

Abstract
The role of Ca2+-calmodulin (CaM) signalling cascades in modulating glutamatergic synaptic transmission on CA1 non-pyramidal fast-spiking neurons was investigated using whole-cell recording and perfusion in rat hippocampal slices.
Paired stimuli (PS), consisting of postsynaptic depolarization to 0 mV and presynaptic stimulation at 1 Hz for 30 s, enhanced excitatory postsynaptic currents (EPSCs) on non-pyramidal neurons in the stratum pyramidale (SP). The potentiation was reduced by the extracellular application of d-amino-5-phosphonovaleric acid (DAP-5, 40 μm), and blocked by the postsynaptic perfusion of 1,2-bis(2-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid (BAPTA, 10 mm), a CaM-binding peptide (100 μm) or CaMKII (281–301) (an autoinhibitory peptide of CaM-dependent protein kinases, 100 μm).
The application of adenophostin, an agonist of inositol trisphosphate receptors (IP3Rs) that evokes Ca2+ release, into SP non-pyramidal neurons via the patch pipette (1 μm) enhanced EPSCs and occluded PS-induced synaptic potentiation. The co-application of BAPTA (10 mm) with adenophostin blocked synaptic potentiation. In addition, Ca2+-CaM (40:10 μm) induced synaptic potentiation, which occluded PS-induced potentiation and was attenuated by introducing CaMKII (281–301) (100 μm). EPSCs were sensitive to an antagonist of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPAR).
Application of Ca2+-CaM into SP non-pyramidal neurons induced the emergence of AMPAR-mediated EPSCs that were not evoked by low stimulus intensity before perfusion. Ca2+-CaM also increased the amplitude and frequency of spontaneous EPSCs. A scavenger of nitric oxide, carboxy-PTIO (30 μm in slice-perfusion solution), did not affect these increases in sEPSCs.
The magnitude of PS-, adenophostin- or Ca2+-CaM-induced synaptic potentiation in SP non-pyramidal neurons increased during postnatal development.
These results indicate that Ca2+-CaM signalling pathways in CA1 SP non-pyramidal neurons up-regulate glutamatergic synaptic transmission probably through the conversion of inactive-to-active synapses.
Interneurons in the central nervous system integrate feedforward and feedback inputs, and control the excitation and synchronous activity of the principal neurons (Wong et al. 1986; Lacaille et al. 1987; Alger, 1991; Freund & Buzsaki, 1996). The up-regulation of glutamatergic synapses on an interneuron increases its activity, enabling the interneuron to limit principal neuron hyperactivity more effectively. Several publications indicate that excitatory synapses on hippocampal interneurons express tetanus-induced long-term potentiation (Buzsaki & Eidelberg, 1982; Taube & Schwartzkroin, 1987; Ouardouz & Lacaille, 1995; Maccaferri & McBain, 1996). However, little is known about how intracellular signalling cascades modulate synaptic plasticity in interneurons.
The activation of glutamate receptors raised Ca2+ levels in hippocampal CA1 interneurons (Carmant et al. 1997). Synaptic potentiation in these interneurons required an increase in intracellular Ca2+ (Ouardouz & Lacaille, 1995). We are interested in determining whether the increase of intracellular Ca2+-CaM and the activity of glutamatergic synapses are inter-dependent. The postsynaptic perfusions of an IP3R agonist or Ca2+-CaM (Wang & Kelly, 1995) and the paired stimuli (Kelso et al. 1986; Maccaferri & McBain, 1996) were used to activate Ca2+-CaM signalling cascades in hippocampal CA1 non-pyramidal neurons. Paired stimuli consisted of postsynaptic depolarization to 0 mV and presynaptic stimulation at 1 Hz for 30 s. The use of these protocols should shed light on investigating monosynaptic plasticity, since tetanic stimulation increased the probability of firing action potentials in pyramidal neurons (Andersen et al. 1980), thereby activating more synapses of recurrent axons onto interneurons (Maccaferri & McBain, 1996).
Axon arbors of CA1 interneurons in the stratum pyramidale (SP) mainly synapse on the soma and proximal dendrites of pyramidal neurons (Freund & Buzsaki, 1996). This subcellular architecture enables SP-interneurons to inhibit pyramidal neurons more efficiently. In view of this functional importance, we have studied intracellular signalling mechanisms of synaptic plasticity in CA1 SP non-pyramidal neurons. Our results indicate that excitatory synapses on these neurons express monosynaptic potentiation, in which the postsynaptic Ca2+-CaM signalling pathways and the conversion of inactive-to-active synapses are involved. These mechanisms are enhanced during postnatal development.
METHODS
Hippocampal slices and solution
Slices (400 μm) were prepared from Sprague-Dawley rats in postnatal days (PND) 7-22. Rats were anaesthetized by the inhalation of methoxyflurane (2 ml in a 4 l bell-jar) and then decapitated by a guillotine. Tissue blocks including the hippocampus and partial cortex were quickly isolated in oxygenated (95 % O2 and 5 % CO2) ice-cold artificial cerebrospinal fluid (ACSF), in which 0.5 mm CaCl2 and 4 mm MgSO4 were used to reduce excitation. Slices were cut with a Vibratome, and then held in oxygenated standard ACSF (mm): 124 NaCl, 3 KCl, 1.2 NaH2PO4, 2.4 CaCl2, 1.3 MgSO4, 10 dextrose, and 10 Hepes at 25 °C for 1-2 h. A slice was transferred to a submersion chamber (Warner RC-26G) and perfused with oxygenated standard ACSF at 31 °C for electrophysiological recordings. The concentration of KCl was raised to 4.5 mm to increase the basal level of spontaneous synaptic activity in studying the effect of Ca2+-CaM on sEPSCs (spontaneous excitatory postsynaptic currents).
Electrical stimulation
Bipolar tungsten electrodes (12 MΩ) were used to stimulate Schaffer collateral and/or commissural (S/C) fibres in area CA1. They were located away from the recording neurons to prevent direct triggering of long and/or irregular arbors of interneurons and to reduce the possibility of evoking polysynaptic activity. Stimulus frequency was 0.1 Hz. Paired stimuli for inducing synaptic potentiation were postsynaptic depolarization to 0 mV and 1 Hz presynaptic stimulation for 30 s. Stimulus intensity for studying inactive synapses was set just below the values to evoke EPSCs at the first stimulus in paired pulses when the standard solution was in the pipette tip.
Neuron selection
Recording neurons in the hippocampal area CA1 were initially selected based on their morphology under DIC microscope (Nikon E600FN or Olympus BX50) and electrophysiological properties. Compared with pyramidal neurons, the selected neurons appeared small (10-15 μm) with round or irregular soma and multipolar processes, i.e. non-pyramidal. The membrane of these non-pyramidal neurons displayed higher input resistances and smaller decay-time constants in response to hyperpolarization pulses. Depolarization pulses (60 ms) induced high frequency discharges (fast spiking), in which action potentials appeared as short-duration, deep fast after-hyperpolarization with little frequency and amplitude accommodation (see waveforms in Fig. 1D). These properties match the criteria of interneurons (Lacaille et al. 1987; Freund & Buzsaki, 1996; McBain et al. 1999). Some recording neurons in each experimental group were labelled by perfusing neurobiotin for further identification (Figs 1, 4–6 and 9–10), and none of them appeared pyramidal-like. Neurons in our studies were non-pyramidal and fast spiking.
Figure 1. Paired stimuli enhanced EPSCs in hippocampal CA1 SP non-pyramidal neurons (PND 18-22).
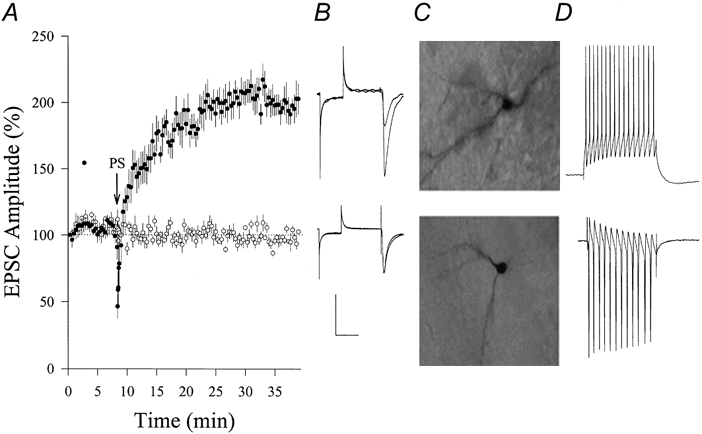
Paired stimuli (PS) were composed of postsynaptic depolarization to 0 mV and 1 Hz presynaptic stimulation for 30 s. A, PS induced increases in EPSCs (•, n = 8) compared with control experiments (no PS; ○, n = 6). Arrow shows PS application. B, waveforms from top to bottom represent EPSCs in potentiation and control, respectively. Calibration bars, 150 pA and 50 ms. C, neurobiotin-labelled non-pyramidal neurons were from potentiation (top) and control (bottom) experiments. Neurobiotin was perfused into neurons through the recording pipette. D, fast-spiking action potentials in non-pyramidal neurons in response to a depolarization pulse (60 ms) during current-clamp (top) and voltage-clamp (bottom) recordings.
