and cultures (Chen & van den Pol, 1998). The present set of experiments serves to elucidate some of the cellular actions of MCH; it will be of great interest to examine the effects of MCH on specific neuronal pathways within and outside the hypothalamus using slices and intact brains in future studies. Two other peptides are cleaved from the same precursor as MCH, peptides NEI and NGE (Bittencourt et al. 1992). These may be inactive fragments of the precursor, but limited work has been unable to ascertain the functional attributes of these other peptides (Shimada et al. 1998) suggesting MCH may be the primary neuropeptide in these neurons.
In conclusion, we find that MCH exerts a strong inhibition of synaptic activity in LH neurons. The mechanism appears to be based on a pertussis toxin-sensitive Gi/Go protein coupling that may reduce calcium currents at VDCCs, decrease cation currents at glutamate receptors and reduce transmitter release. The inhibition of both excitatory and inhibitory synaptic activity by MCH underlines the importance of elucidating the precise synaptic relations of MCH, GABA, and glutamatergic cells to fully understand the role of MCH within the brain. This is further underscored by the opposing excitatory actions of hypocretin. Complex behaviours such as feeding that involve neurons of the LH may not be amenable to simple explanations coupling single neuropeptides with a monotonic function.
Acknowledgments
Grant support was provided by NIH NS31573, NS10174, NS34887 and the National Science Foundation.
References
- Anand BK, Brobeck JR. Hypothalamic control of food intake in rats and cats. Yale Journal of Biological Medicine. 1951;24:123–140. [PMC free article] [PubMed] [Google Scholar]
- Bächner D, Kreienkamp HJ, Weise C, Buck F, Richter D. Identification of melanin concentrating hormone (MCH) as the natural ligand for the orphan somatostatin-like receptor 1 (SLC-1) FEBS Letters. 1999;457:522–524. doi: 10.1016/s0014-5793(99)01092-3. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Quantal analysis of EPSCs recorded from small numbers of synapses in hippocampal culture. Journal of Neurophysiology. 1995;73:1145–1156. doi: 10.1152/jn.1995.73.3.1145. [DOI] [PubMed] [Google Scholar]
- Bittencourt JC, Presse F, Arias C, Peto C, Vaughan J, Nahon J-L, Vale W, Sawchenko PE. The melanin-concentrating hormone system of the rat brain: an immuno- and hybridization histochemical characterization. Journal of Comparative Neurology. 1992;319:218–245. doi: 10.1002/cne.903190204. [DOI] [PubMed] [Google Scholar]
- Boehm S, Betz H. Somatostatin inhibits excitatory transmission at rat hippocampal synapses via presynaptic receptors. Journal of Neuroscience. 1997;17:4066–4075. doi: 10.1523/JNEUROSCI.17-11-04066.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broberger C, de lecea L, Sutcliffe JG, Hökfelt T. Hypocretin/orexin and melanin-concentrating hormone-expressing cells form distinct populations in the rodent lateral hypothalamus: relationship to the neuropeptide Y and Agouti gene-related protein systems. Journal of Comparative Neurology. 1998;402:460–474. [PubMed] [Google Scholar]
- Carroll RC, Lissin DV, von zastrow M, Nicoll RA, Malenka RC. Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nature Neuroscience. 1999;2:454–460. doi: 10.1038/8123. [DOI] [PubMed] [Google Scholar]
- Chambers J, Ames RS, Bergsma D, Muir A, Fitzgerald LR, Hervieu G, Dytko GM, Foley JJ, Martin J, Liu WS, Park J, Ellis C, Ganguly S, Konchar S, Cluderay J, Leslie R, Wilson S, Sarau HM. Melanin-concentrating hormone is the cognate ligand for the orphan G-protein-coupled receptor SLC-1. Nature. 1999;400:261–265. doi: 10.1038/22313. [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammel T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Chen G, van den Pol AN. Multiple NPY receptors coexist in pre- and postsynaptic sites: inhibition of GABA release in isolated self-innervating SCN neurons. Journal of Neuroscience. 1996;16:7711–7724. doi: 10.1523/JNEUROSCI.16-23-07711.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, van den Pol AN. Adenosine modulation of calcium currents and presynaptic inhibition of GABA release in suprachiasmatic and arcuate nucleus neurons. Journal of Neurophysiology. 1997;77:3035–3047. doi: 10.1152/jn.1997.77.6.3035. [DOI] [PubMed] [Google Scholar]