Figure 7. Carboxy-PTIO, a nitric oxide scavenger, did not affect Ca2+-CaM-induced increases in the frequency and amplitude of sEPSCs on CA1 SP non-pyramidal neurons (PND 18-22).
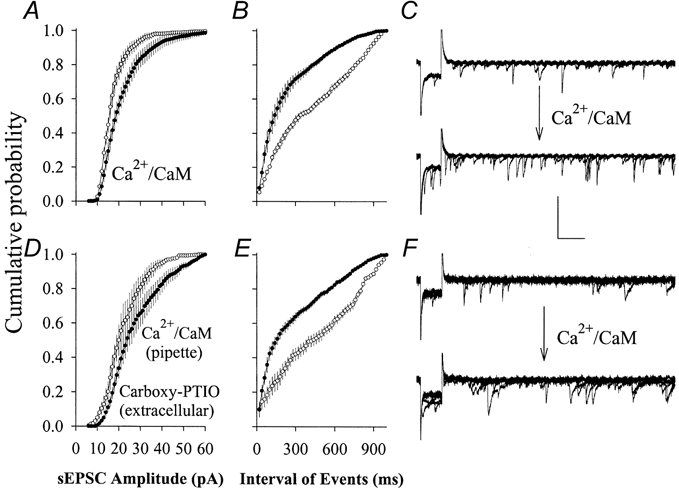
A-C, Ca2+-CaM increased the frequency and amplitude of sEPSCs. Cumulative probability is plotted as a function of EPSC amplitudes and inter-EPSC intervals. Perfusing Ca2+-CaM into non-pyramidal neurons (•, n = 5) shifted EPSC amplitudes larger (A) and inter-EPSC intervals shorter (B). C, sEPSC waveforms before (top) and after perfusing with Ca2+-CaM (bottom); each shows six consecutive superimposed traces. D-F, the postsynaptic application of Ca2+-CaM increased the frequency and amplitude of sEPSCs in the presence of extracellular carboxy-PTIO (30 μm). Cumulative probability is plotted as a function of EPSC amplitudes (D) and inter-EPSC intervals (E). F, sEPSC waveforms before (top) and after perfusing with Ca2+-CaM (bottom); each shows six consecutive superimposed traces. Calibration bars are 60 ms (horizontal), 50 pA for C and 35 pA for F (vertical).
Figure 4. Adenophostin enhanced glutamatergic EPSCs in hippocampal CA1 SP non-pyramidal neurons (PND 18-22) and BAPTA blocked the potentiation.
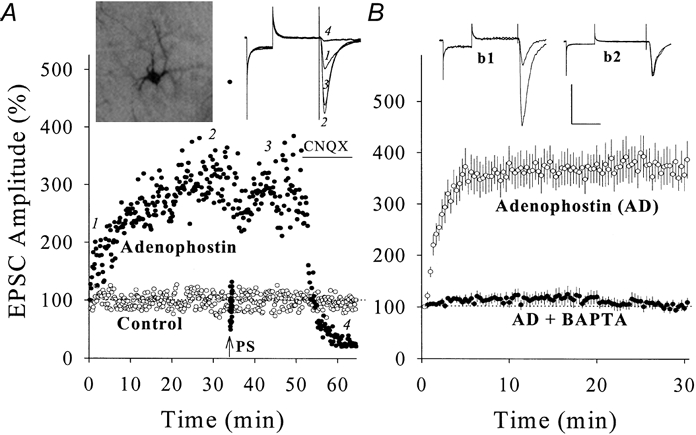
A, the postsynaptic perfusion of adenophostin (1 μm in pipette solution) enhanced EPSCs (•) compared with a control (○). After 34 min, PS (arrow) could not induce further potentiation, and CNQX (10 μm, horizontal bar) blocked EPSCs. Insets show neurobiotin/adenophostin-perfused neuron (left) and EPSC waveforms at time points 1, 2, 3 and 4 (right). B, the co-perfusion of adenophostin with BAPTA (10 mm in pipette) could not induce EPSC increase (•, n = 7) compared with adenophostin alone (○, n = 9). Insets show EPSCs of adenophostin-induced potentiation (b1) and adenophostin + BAPTA-induced potentiation effect (b2). Calibration bars are 150 pA and 50 ms.
Figure 6. The postsynaptic perfusion of Ca2+-CaM causes the emergence of evoked-EPSCs in CA1 SP non-pyramidal neurons (PND 18-22).
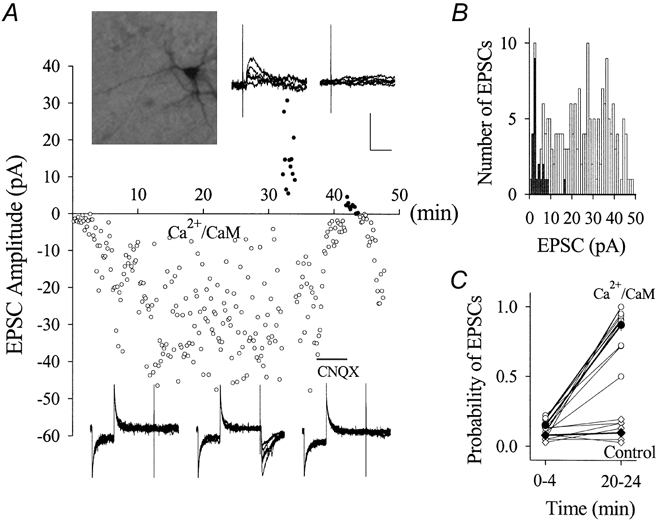
The stimulus intensity for studying inactive synapses was set to evoke no EPSCs in stimulus 1 of paired pulses when the standard solution was in the pipette tip. A, the plot of EPSC amplitudes vs. time shows an example of the effect of perfusing Ca2+-CaM on EPSCs when the membrane potentials were held at -70 and +50 mV. Values of EPSCs are represented as negative at -70 mV (○) and positive at +50 mV (•). EPSCs were not detected in the initial 2 min while the pipette tip was filled with the standard solution. When Ca2+-CaM (40:10 μm back-filled in the pipette) diffused into the neuron, EPSCs emerged over time. Both inward EPSCs at -70 mV and outward EPSCs at +50 mV were blocked by CNQX (10 μm, horizontal bar). Insets on top show a neurobiotin-labelled recording neuron and outward EPSCs recorded at +50 mV before and after applying CNQX. Bottom waveforms (insets, left to right) show EPSCs at -70 mV in control, perfusing Ca2+-CaM and washing-on CNQX. Six consecutive traces are superimposed in each of the insets. Calibration bars are 40 pA and 50 ms (bottom) and 40 pA and 20 ms (top). B, histogram of EPSC amplitudes vs. event number from the example in A. ▪ show EPSCs recorded in the initial 4 min and □ show those during Ca2+-CaM perfusion (4-30 min). C, the probability of detecting evoked EPSCs increased during Ca2+-CaM perfusion (20-24 min) compared with initial 4 min (○, n = 12). Perfusion with standard pipette solution (control) did not change the probability of evoked EPSCs (⋄, n = 6). ♦ and • and thick lines show mean data.
Figure 9. The magnitude of PS-induced synaptic potentiation in CA1 SP non-pyramidal neurons increased during postnatal development.
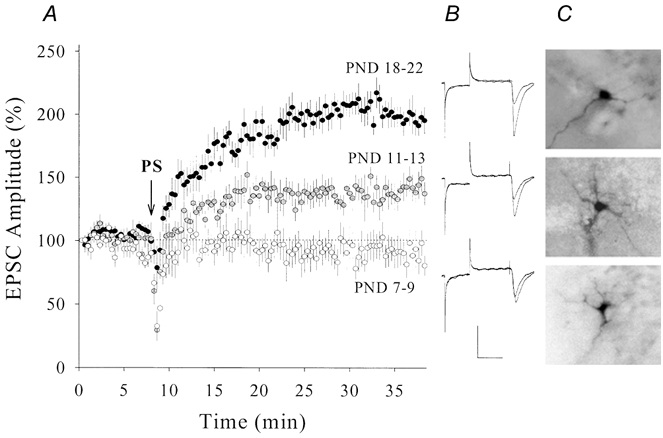
A, synaptic potentiation was higher in postnatal days (PND) 18-22 (•, n = 8) than PND 11-13 ( , n = 7; P < 0.01). Potentiation was not observed in PND 7-9 (○, n = 7). Arrow denotes PS application. B, waveforms from top to bottom represent EPSCs from an example in each group, PND 18-22, 11-13 and 7-9, respectively. Calibration bars are 150 pA and 50 ms. C, neurobiotin-labelled recording neurons from PND 18-22 (top), 11-13 (middle) and 7-9 (bottom).
, n = 7; P < 0.01). Potentiation was not observed in PND 7-9 (○, n = 7). Arrow denotes PS application. B, waveforms from top to bottom represent EPSCs from an example in each group, PND 18-22, 11-13 and 7-9, respectively. Calibration bars are 150 pA and 50 ms. C, neurobiotin-labelled recording neurons from PND 18-22 (top), 11-13 (middle) and 7-9 (bottom).
Figure 10. The magnitude of adenophostin-induced synaptic potentiation in CA1 SP non-pyramidal neurons increased during postnatal development.
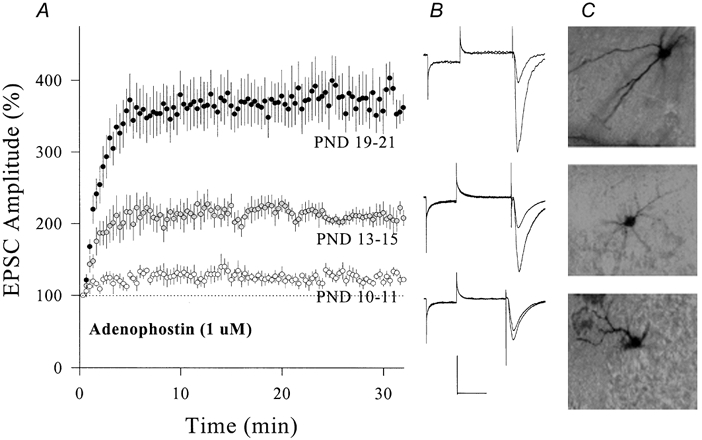
Adenophostin (1 μm) was applied through recording patch pipettes. A, the comparison of adenophostin-induced synaptic potentiation in PND 19-21 (•, n = 9), 13-15 (, n = 6) and 10-11 (○, n = 5; P < 0.01). B, waveforms from top to bottom represent EPSCs from an example in each group PND 19-21, 13-15 and 10-11, respectively. Calibration bars are 150 pA and 50 ms. C, neurobiotin-labelled non-pyramidal neurons from PND 19-21 (top), 13-15 (middle) and 10-11 (bottom) groups.
Electrophysiological recordings
EPSCs (evoked or spontaneous) were recorded in non-pyramidal neurons by whole-cell voltage clamp (Axopatch-1D amplifier, Axon Instrument Inc.) with holding potentials between -70 and -72 mV. At the end of experiments, some slices were perfused with 10 μm 6-cyano-7-nitroquinoxaline-2,3-(1H,4H)-dione (CNQX) to examine whether EPSCs were mediated by the AMPA-type glutamate receptors (Fig. 4–6A and 11). Standard pipette solution (control) contained (mm): 135 potassium gluconate, 15 KCl, 5 NaCl, 10 Hepes, 0.5 EGTA, 4 Mg-ATP and 0.5 Tris-GTP, and was filtered with a centrifuge filter (0.1 μm pores). Care was taken to maintain the osmolarity of pipette solution as 295-310 mosmol l−1. The resistance of the whole-cell pipettes was 6-8 MΩ.
Figure 11. Ca2+-CaM-induced emergence of EPSCs increased during postnatal development.
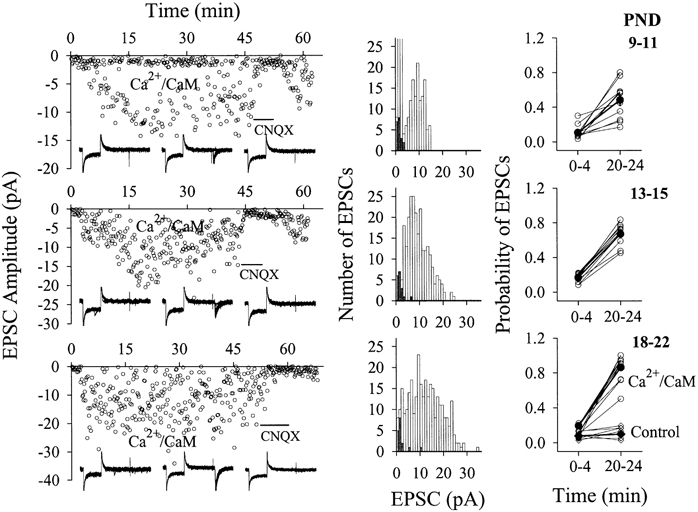
The stimulus intensity for studying inactive synapses was set to evoke no EPSCs in stimulus 1 of paired pulses when the standard solution was in the pipette tip. Panels show results of Ca2+-CaM perfusion into CA1 SP non-pyramidal neurons from rats in PND 9-11 (top row), 13-15 (middle row) and 18-22 (bottom row). Left panels show an example from each group. EPSCs were not detected in the initial 2 min while the pipette tip was filled the standard solution. While Ca2+-CaM (40:10 μm back-filled in pipette) diffused into the recording neuron, EPSCs emerged over time, and were blocked by CNQX (10 μm, horizontal bar). Insets show representative EPSCs from control (left), perfusing Ca2+-CaM (middle) and washing with CNQX (right), each waveform contains ten consecutive superimposed traces. Middle panels show histograms of EPSC amplitudes vs. event number from the examples in the left panels. ▪ show data points in the initial 4 min and □ show EPSCs recorded during Ca2+-CaM perfusion (4-30 min). Right panels show the probability of detecting evoked-EPSCs before (0-4 min) and after Ca2+-CaM perfusion (20-24 min), which significantly increases after Ca2+-CaM perfusion. Comparing the results in these three ages (PND 9-11, n = 11; 13-15, n = 11 and 18-22, n = 12), the maximum amplitudes and probabilities of Ca2+-CaM-induced emergence of EPSCs increase during postnatal development. The perfusion of standard pipette solution (control) did not change EPSC amplitudes and probabilities (right-bottom panel, n = 6). • and ♦ connected by thick lines show mean data, and ○ and ⋄ are from individual experiments.
Recording pipettes were also used to perfuse modulators of signalling pathways into non-pyramidal neurons (postsynaptic neurons) to examine their effects on glutamatergic synaptic transmission. The stock solution of modulators was prepared with distilled water (100 times higher than the final concentration), and diluted with the standard pipette solution before use. Ca2+-CaM was made by mixing Ca2+ and CaM at a molar ratio of 4:1 (Wang & Kelly, 1995). The concentration of inhibitors was above their IC50 but this did not appear to affect basal synaptic transmission under control conditions, and thus the effects of the inhibitors on synaptic potentiation during co-perfusion experiments were probably not due to their side-effects. Whole-cell perfusion was done by filling pipette tips with standard solution and the back-filled pipette with modulator-containing solution, which allows comparisons of the effect of modulators on synaptic transmission with control. The success of this method has been examined by perfusing neurobiotin and examining the resultant fluorescence in the recording neurons 2-4 min after the formation of the whole-cell configuration.
Data analysis
Data were collected and analysed only if neurons displayed stable membrane potentials in the range -65 to -70 mV with no significant changes in series and input resistances throughout each experiment. Series and input resistances were monitored in all experiments by measuring responses to voltage pulses of 5 mV and 50 ms (see the first part of representative waveforms in the figures). We started recording EPSCs when whole-cell access was established and where instantaneous currents were at least threefold higher than steady-state currents (Marty & Neher, 1995). We measured the amplitudes of population, emerged and spontaneous EPSCs. The first population of EPSC was defined as baseline (100 %) and used to normalize the remaining monosynaptic EPSCs. As adenophostin- or Ca2+-CaM-induced synaptic potentiation developed quickly (see Results), the use of the first EPSC as the baseline should minimize the effects of modulators on baseline values. Spontaneous EPSCs were accounted for only when the ratio of sEPSC to baseline noise was above three. Synaptic strengths are presented as means ± standard error of the mean. Values for comparison among groups were selected at the same time (30 min) and tested by ANOVA. Representative waveforms of population EPSCs were averaged from four consecutive responses. Those of emerged and sEPSCs were superimposed from 10 and 6 traces, respectively.
Morphology of recording neurons
Neurobiotin was dissolved in pipette solutions at a final concentration of 0.25-1 %, and back-filled into recording pipettes whose tips contained the standard solution. After recordings, hippocampal slices were perfused with oxygenated ACSF for an additional 30 min to allow neurobiotin filling of fine processes. Then, slices were fixed with 4 % paraformaldehyde, 0.25 % glutaldehyde and 1 % picric acid in 0.1 m phosphate buffer (PBS) at 4 °C for 24-72 h. Slices were incubated in avidin and/or horseradish peroxidase (Vectastain ABC) for 3 h, and in 1 % DAB-CoCl2 (Sigma) 1 min for staining neurobiotin-filled neurons. The reaction was stopped by PBS. Neurobiotin-stained neurons were photographed through an inverted microscope (Nikon Diaphot, × 20 and × 40 objectives).