- Chen G, van den Pol AN. Presynaptic GABAB autoreceptor modulation of P/Q-type calcium channels and GABA release in rat suprachiasmatic nucleus neurons. Journal of Neuroscience. 1998;18:1913–1922. doi: 10.1523/JNEUROSCI.18-05-01913.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM. Detection of spontaneous synaptic events with an optimally scaled template. Biophysical Journal. 1997;73:220–229. doi: 10.1016/S0006-3495(97)78062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X-B, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FSII, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proceedings of the National Academy of Sciences of the USA. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle AN. Melanin-concentrating hormone. In: Eberle AN, editor. The Melanotropins. Basel: Karger; 1988. pp. 321–332. [Google Scholar]
- Edwards CMB, Abusnana S, Sunter D, Murphy KG, Ghatel MA, Bloom SR. The effect of the orexins on food intake: comparison with neuropeptide Y, melanin-concentrating hormone and galanin. Journal of Endocrinology. 1999;160:7–12. doi: 10.1677/joe.0.160r007. R. [DOI] [PubMed] [Google Scholar]
- Elias CF, Saper CB, Maratos-Flier E, Tritos NA, Lee C, Kelly J, Tatro JB, Hoffman GE, Ollmann MM, Barsh GS, Sakurai T, Yanagisawa M, Elmquist JK. Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. Journal of Comparative Neurology. 1998;402:442–459. [PubMed] [Google Scholar]
- Gallistel CR, Gomita Y, Yadin E, Compbell KA. Forebrain origins and terminations of the medial forebrain bundle metabolically activated by rewarding stimulation or by reward-blocking doses of pimozide. Journal of Neuroscience. 1985;5:1246–1261. doi: 10.1523/JNEUROSCI.05-05-01246.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XB, Chen G, van den Pol AN. GABA-dependent firing of glutamate-evoked action potentials at AMPA/Kainate receptors in developing hypothalamic neurons. Journal of Neurophysiology. 1998;79:716–726. doi: 10.1152/jn.1998.79.2.716. [DOI] [PubMed] [Google Scholar]
- Gao XB, van den Pol AN. Neurotrophin-3 potentiates excitatory GABAergic synaptic transmission in cultured developing hypothalamic neurones of the rat. Journal of Physiology. 1999;518:81–95. doi: 10.1111/j.1469-7793.1999.0081r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath RG. Pleasure and brain activity in man. Journal of Nervous and Mental Disease. 1972;154:3–18. doi: 10.1097/00005053-197201000-00002. [DOI] [PubMed] [Google Scholar]
- Hille B. Modulation, slow synaptic action, and second messengers. In: Hille B, editor. Ionic Channels of Excitable Membranes. Sunderland MA USA: Sinauer Associates, Inc; 1992. pp. 170–201. [Google Scholar]
- Horvath TL, Diano S, van den Pol AN. Synaptic interaction between hypocretin (orexin) and neuropeptide Y cells in the rodent and primate hypothalamus: a novel circuit implicated in metabolic and endocrine regulations. Journal of Neuroscience. 1999;19:1072–1087. doi: 10.1523/JNEUROSCI.19-03-01072.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Viale A, Picard F, Nahon J, Richard D. Effects of leptin on melanin-concentrating hormone expression in the brain of lean and obese Lepob/Lepob mice. Neuroendocrinology. 1999;69:145–153. doi: 10.1159/000054413. [DOI] [PubMed] [Google Scholar]
- Kaneko S, Yada N, Fukuda K, Kikuwaka M, Akaike A, Satoh M. Inhibition of Ca2+ channel current by μ- and κ- opioid receptors coexpressed in Xenopus oocytes: desensitization dependence on Ca2+ channel α1 subunits. British Journal of Pharmacology. 1997;121:806–812. doi: 10.1038/sj.bjp.0701181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Akaike A, Satoh M. Receptor-mediated modulation of voltage-dependent Ca2+ channels via heterotrimeric G-proteins in neurons. Japanese Journal of Pharmacology. 1999;8:324–331. doi: 10.1254/jjp.81.324. [DOI] [PubMed] [Google Scholar]
- Kawauchi H, Kawazoe I, Tsubokawa M, Kishida M, Baker BI. Characterization of melanin-concentrating hormone in chum salmon pituitaries. Nature. 1983;305:321–323. doi: 10.1038/305321a0. [DOI] [PubMed] [Google Scholar]