Chemicals
1,2-bis(2-Aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid (BAPTA), d-amino-5-phosphonovaleric acid (DAP-5), CNQX and carboxy-PTIO (2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide) were purchased from Sigma. Calmodulin (CaM), CaM-binding peptide and CaMKII (281-301) were from Calbiochem. Other chemicals were from Fisher Scientific. Adenophostin was a gift from Dr Masaaki Takahashi (Biological Research Laboratory, Sankyo Co., Ltd, Japan).
RESULTS
Paired stimuli induce synaptic potentiation on CA1 SP non-pyramidal neurons
We studied synaptic plasticity on non-pyramidal neurons in CA1 stratum pyramidale (SP) using paired stimuli (PS, see Methods), which presumably prevents tetanus-induced polysynaptic plasticity in interneurons (Maccaferri & McBain, 1996). Recording pipettes contained the standard solution, and some were back-filled with neurobiotin for labelling neurons. We did not observe significant changes in excitatory postsynaptic currents (EPSCs) in 40 min (98 ± 3 %, n = 6; open symbols in Fig. 1a). In another group with the same age (postnatal days 18-22), PS was applied after 8 min of whole-cell recording. The amplitude of EPSCs gradually increased, and the average level of potentiation at 30 min was 199 ± 15 % (n = 8; filled symbols in Fig. 1a). These results indicate that PS induces synaptic potentiation in CA1 SP non-pyramidal neurons.
In addition to selecting neurons based on their non-pyramidal shape as viewed under a DIC microscope and fast-spiking in response to depolarization (Fig. 1D), we labelled some of them with neurobiotin. None of these stained neurons appears pyramidal-like, and Fig. 1C shows two examples. Hippocampal CA1 SP-neurons in our studies are non-pyramidal and fast spiking, i.e. interneurons (Freund & Buzsaki, 1996).
PS-induced synaptic potentiation requires a Ca2+-CaM signalling pathway
The PS protocol may activate N-methyl-d-aspartate receptors (NMDARs; Mayer et al. 1989; Jahr & Stevens, 1990) and voltage-gated calcium channels (Fox et al. 1987; Tsien et al. 1988). Ca2+ inflow may evoke Ca2+ release (Ehrlich, 1995; Berridge, 1998). To examine an involvement of postsynaptic Ca2+ increases in PS-induced synaptic potentiation, we preloaded BAPTA (10 mm in pipettes), a quick chelator of Ca2+ (Tsien, 1980), into SP non-pyramidal neurons (PND 18-22). The PS failed to induce synaptic potentiation (99 ± 6 %, n = 7, filled symbols in Fig. 2a) compared to control with the standard pipette solution (199 ± 15 %, n = 8, open symbols). These results indicate that postsynaptic Ca2+ increases are required for PS-induced synaptic potentiation in SP non-pyramidal neurons.
Figure 2. Effects of BAPTA or DAP-5 on PS-induced potentiation of EPSCs in hippocampal CA1 SP non-pyramidal neurons (PND 18-22).
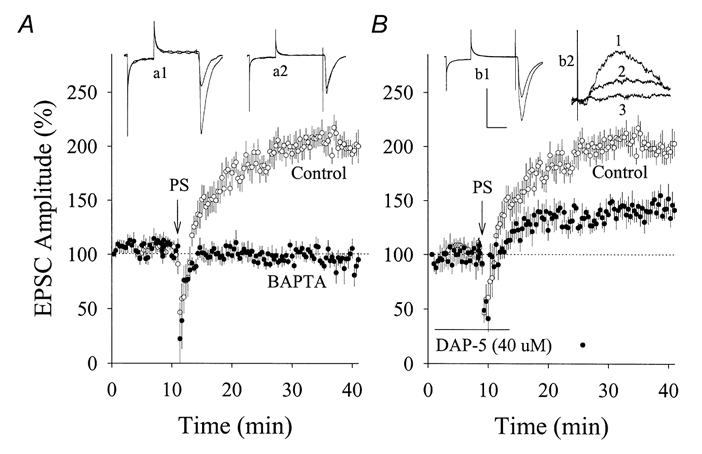
A, compared with the standard pipette solution (○, n = 8), pre-loading with BAPTA (10 mm in pipette solution; •, n = 7) in postsynaptic neurons prevented PS-induced synaptic potentiation. EPSC waveforms represent synaptic potentiation (a1) and BAPTA effect (a2). B, PS induced a partial increase of EPSCs in the presence of DAP-5 (40 μm; •, n = 5) compared with controls (○). EPSC waveforms in inset (b1) represent the effect of DAP-5 on PS-induced potentiation. Inset (b2) shows EPSCs at +50 mV that emerged after Ca2+-CaM perfusion (trace 1) and effects of 10 μm CNQX (trace 2) or CNQX plus 40 μm DAP-5 (trace 3). Arrows indicate PS application in A and B. Calibration bars are 150 pA and 30 ms for a1 and 2 and b1; 20 pA and 10 ms for b2. Control data in A and B are the same as those used in Fig. 1.
We have examined the role of NMDARs in PS-induced synaptic potentiation. An NMDAR antagonist, DAP-5 (40 μm), was perfused to hippocampal slices immediately after getting whole-cell recordings of EPSCs in SP non-pyramidal neurons (PND 18-22). After adding DAP-5 for 10 min, PS still enhanced the amplitude of EPSCs (142 ± 8 %, n = 5; filled symbols in Fig. 2B), but to a lesser extent than PS-induced synaptic potentiation under control conditions (199 ± 15 %, n = 8; open symbols; P = 0.006). It is noteworthy that 40 μm DAP-5 blocked EPSCs recorded at +50 mV in 10 μm CNQX (inset b2 of Fig. 2B). These results indicate that the Ca2+ requirement for PS-induced synaptic potentiation in SP non-pyramidal neurons is partially due to the activation of NMDARs. We did not examine the contribution of voltage-gated calcium channels because blocking these channels affected synaptic transmission (data not shown).
As Ca2+ binds CaM with a high affinity (Cohen, 1988; Klee & Cohen, 1988), we examined whether Ca2+-CaM is required for synaptic potentiation. A CaM-binding peptide (CBP, 100 μm in pipettes), an antagonist of Ca2+-CaM (Ocorr & Schulman, 1991; Hanson et al. 1994), was pre-loaded into SP non-pyramidal neurons (PND 18-22). CBP prevented PS-induced synaptic potentiation (96 ± 5 %, n = 5, filled symbols in Fig. 3a), indicating the requirement of Ca2+-CaM. Moreover, we examined whether CaM-dependent protein kinases are involved in PS-induced synaptic potentiation since CaMKII, a target of Ca2+-CaM, is enriched in postsynaptic densities (Kelly et al. 1984; Cohen, 1988; Klee & Cohen, 1988). The pre-loading of an autoinhibitory peptide of these kinases, CaMKII (281-301) (Hanson & Schulman, 1992; 100 μm in pipettes), into postsynaptic neurons attenuated PS-induced synaptic potentiation (109 ± 11 %, n = 5; filled symbols in Fig. 3B). These results indicate that a postsynaptic Ca2+-CaM signalling pathway is involved in PS-induced synaptic potentiation in SP non-pyramidal neurons.
Figure 3. Calmodulin-binding peptide (CBP) or CaMKII (281-301) blocked PS-induced potentiation of EPSCs in hippocampal CA1 SP non-pyramidal neurons (PND 18-22).
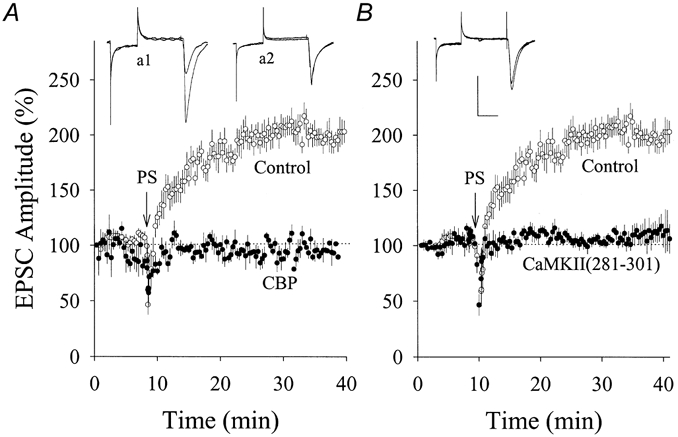
A, compared with the standard pipette solution (○, n = 8), pre-loading CBP (100 μm in pipette solution) in postsynaptic neurons prevented PS-induced synaptic potentiation (•, n = 5). EPSC waveforms represent synaptic potentiation (a1) and CBP effect (a2). B, CaMKII (281-301) (100 μm in pipette) attenuated PS-enhanced EPSCs (•, n = 5) compared with controls (○). EPSC waveforms represent CaMKII (281-301) effect. Arrows in A and B indicate PS application, and calibration bars are 150 pA and 30 ms. Control data in A and B are the same as those in Fig. 1.
Raising postsynaptic Ca2+ or CaM increases EPSCs on CA1 SP non-pyramidal neurons
If postsynaptic Ca2+-CaM is essential to synaptic potentiation, raising Ca2+ or Ca2+-CaM should enhance synaptic strength. We increased intracellular Ca2+ by perfusing adenophostin (a potent agonist of IP3Rs that evokes Ca2+ release; Takahashi et al. 1994; Delisle et al. 1997) into non-pyramidal neurons (PND 18-22). Compared with control (open symbols), adenophostin (1 μm in pipettes) increased the amplitude of EPSCs (filled symbols in Fig. 4a) that were subsequently blocked by 10 μm CNQX, i.e. glutamatergic. Mean data of adenophostin-induced synaptic potentiation are shown in Fig. 4B (373 ± 31 %, n = 9, open symbols). Similarly, the postsynaptic perfusion of thapsigargin, which increases cytosolic Ca2+ accumulation by inhibiting Ca2+-ATPase (Treiman et al. 1998), enhanced EPSCs (n = 3, data not shown). However, co-perfusing adenophostin with BAPTA (10 mm) failed to induce synaptic potentiation (104 ± 10 %, n = 7, filled symbols in Fig. 4B). These results indicate that postsynaptic Ca2+ enhances synaptic function in non-pyramidal neurons. It is noteworthy that PS could not enhance EPSC amplitudes after adenophostin-induced potentiation (n = 7; an example in Fig. 4a), indicating that PS- and adenophostin-induced synaptic potentiation share similar mechanisms.
Next, we raised Ca2+-CaM in non-pyramidal neurons (PND 18-22) by whole-cell perfusion. Compared with control (open symbols), Ca2+-CaM (40:10 μm in pipettes) increased the amplitude of EPSCs (filled symbols in Fig. 5a) that were subsequently blocked by 10 μm CNQX. Mean data of Ca2+-CaM-induced synaptic potentiation are shown in Fig. 5B (open symbols; 277 ± 25 %, n = 11). Ca2+-CaM-induced synaptic potentiation occluded the PS effect (n = 4, an example in Fig. 5a), indicating that they share similar mechanisms. To examine if Ca2+-CaM activates CaM-dependent protein kinases to enhance EPSCs, we co-perfused CaMKII (281-301) (100 μm in pipettes), which has no CaM-binding sequence (Kelly, 1992), with Ca2+-CaM into non-pyramidal neurons. This co-perfusion blocked Ca2+-CaM-induced synaptic potentiation (106 ± 6 %, n = 13, filled symbols in Fig. 5B). The perfusion of 100 μm CaMKII (281-301) alone did not affect basal synaptic transmission (n = 3, grey symbols in Fig. 5B). These results indicate that CaM-dependent protein kinases involve Ca2+-CaM-induced synaptic potentiation.
Figure 5. Ca2+-CaM enhanced glutamatergic EPSCs in hippocampal CA1 SP non-pyramidal neurons (PND 18-22) and CaMKII (281-301) blocked the potentiation.
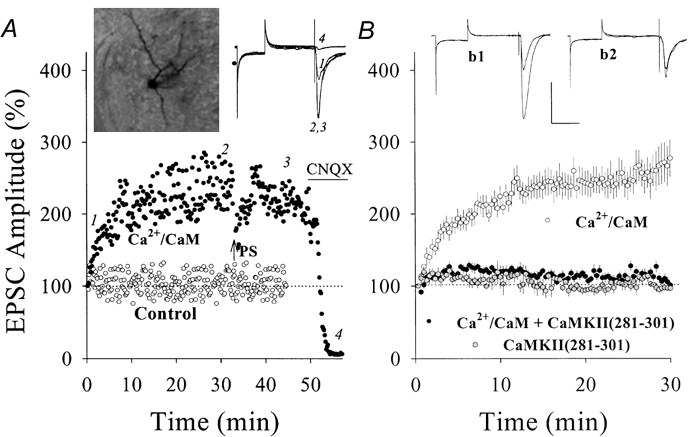
A, the postsynaptic perfusion of Ca2+-CaM (40:10 μm in pipette solution) enhanced EPSCs (•) compared with a control (○). After 34 min, PS (arrow) could not induce further potentiation and CNQX (10 μm, horizontal bar) blocked EPSCs. Insets show neurobiotin/Ca2+-CaM-perfused neuron (left) and EPSC waveforms at time points 1, 2, 3 and 4 (right). B, the co-perfusion of Ca2+-CaM with 100 μm CaMKII (281-301) could not enhance EPSCs (•, n = 13) compared with Ca2+-CaM alone (○, n = 11). Grey symbols indicate the perfusion of CaMKII (281-301) alone (n = 3). Insets show EPSCs of Ca2+-CaM-induced potentiation (b1) and CaMKII (281-301) effect (b2). Calibration bars are 150 pA and 50 ms.
The results above indicate that Ca2+-CaM signalling cascades enhance monosynaptic function in non-pyramidal neurons in hippocampal CA1 stratum pyramidale. Increases in EPSCs recorded from a population of synapses may be caused by the conversion of some inactive synapses to active ones, for which signalling molecules may enhance receptor function and/or transmitter release. CaMKII has been shown to phosphorylate glutamate receptors during long-term potentiation (Barria et al. 1997), so we examined whether Ca2+-CaM signalling cascades modulate synaptic targets to convert inactive synapses to active ones.
Ca2+-CaM converts inactive to active synapses on CA1 SP non-pyramidal neurons
If synapses display an extremely low probability of responses to single stimuli, we define them as inactive synapses, which presumably can be activated by multipulse stimuli (facilitation, Zucker, 1989) or signalling cascades. To study the inactive synapse, we need a stimulus protocol that evokes EPSCs at low probability and facilitates them. Paired-pulse stimuli (Wang & Kelly, 1996) were used. Here the stimulus intensity did not evoke EPSCs in pulse one but evoked low amplitude EPSCs in pulse two when the standard solution was in the pipette tip. After a signalling molecule is perfused into the recording neurons, the emergence of EPSCs in the first pulse indicates that this molecule converts inactive synapses to active ones. To perfuse Ca2+-CaM, we filled the pipette tip with standard solution and back-filled with Ca2+-CaM (40:10 μm).
After Ca2+-CaM diffused into non-pyramidal neurons (PND 18-22), the first stimuli evoked EPSCs that were subsequently blocked by 10 μm CNQX (Fig. 6a). The distribution of EPSC amplitudes (same data as Fig. 6a) recorded in 5-30 min (open bars) and 0-4 min (filled bars) is plotted in Fig. 6B. The probability of detecting evoked EPSCs in Fig. 6C was calculated from events in 0-4 and 20-24 min, and the mean values (filled circles) were 0.15 ± 0.02 vs. 0.87 ± 0.04, respectively (n = 12, P < 0.001). When pipettes were back-filled with the standard solution, the mean values of probability in 0-4 and 20-24 min were 0.09 ± 0.01 vs. 0.1 ± 0.03, respectively (n = 6, P = 0.61; filled diamonds in Fig. 6C). These results indicate that the emergence of glutamatergic EPSCs in the first stimulus was due to postsynaptic Ca2+-CaM application, i.e. Ca2+-CaM in non-pyramidal neurons converts inactive synapses to active ones. This Ca2+-CaM-induced conversion was also seen at unitary synapses in pyramidal-to-interneuron pairs (n = 4; J.-H.Wang & J. Wei, unpublished data).
We switched the holding potential from -70 to +50 mV in four experiments of Ca2+-CaM perfusion and observed outward EPSCs at +50 mV. CNQX (10 μm) blocked EPSCs at -70 mV in all four experiments and outward EPSCs at +50 mV in three experiments (an example in Fig. 6a). However, one neuron in CNQX displayed outward EPSCs at +50 mV that were sensitive to 40 μm DAP-5 (inset in b2 of Fig. 2B). It is not clear why NMDAR-mediated EPSCs were not detected in most of the non-pyramidal neurons in our studies, and this was different from the results in pyramidal neurons (Isaac et al. 1995; Liao et al. 1995; Durand et al. 1996). We did not record EPSCs at +50 mV in all our experiments of Ca2+-CaM perfusion. In addition to a low chance of detecting NMDAR-mediated EPSCs, membrane currents fluctuated at +50 mV, presumably due to activation of voltage-gated ion channels. It took a few minutes for inactivation of those channels and the disappearance of the fluctuation. As Ca2+-CaM-induced emergence of EPSCs usually occurred in 4 min (Fig. 6a and 11), we were able to record baseline values of EPSCs at -70 mV.
Ca2+-CaM increases the amplitude and frequency of spontaneous EPSCs in CA1 SP non-pyramidal neurons
We examined the effect of Ca2+-CaM on sEPSCs. Ca2+-CaM were perfused into SP non-pyramidal neurons (PND 18-22), when pipette tips were filled with the standard solution and back-filled with Ca2+-CaM (40:10 μm). sEPSCs were recorded for 4 min immediately after the formation of the whole-cell configuration. After 15 min for the perfusion of Ca2+-CaM into neurons, we recorded sEPSCs for another 4 min. Figure 7 shows the relationship between cumulative probability and sEPSC amplitudes (Fig. 7a) or inter-sEPSC intervals (Fig. 7B). After perfusing Ca2+-CaM, sEPSC amplitudes increased and inter-EPSC intervals shortened (n = 5, filled symbols). Standard error bars show the variation of the cumulative probability in sEPSC amplitude and the intervals among neurons. These results indicate that Ca2+-CaM-induced synaptic potentiation in SP non-pyramidal neurons may require post- and presynaptic mechanisms because a change in sEPSC frequency is believed to be presynaptic in origin and amplitude is a postsynaptic index (Redman, 1990).