- Kim YI, Dudek FE. Intracellular electrophysiological study of suprachiasmatic nucleus neurons in rodents: inhibitory synaptic mechanisms. Journal of Physiology. 1992;458:247–260. doi: 10.1113/jphysiol.1992.sp019416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Marty A. Protein kinase A-mediated enhancement of miniature IPSC frequency by noradrenaline in rat cerebellar stellate cells. Journal of Physiology. 1997;498:165–176. doi: 10.1113/jphysiol.1997.sp021849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Lembo PMC, Grazzini E, Cao J, Hubatsch DA, Pelletier M, Hoffert C, St-Onge S, Pou C, Labrecque J, Groblewski T, O'donnell D, Payza K, Ahmad S, Walker P. The receptor for the orexigenic peptide melanin-concentrating hormone is a G-protein-coupled receptor. Nature Cell Biology. 1999;1:267–271. doi: 10.1038/12978. [DOI] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li H, Kadotani R, Rogers W, Lin X, Qui X, Dejong PJ, Nishino S, Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- Lipscombe D, Kongsamut S, Tsien RW. Alpha-adrenergic inhibition of sympathetic neurotransmitter release mediated by modulation of N-type calcium-channel gating. Nature. 1989;340:639–642. doi: 10.1038/340639a0. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Bean BP. GABAB receptor inhibition of P-type Ca2+ channels in central neurons. Neuron. 1993;10:889–898. doi: 10.1016/0896-6273(93)90204-5. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Monzon ME, de souza MM, Izquierdo LA, Izquierdo I, Barros DM, de barioglio SR. Melanin-concentrating hormone (MCH) modifies memory retention in rats. Peptides. 1999;20:1517–1519. doi: 10.1016/s0196-9781(99)00164-3. [DOI] [PubMed] [Google Scholar]
- Moran TH, Lew MF, Blass EM. Intracranial self-stimulation in 3 day old rats. Science. 1981;214:1366–1368. doi: 10.1126/science.6975999. [DOI] [PubMed] [Google Scholar]
- Nahon JL, Presse F, Bittencourt JC, Sawchenko P, Vale W. The rat melanin-concentrating hormone mRNA encodes multiple putative neuropeptides coexpressed in the dorsolateral hypothalamus. Endocrinology. 1989;125:2056–2065. doi: 10.1210/endo-125-4-2056. [DOI] [PubMed] [Google Scholar]
- Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet. 2000;355:39–40. doi: 10.1016/S0140-6736(99)05582-8. [DOI] [PubMed] [Google Scholar]
- Obrietan K, van den Pol AN. Neuropeptide Y depresses GABA-mediated calcium transients in developing suprachiasmatic nucleus neurons: a novel form of calcium long-term depression. Journal of Neuroscience. 1996;16:3521–3333. doi: 10.1523/JNEUROSCI.16-10-03521.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, Li R, Hungs M, Pedrazzoli M, Padigaru M, Kucherlapati M, Fan J, Maki R, Lammers Gj, Bouras C, Kucherlapati R, Nishino S, Mignot E. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nature Medicine. 2000;6:991–997. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. Journal of Neuroscience. 1998;18:9996–10008. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu D, Ludwig DS, Grammeltpft S, Piper M, Pelleymounter MA, Cullen MJ, Mathes WF, Przypek J, Kanarek R, Maratos-Flier E. A role of melanin-concentrating hormone in the central regulation of feeding behavior. Nature. 1996;380:243–247. doi: 10.1038/380243a0. [DOI] [PubMed] [Google Scholar]
- Raymond LA, Blackstone CD, Huganir RL. Phosphorylation and modulation of recombinant GluR6 glutamate receptors by cAMP-dependent protein kinase. Nature. 1993;361:637–641. doi: 10.1038/361637a0. [DOI] [PubMed] [Google Scholar]
- Rhim H, Kinney GA, Emmerson PJ, Miller RJ. Regulation of neurotransmission in the arcuate nucleus of the rat by different neuropeptide Y receptors. Journal of Neuroscience. 1997;17:2980–2989. doi: 10.1523/JNEUROSCI.17-09-02980.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls ET. The neurophysiology of feeding. International Journal of Obesity. 1984;8:139–150. [PubMed] [Google Scholar]
- Rossi M, Beak SA, Choi SJ, Small CJ, Morgan DGA, Ghatei MA, Smith DM, Bloom SR. Investigation of the feeding effects of melanin concentrating hormone on food intake - action independent of galanin and the melanocortin receptors. Brain Research. 1999;846:164–170. doi: 10.1016/s0006-8993(99)02005-3. [DOI] [PubMed] [Google Scholar]
- Saito Y, Nothacker HP, Wang ZW, Lin SHS, Leslie F, Civelli O. Molecular characterization of the melanin-concentrating-hormone receptor. Nature. 1999;400:265–269. doi: 10.1038/22321. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Mishii I, Matsuzaki RM, Chemelli H, Tanaka SC, Williams JA, Richardson GP, Kozlowski S, Wilson JRS, Arch RE, Buckingham AC, Haynes SA, Carr RS, Annan mcnulty DE, Liu W-S, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: A family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- Sawchenko PE. Toward a new neurobiology of energy balance, appetite, and obesity: The anatomists weigh in. Journal of Comparative Neurology. 1998;402:435–441. [PubMed] [Google Scholar]