If increases in transmitter release occur during the perfusion of Ca2+-CaM into postsynaptic neurons, retrograde messengers are required for post- to presynaptic communication. We examined the possibility that NO acts as a retrograde messenger because Ca2+-CaM activates nitric oxide synthase (NOS; Griffith & Stuehr, 1995). Carboxy-PTIO (30 μm), a NO scavenger (Rand & Li, 1995), was added to the solution of slice perfusion to block post- to presynaptic NO diffusion when we introduced Ca2+-CaM into SP non-pyramidal neurons. This concentration has been shown to block NO action and long-term potentiation (Ko & Kelly, 1999). Ca2+-CaM-induced increases in sEPSC frequency and amplitude were not attenuated by carboxy-PTIO (Fig. 7D–F, n = 5). These results imply that NO is not a retrograde messenger in Ca2+-CaM-induced synaptic potentiation, and the increase in sEPSC frequency may be due to the conversion of inactive synapses to active ones instead of purely presynaptic changes.
Ca2+-CaM signals differentially increase EPSCs on SP and SR non-pyramidal neurons
Tetanic stimulation induced synaptic potentiation in hippocampal CA1 oriens interneurons (Taube & Schwartzkroin, 1987; Ouardouz & Lacaille, 1995) but depression in SR interneurons (McMahon & Kauer, 1997). We have compared the levels of synaptic potentiation induced by adenophostin or Ca2+-CaM in SP and SR non-pyramidal neurons. Adenophostin-induced synaptic potentiation was larger in SP non-pyramidal neurons (377 ± 31 %, n = 9; filled symbols) than in SR (214 ± 13 %, n = 6; Fig. 8a). Similarly, Ca2+-CaM induced larger synaptic potentiation in SP non-pyramidal neurons (277 ± 25 %, n = 11) compared with that in SR (160 ± 9 %, n = 6; open symbols Fig. 8B). These results indicate that the Ca2+-CaM signalling pathway differentially modulates synaptic function in SP and SR non-pyramidal neurons. We have not observed PS-induced synaptic potentiation in SR non-pyramidal neurons (n = 5, data not shown), consistent with a report of Maccaferri & McBain, 1996.
Figure 8. Adenophostin or Ca2+-CaM induced differential potentiation at glutamatergic synapses on SP vs. SR non-pyramidal neurons in hippocampal area CA1.
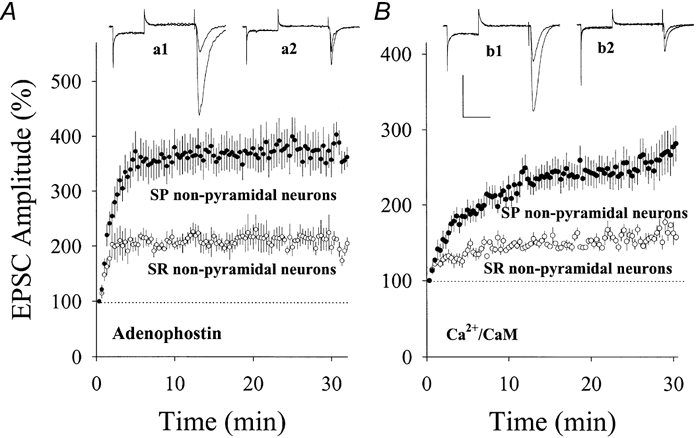
A, the comparison of synaptic potentiation induced by postsynaptic applications of 1 μm adenophostin in two groups of neurons. The potentiated EPSCs is higher in SP (•, n = 9) than SR non-pyramidal neurons (○, n = 6). Insets show EPSCs recorded from SP (a1) and SR neurons (a2). B, the comparison of synaptic potentiation induced by applying Ca2+-CaM (40:10 μm) into SP and SR non-pyramidal neurons. The magnitude of synaptic potentiation is higher in SP (•, n = 11) compared with SR non-pyramidal neurons (○, n = 6). Insets show EPSCs recorded from SP (b1) and SR neurons (b2). Calibration bars are 200 pA and 45 ms for EPSCs in SP non-pyramidal neurons and 100 pA and 45 ms for SR in A and B. The kinetics of EPSCs are faster in SR non-pyramidal neurons (a2 and b2) compared with SP neurons.
The kinetics of EPSCs in SR non-pyramidal neurons were faster and the amplitude was lower than those in SP (waveforms in Fig. 8). Rising times to EPSC peak in SP and SR non-pyramidal neurons were 3.55 ± 0.07 ms (n = 12) and 1.82 ± 0.09 ms (n = 10, P < 0.001), respectively. In addition, the duration of EPSCs in SR non-pyramidal neurons was shorter (waveforms in Fig. 8). We did not measure decay-time constants since EPSCs recorded in each neuron were generated from a population of synapses and the decay of unitary EPSCs in pyramidal-to-interneuron pairs was variable among synapses (n = 10; J.-H. Wang & J. Wei, unpublished observations, data not shown). Differences in EPSC kinetics and amplitudes imply that these two groups of non-pyramidal neurons contain different subtypes of synaptic AMPARs and/or densities of synapses.
Postnatal changes of synaptic potentiation induced by PS, adenophostin or Ca2+-CaM
We compared levels of synaptic potentiation in CA1 SP non-pyramidal neurons during postnatal development. Figure 9a shows the effect of PS on EPSCs in PND 7-9, 11-13 and 18-22. The level of potentiation was higher in PND 18-22 (199 ± 15 %, n = 8, filled symbols) than in PND 11-13 (135 ± 10 %, n = 7, grey symbols). PS did not induce synaptic potentiation in PND 7-9 (94 ± 10 %, n = 7, open symbols). Similarly, adenophostin-induced synaptic potentiation shows postnatal changes in magnitude (Fig. 10). Levels of synaptic potentiation at 30 min were 126 ± 7 % in PND 10-11 (open symbols, n = 5), 212 ± 16 % in PND 13-15 (grey symbols, n = 6) and 373 ± 31 % in PND 19-21 (filled symbols, n = 9). The magnitude of PS- or adenophostin-induced synaptic potentiation was significantly different among the three ages (P < 0.01).
In addition to PND 18-22, we examined whether Ca2+-CaM could induce the emergence of EPSCs in young rats using the same protocol as Fig. 6. Figure 11 shows that EPSCs emerged during perfusing Ca2+-CaM. Probabilities of evoked EPSCs (before vs. after) perfusion were 0.11 ± 0.02 vs. 0.48 ± 0.06 in PND 9-11 and 0.17 ± 0.02 vs. 0.67 ± 0.04 in PND 13-15, respectively. Increases in probability were significant in each of these ages (P < 0.001), indicating that Ca2+-CaM converts inactive to active synapses in young rats. We also compared the maximum amplitude and probability of emerged EPSCs among the three ages. By analysing data in PND 9-11 (top panels, n = 11), 13-15 (middle panels, n = 11) and 18-22 (bottom panels, n = 12), we found that the maximum amplitudes of emerged EPSCs were 24.9 ± 2.7, 40 ± 4.7 and 62 ± 5.1 pA, respectively (P < 0.05). The probability of emerged EPSCs are significantly different in the three groups (P < 0.05, Fig. 11). These results indicate that postsynaptic Ca2+-CaM increases the conversion rate of inactive-to-active synapses during postnatal development.
DISCUSSION
We have investigated the influence of the Ca2+-CaM signalling pathway on synaptic transmission in hippocampal CA1 non-pyramidal neurons, and used paired stimuli, adenophostin (IP3R agonist) and Ca2+-CaM to activate this pathway. These three protocols induced synaptic potentiation in SP non-pyramidal neurons, and underlying mechanisms included the conversion of inactive to active synapses and developmental maturation.
It has been suggested that tetanic stimulation induced synaptic potentiation in hippocampal CA1 interneurons through polysynaptic mechanisms (Maccaferri & McBain, 1996). However, we used paired stimuli (Kelso et al. 1986; Maccaferri & McBain, 1996) which are believed to change synaptic strength in recording neurons. Taking this together with the short latency of EPSCs (waveforms in Figs 1–3), we suggest that the potentiation in SP non-pyramidal neurons is monosynaptic.
Paired stimuli-induced synaptic potentiation required an increase in postsynaptic calcium (Fig. 2a). Intracellular Ca2+ may rise through the activation of NMDAR-channels (Mayer et al. 1989; Jahr & Stevens, 1990), voltage-gated calcium channels (Fox et al. 1987; Tsien et al. 1988) and/or Ca2+ releases (Ehrlich, 1995; Berridge, 1998; Fagni et al. 2000). We observed that DAP-5 (40 μm) partially attenuated PS-induced synaptic potentiation, indicating that NMDARs are not the sole Ca2+ provider for the potentiation in non-pyramidal neurons. The NMDAR-independent portion of PS-induced potentiation may depend upon Ca2+ released from intracellular stores, because adenophostin (an agonist of IP3Rs) induced synaptic potentiation and occluded the effect of paired stimuli (Fig. 4).
Paired stimuli-, adenophostin- or Ca2+-CaM-induced synaptic potentiation was blocked by introducing inhibitors of the Ca2+-CaM signalling pathway (Figs 3–5) into postsynaptic neurons, indicating an involvement of CaM-dependent protein kinases. Adenophostin- or Ca2+-CaM-induced synaptic potentiation occluded PS-induced potentiation (Figs 4–5). Therefore, the Ca2+-CaM signalling pathway is one of the essential signalling cascades to synaptic potentiation in SP non-pyramidal neurons. However, compared with PS- and Ca2+-CaM-induced synaptic potentiation, the level of adenophostin-induced potentiation was higher and the rising phase was shorter. Our interpretation for these differences is that adenophostin may greatly increase Ca2+ levels that activate other Ca2+ signalling pathways besides CaM. It is noteworthy that PS-induced synaptic potentiation developed slowly (Fig. 1 and 9), because the potentiation did not contain a component of post-tetanic potentiation, a short-term synaptic plasticity (Zucker, 1989).
We first demonstrate the presence of Ca2+-CaM signalling pathways in hippocampal CA1 non-pyramidal neurons using physiological methods. The α-subunit of CaM-KII was not detected in hippocampal CA1 GABAergic interneurons with immunocytochemistry (Liu & G, 1996; Sik et al. 1998), but small amounts of enzymes below detection threshold might still be functional (McBain et al. 1999). Compared to electrophysiology, immunocytochemistry may be a less sensitive method. For example, the maturation of AMPAR-containing synapses was detected by a physiological approach in PND 6 (Durand et al. 1996) whereas AMPARs were detected by immunocytochemistry after postnatal week 2 (Petralia et al. 1999). On the other hand, if αCaMKII is not present in interneurons, other types of CaM-dependent protein kinases, βCaM-KII, CaMKI and CaMKIV (Schulman & Lou, 1989), may be involved in synaptic potentiation. Being unable to identify if non-pyramidal neurons in our studies were GABAergic, we cannot conclude whether our results differ from those in previous immunocytochemical studies (Liu & Jones, 1996; Sik et al. 1998). However, we believe that the most of hippocampal interneurons are GABAergic (Freund & Buzsaki, 1996).
Our results indicate that Ca2+-CaM signalling pathways in CA1 SP non-pyramidal neurons act on postsynaptic targets and convert inactive synapses to active ones. As AMPARs are enriched in the postsynaptic membrane of hippocampal interneurons (Nusser et al. 1998), the inactivity of synapses may be due to the non-functional status of receptors. Ca2+-CaM signalling cascades enhance the function of AMPARs and then convert inactive to active synapses in non-pyramidal neurons. The conversion of silent synapses to functional ones was suggested as one of mechanisms in long-term potentiation (Isaac et al. 1995; Liao et al. 1995), in which AMPAR-mediated EPSCs emerged in NMDAR-containing synapses. However, inactive synapses in our studies seem to be different from silent synapses in pyramidal neurons. Most of the inactive synapses in SP non-pyramidal neurons did not show NMDAR-mediated EPSCs (Fig. 6). Their functional status was facilitated by paired pulses, and such a protocol was not used in previous studies (Isaac et al. 1995; Liao et al. 1995). Although the receptor composition of inactive synapses in non-pyramidal neurons may be different from that of silent synapses in pyramidal neurons, they appear to be similar in the emergence of AMPAR-mediated EPSCs during synaptic potentiation.
Changes in sEPSC frequency usually indicate presynaptic phenomena (Redman, 1990). Increases in sEPSC frequency during the postsynaptic perfusion of Ca2+-CaM could be interpreted as a presynaptic involvement. However, a NO scavenger had little effect on Ca2+-CaM-induced increases in sEPSC frequency (Fig. 7), implying that NO and presynaptic mechanisms are not involved. An alternative postsynaptic mechanism might be the conversion of inactive to active synapses because increases in the number of postsynaptic AMPARs were associated with an increase in sEPSC frequency (Petralia et al. 1999). The addition of more functional receptors increases the chance for a transmitter to evoke more synaptic events such that the probability of evoked EPSCs and the frequency of sEPSCs increase. Changes in sEPSC amplitude and frequency may also be an index of inactive-to-active synapse conversion that is based on postsynaptic mechanisms.
The magnitude of PS-, adenophostin- and Ca2+-CaM-induced synaptic potentiation on SP non-pyramidal neurons in hippocampal area CA1 increased during postnatal development (Figs 9–11). As underlying mechanisms appear postsynaptic in origin, postnatal changes may result from quantitative increases in intracellular Ca2+-CaM targets and/or postsynaptic receptors. It is not clear whether the number of synaptic AMPARs in non-pyramidal neurons changes during development, similar to that in pyramidal neurons (Durand et al. 1996; Liao et al. 1999; Petralia et al. 1999). As the expression of protein kinase development rises (Huang, 1989; Kelly, 1992), postnatal changes appear, probably due to the quantitative increase in Ca2+-CaM signalling cascades. AMPARs in non-pyramidal neurons may primarily be at a low functional status. When Ca2+-CaM signalling cascades mature, their modulation of synaptic targets may increase the ratio of functional to non-functional AMPARs and the conversion rate of inactive-to-active synapses over early postnatal weeks.
We investigated synaptic plasticity in hippocampal CA1 SP non-pyramidal neurons that are important in controlling the excitatory activity of pyramidal neurons (Freund & Buzsaki, 1996). The activation of postsynaptic Ca2+-CaM signalling cascades by pairied stimuli, evoking Ca2+ release or perfusing Ca2+-CaM induces monosynaptic potentiation in these neurons. The physiological consequences of the up-regulation of glutamatergic synapses in interneurons would be to increase their activity and enable them to inhibit principal neurons more efficiently.
Acknowledgments
We thank Dr M. Takahashi for adenophostin, Ms J. Wei for experimental assistance and Dr E. Floor for reading the final version. We especially thank the editors of The Journal of Physiology for help with correcting the writing. This study was supported by University of Kansas startup funds to J.H.W. and NS32470 to P.T.K.