- Schofield GG. Norepinephrine inhibits a Ca2+ current in rat sympathetic neurons via a G-protein. European Journal of Pharmacology. 1991;207:195–207. doi: 10.1016/0922-4106(91)90031-c. [DOI] [PubMed] [Google Scholar]
- Shapiro MS, Hille B. Substance P and somatostatin inhibits calcium channels in rat sympathetic neurons via different G protein pathways. Neuron. 1993;10:11–20. doi: 10.1016/0896-6273(93)90237-l. [DOI] [PubMed] [Google Scholar]
- Shimada M, Tritos NA, Lowell BB, Flier LS, Maratos-Flier E. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature. 1998;396:670–674. doi: 10.1038/25341. [DOI] [PubMed] [Google Scholar]
- Shimomura Y, Mori M, Sugo T, Ishibashi Y, Abe M, Kurokawa T, Onda H, Nishimura O, Sumino Y, Fujino M. Isolation and identification of melanin-concentrating hormone as the endogenous ligand of the SLC-1 receptor. Biochemical and Biophysical Research Communications. 1999;261:622–626. doi: 10.1006/bbrc.1999.1104. [DOI] [PubMed] [Google Scholar]
- Skofitsch G, Jacobowitz DM, Zamir N. Immunohistochemical localization of a melanin concentrating hormone-like peptide in the rat brain. Brain Research Bulletins. 1985;15:635–649. doi: 10.1016/0361-9230(85)90213-8. [DOI] [PubMed] [Google Scholar]
- Stellar E. The physiology of motivation. Psychological Reviews. 1954;101:301–311. doi: 10.1037/0033-295x.101.2.301. [DOI] [PubMed] [Google Scholar]
- Sticker EM, Swerdloff AF, Zigmond MJ. Intrahypothalamic injections of kainic acid produce feeding and drinking deficits in rats. Brain Research. 1978;158:470–473. doi: 10.1016/0006-8993(78)90692-3. [DOI] [PubMed] [Google Scholar]
- Thannickal R, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM. Reduced number of hyocretin neurons in human epilepsy. Neuron. 2000;27:469–474. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TJ, Dunlap K. Pharmacological characterization of presynaptic calcium channels using subsecond biochemical measurements of synaptosomal neurosecretion. Neuropharmacology. 1995;34:1469–1478. doi: 10.1016/0028-3908(95)00133-q. [DOI] [PubMed] [Google Scholar]
- Valenstein ES, Campbell JF. Medial forebrain bundle-lateral hypothalamic area and reinforcing brain stimulation. American Journal of Physiology. 1966;210:270–274. doi: 10.1152/ajplegacy.1966.210.2.270. [DOI] [PubMed] [Google Scholar]
- van den Pol AN. Hypothalamic hypocretin (orexin): robust innervation of the spinal cord. Journal of Neuroscience. 1999;19:3171–3182. doi: 10.1523/JNEUROSCI.19-08-03171.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN. Narcolepsy, a neurodegenerative disease of the hypocretin system. Neuron. 2000;27:415–418. doi: 10.1016/s0896-6273(00)00050-7. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Gao XB, Obrietan K, Kilduff T, Belousov A. Pre- and postsynaptic actions and modulation of neuroendocrine neurons by a new hypothalamic peptide, hypocretin/orexin. Journal of Neuroscience. 1998;18:7962–7971. doi: 10.1523/JNEUROSCI.18-19-07962.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Obrietan K, Chen G, Belousov AB. Neuropeptide Y-mediated long-term depression of excitatory activity in suprachiasmatic nucleus neurons. Journal of Neuroscience. 1996;16:5883–5895. doi: 10.1523/JNEUROSCI.16-18-05883.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Trombley PQ. Glutamate neurons in hypothalamus regulate excitatory transmission. Journal of Neuroscience. 1993;13:2829–2836. doi: 10.1523/JNEUROSCI.13-07-02829.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Wuarin JP, Dudek FE. Glutamate, the dominant excitatory transmitter in neuroendocrine regulation. Science. 1990;250:1276–1278. doi: 10.1126/science.1978759. [DOI] [PubMed] [Google Scholar]
- Viale A, Kerdelhue B, Nahon JL. 17 Beta-estradiol regulation of melanin-concentrating hormone and neuropeptide-E-I contents in cynomolgus monkeys: a preliminary study. Peptides. 1999;20:553–559. doi: 10.1016/s0196-9781(99)00007-8. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus. Neuron. 1994;12:1139–48. doi: 10.1016/0896-6273(94)90321-2. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends in Neurosciences. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- Zeilhofer HU, Muller TH, Swandulla D. Calcium channel types contributing to excitatory and inhibitory synaptic transmission between individual hypothalamic neurons. Pflügers Archiv. 1996;432:248–257. doi: 10.1007/s004240050131. [DOI] [PubMed] [Google Scholar]