References
- Alger BE. Gating of GABAergic inhibition in hippocampal pyramidal cells. Annals of the New York Academy of Sciences. 1991;627:249–263. doi: 10.1111/j.1749-6632.1991.tb25929.x. [DOI] [PubMed] [Google Scholar]
- Andersen P, Sundberg SH, Sveen O, Swann JW, Wigstrom H. Possible mechanisms for long-lasting potentiation of synaptic transmission in hippocampal slices from guinea-pigs. Journal of Physiology. 1980;302:463–482. doi: 10.1113/jphysiol.1980.sp013256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, Muller D, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by Ca2+/calmodulin-dependent protein kinase II during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Eidelberg E. Direct afferent excitation and long-term potentiation of hippocampal interneurons. Journal of Neurophysiology. 1982;48:597–607. doi: 10.1152/jn.1982.48.3.597. [DOI] [PubMed] [Google Scholar]
- Carmant L, Woodhall G, Ouardouz M, Robitaille R, Lacaille JC. Interneuron-specific Ca2+ responses linked to metabotropic and ionotropic glutamate receptors in rat hippocampal slices. European Journal of Neuroscience. 1997;9:1625–1635. doi: 10.1111/j.1460-9568.1997.tb01520.x. [DOI] [PubMed] [Google Scholar]
- Cohen P. The calmodulin-dependent multiprotein kinase. In: Cohen P, Klee CB, editors. Calmodulin. Amsterdam: Elsevier Science Publishers B. V; 1988. pp. 145–193. [Google Scholar]
- Delisle S, Marksberry EW, Bonnett C, Jenkins DJ, Potter BVL, Takahashi M, Tanzawa K. Adenophostin A can stimulate Ca2+ influx without depleting the inositol 1,4,5,-trisphosphate-sensitive Ca2+ stores in Xenopus oocyte. Journal of Biological Chemistry. 1997;272:9956–9961. doi: 10.1074/jbc.272.15.9956. [DOI] [PubMed] [Google Scholar]
- Durand GM, Kovalchuk Y, Konnerth A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature. 1996;381:71–75. doi: 10.1038/381071a0. [DOI] [PubMed] [Google Scholar]
- Ehrlich BE. Functional properties of intracellular calcium-release channels. Current Opinion in Neurobiology. 1995;5:304–309. doi: 10.1016/0959-4388(95)80042-5. [DOI] [PubMed] [Google Scholar]
- Fagni L, Chavis P, Ango F, Bockaert J. Complex interactions between mGluRs, intracellular Ca2+ stores and ion channels in neurons. Trends in Neurosciences. 2000;23:80–88. doi: 10.1016/s0166-2236(99)01492-7. [DOI] [PubMed] [Google Scholar]
- Fox AP, Nowycky MC, Tsien RW. Single-channel recordings of three types of calcium channels in chick sensory neurones. Journal of Physiology. 1987;394:173–200. doi: 10.1113/jphysiol.1987.sp016865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Griffith OW, Stuehr DJ. Nitric oxide synthases: properties and catalytic mechanism. Annual Review of Physiology. 1995;57:707–736. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Meyer T, Stryer L, Schulman H. Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron. 1994;12:943–956. doi: 10.1016/0896-6273(94)90306-9. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Schulman H. Inhibitory autophosphorylation of multifunctional Ca2+/calmodulin-dependent protein kinase analyzed by site-directed mutagenesis. Journal of Biological Chemistry. 1992;267:17216–17224. [PubMed] [Google Scholar]
- Huang KP. The mechanism of protein kinase C activation. Trends in Neurosciences. 1989;12:425–432. doi: 10.1016/0166-2236(89)90091-x. [DOI] [PubMed] [Google Scholar]
- Isaac JTR, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Jahr CE, Stevens CF. A quantitative description of NMDA receptor-channel kinetic behavior. Journal of Neuroscience. 1990;10:1830–1837. doi: 10.1523/JNEUROSCI.10-06-01830.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly PT. Calmodulin-dependent protein kinase II. Molecular Neurobiology. 1992;5:153–177. doi: 10.1007/BF02935544. [DOI] [PubMed] [Google Scholar]
- Kelly PT, Mcguinness TL, Greengard P. Evidence that the major postsynaptic density protein is a component of a Ca2+/calmodulin-dependent protein kinase. Proceedings of the National Academy of Sciences of the USA. 1984;81:945–949. doi: 10.1073/pnas.81.3.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso SR, Ganong AH, Brown TH. Hebbian synapses in hippocampus. Proceedings of the National Academy of Sciences of the USA. 1986;83:5326–5330. doi: 10.1073/pnas.83.14.5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klee CB, Cohen P. The calmodulin-regulated protein phosphatase. In: Cohen P, Klee CB, editors. Calmodulin. Amsterdam: Elsevier Science Publishers B.V; 1988. pp. 225–248. [Google Scholar]
- Ko G, Kelly PT. Nitric oxide acts as a postsynaptic signaling molecule in calcium/calmodulin-induced synaptic potentiation in hippocampal CA1 pyramidal neurons. Journal of Neuroscience. 1999;19:6784–6794. doi: 10.1523/JNEUROSCI.19-16-06784.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacaille JC, Mueller AL, Kunkel DD, Schwartzkroin PA. Local circuit interactions between oriens/alveus interneurons and CA1 pyramidal cells in hippocampal slices: electrophysiology and morphology. Journal of Neuroscience. 1987;7:1979–1993. doi: 10.1523/JNEUROSCI.07-07-01979.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Zhang X, O'brien R, Ehlers MD, Huganir RL. Regulation of morphological postsynaptic silent synapses in developing hippocampal neurons. Nature Neuroscience. 1999;2:37–43. doi: 10.1038/4540. [DOI] [PubMed] [Google Scholar]
- Liao D-Z, Hessler NA, Malinow R. Activation of postsynaptic silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Liu X-B, Jones EG. Localization of alpha type II calcium calmodulin-dependent protein kinase at glutamatergic but not γ-aminobutyric acid GABAergic synapses in thalamus and cerebral cortex. Proceedings of the National Academy of Sciences of the USA. 1996;93:7332–7336. doi: 10.1073/pnas.93.14.7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBain CJ, Freund TF, Mody I. Glutamatergic synapses onto hippocampal interneurons: precision timing without lasting plasticity. Trends in Neurosciences. 1999;22:228–235. doi: 10.1016/s0166-2236(98)01347-2. [DOI] [PubMed] [Google Scholar]
- Maccaferri G, Mcbain CJ. Long-term potentiation in distinct subtypes of hippocampal non-pyramidal neurons. Journal of Neuroscience. 1996;16:5334–5343. doi: 10.1523/JNEUROSCI.16-17-05334.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon LL, Kauer JA. Hippocampal interneurons express a novel form of synaptic plasticity. Neuron. 1997;18:295–305. doi: 10.1016/s0896-6273(00)80269-x. [DOI] [PubMed] [Google Scholar]
- Marty A, Neher E. Tight-seal whole-cell recording. In: Sakmann B, Neher E, editors. Single Channel Recording. New York, London: Plenum Press; 1995. pp. 31–52. [Google Scholar]
- Mayer ML, Vyklicky LJ, Clements J. Regulation of NMDA receptor desensitization in mouse hippocampal neurons by glycine. Nature. 1989;338:425–427. doi: 10.1038/338425a0. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Lujan R, Laube G, Roberts JDB, Molnar E, Somogyi P. Cell type and pathway dependence of synaptic AMPA receptor number and variability in hippocampus. Neuron. 1998;21:545–559. doi: 10.1016/s0896-6273(00)80565-6. [DOI] [PubMed] [Google Scholar]
- Ocorr KA, Schulman H. Activation of multifunctional Ca2+/calmodulin-dependent kinase in intact hippocampal slices. Neuron. 1991;6:907–914. doi: 10.1016/0896-6273(91)90231-n. [DOI] [PubMed] [Google Scholar]
- Ouardouz M, Lacaille J-C. Mechanisms of selective long-term potentiation of excitatory synapses in stratum oriens/alveus interneurons of rat. Journal of Neurophysiology. 1995;73:810–819. doi: 10.1152/jn.1995.73.2.810. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Esteban JA, Wang Y-X, Partridge JG, Zhao H-M, Wenthold RJ, Malinow R. Selective acquisition of AMPA receptors over postnatal development suggests a molecular basis for silent synapses. Nature Neuroscience. 1999;2:31–36. doi: 10.1038/4532. [DOI] [PubMed] [Google Scholar]
- Rand MJ, Li CG. Discrimination by the NO trapping agent, carboxy-PTIO, between NO and the nitrergic transmitter but not between NO and EDRF. British Journal of Pharmacology. 1995;116:1906–1910. doi: 10.1111/j.1476-5381.1995.tb16681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman S. Quantal analysis of synaptic potentials in neurons of the central nervous system. Physiological Reviews. 1990;70:165–198. doi: 10.1152/physrev.1990.70.1.165. [DOI] [PubMed] [Google Scholar]
- Schulman H, Lou LL. Multifunctional Ca2+/calmodulin-dependent protein kinase: domain structure and regulation. Trends in Biochemical Sciences. 1989;14:62–66. doi: 10.1016/0968-0004(89)90045-5. [DOI] [PubMed] [Google Scholar]
- Sik A, Hajos N, Culacsi A, Mody I, Freund TF. The absence of a major Ca2+ signaling pathway in GABAergic neurons of the hippocampus. Proceedings of the National Academy of Sciences of the USA. 1998;95:3245–3250. doi: 10.1073/pnas.95.6.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Tanzawa K, Takahashi S. Adenophostins, newly discovered metabolites of Penicillium brevicompactum, act as potent agonist of inositol 1,4,5-trisphosphate receptor. Journal of Biological Chemistry. 1994;269:369–372. [PubMed] [Google Scholar]
- Taube JS, Schwartzkroin PA. Intracellular recording from hippocampal CA1 interneurons before and after development of long-term potentiation. Brain Research. 1987;419:32–38. doi: 10.1016/0006-8993(87)90565-8. [DOI] [PubMed] [Google Scholar]
- Treiman M, Caspersen C, Christensen SB. A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases. Trends in Pharmacological Sciences. 1998;19:131–135. doi: 10.1016/s0165-6147(98)01184-5. [DOI] [PubMed] [Google Scholar]
- Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends in Neurosciences. 1988;11:432–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- Tsien RY. New calcium indicators and buffers with high selectivity against magnesium and protons: design, synthesis, and properties of prototype structures. Biochemistry. 1980;19:2396–2404. doi: 10.1021/bi00552a018. [DOI] [PubMed] [Google Scholar]
- Wang JH, Kelly PT. Postsynaptic injection of Ca2+/CaM induces synaptic potentiation requiring CaM-KII and PKC activity. Neuron. 1995;15:443–452. doi: 10.1016/0896-6273(95)90048-9. [DOI] [PubMed] [Google Scholar]
- Wang J-H, Kelly PT. Regulation of synaptic facilitation by postsynaptic Ca2+/CaM pathways in hippocampal CA1 neurons. Journal of Neurophysiology. 1996;76:276–286. doi: 10.1152/jn.1996.76.1.276. [DOI] [PubMed] [Google Scholar]
- Wong RKS, Traub RD, Miles R. Cellular basis of neuronal synchrony in epilepsy. Advances in Neurology. 1986;44:583–592. [PubMed] [Google Scholar]
- Zucker RS. Short-term synaptic plasticity. Annual Review of Neuroscience. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]