

Abstract
Myoadenylate deaminase (AMPD) deficiency is present in 1–2 % of the population. In theory, this deficiency may alter exercise energy metabolism by impairing the purine nucleotide cycle (PNC) and reducing tricarboxylic acid (TCA) cycle anaplerosis. The role of the PNC in TCA cycle anaplerosis is still a debated issue in physiology. Using patients with the AMPD1 mutation will allow a human ‘knockout’ approach to answering this question.
Muscle AMPD activity and genotype (whole blood AMPD1 analysis) was used to classify participants into three groups: n = 3 with absence of AMPD activity and -/- AMPD1 genotype (homozygous); n = 4 with less than 50 % normal AMPD activity and +/- genotype (heterozygous) and n = 12 with normal AMPD activity and +/+ genotype (control). Biopsies were taken from the vastus lateralis muscle before and after incremental cycle ergometry exercise to exhaustion. The muscle biopsies were analysed for AMPD activity, purine nucleotides/nucleosides and bases, creatine, phosphocreatine, amino acids, and the TCA cycle intermediates malate, citrate and fumarate.
Time to exhaustion on the cycle ergometer was not different between groups. Muscle adenosine monophosphate increased significantly with exercise for homozygous subjects as compared with the other groups (P < 0.05). Inosine monophosphate increased significantly after exercise for control (P < 0.05) but not for the homozygous subjects. There were no other between-group differences for any other measured variables.
In summary, complete and partial muscle AMPD deficiency did not affect TCA cycle anaplerosis, phosphocreatine hydrolysis, energy charge or exercise performance.
The most common known genetic abnormality of human skeletal muscle is myoadenylate deaminase (AMPD) deficiency (Norman et al. 1995, 1998). Myoadenylate deaminase catalyses the reaction from adenosine monophosphate (AMP) to inosine monophosphate (IMP) plus ammonia (NH3). A deficiency of myoadenylate deaminase activity was first characterized by Fishbein et al. (1978). AMPD deficiency is an autosomal recessive and has been reported in about 1–2 % of the population (Norman et al. 1995, 1998; Verzijl et al. 1998) with an allelic frequency of about 13 % in Caucasians and African-Americans (Morisaki et al. 1992; Norman et al. 1998; Verzijl et al. 1998). The gene encoding the AMPD protein is termed AMPD1 and is found on chromosome 1 (Morisaki et al. 1992; Sabina et al. 1992). Most patients with primary AMPD1 deficiency have a C/T transition mutation at nucleotide 34 in exon 2 that results in a nonsense mutation at codon 12 and a truncated protein (Morisaki et al. 1992). The diagnosis has been traditionally established using the forearm ischaemic test to demonstrate no ammonia rise with a normal lactate rise (an increase of NH3:lactate < 0.4) and by absent histochemical staining for AMPD on a frozen section of skeletal muscle (Shumate et al. 1979; Fishbein et al. 1990).
The diagnosis of AMPD1 deficiency is complicated by the fact that the clinical phenotype can vary and there are several conditions that can mimic some of the investigative abnormalities described above. For example, in addition to primary AMPD1 deficiency, there have been two other forms of apparent reductions in AMPD1 activity termed ‘coincidental’ and ‘acquired’ AMPD deficiency (Fishbein, 1999). In the coincidental form, AMPD deficiency is thought to co-exist with another neuromuscular disease due to a statistical probability with a prevalent disorder. The acquired form most probably represents the co-existence of a partial reduction in AMPD activity in a heterozygous carrier with a superimposed further reduction from a primary neuromuscular disease (Fishbein et al. 1978, 1999). Fishbein has also described several patients who demonstrated a normal work-up for neuromuscular disease and had normal AMPD activity in skeletal muscle, yet had no NH3 rise in response to forearm ischaemic testing (Fishbein et al. 1990). It was postulated that these ‘functional’ AMPD-deficient patients could have an impaired activation of a mutant enzyme in vivo (Km mutant) or possibly an impairment of NH3 efflux from the muscle (Fishbein et al. 1990). The existence of the different forms of AMPD deficiency has probably contributed to some of the discrepancies in the assessed frequency of the disorder in different patient populations.
There is an ongoing debate as to the potential significance of the primary AMPD1 mutation to skeletal muscle metabolism (Fishbein et al. 1978; Sabina et al. 1980; Norman et al. 1995; Verzijl et al. 1998; Fishbein, 1999). For example, some have found the absence of AMPD activity (Norman et al. 1995) and the presence of the AMPD1 mutation (Norman et al. 1998; Verzijl et al. 1998) in about 2 % of asymptomatic adults. On the other hand, the absence of AMPD activity has been found in 8 % of patients with complaints of myalgia (Kelemen et al. 1982). At the biochemical level, one report found a marked reduction in total purine nucleotides after exercise in a patient with AMPD enzyme deficiency (Sabina et al. 1980), whereas another study found that lactate accumulation, phosphocreatine hydrolysis and cellular energy charge were not differentially affected by ischaemic, isometric exercise in AMPD deficient patients as compared with controls (Sinkeler et al. 1987).
It is possible that the muscle fatigue and weakness reported by some AMPD deficient patients could occur via inhibition of the potential anaplerotic function of the purine nucleotide cycle (PNC) (Sabina et al. 1980; Flanagan et al. 1986; Gibala et al. 1997); a decrease in phosphorylation potential (Sahlin & Broberg, 1990); an increase in adenosine concentration (Blazev & Lamb, 1999); and/or acceleration of phosphocreatine hydrolysis and glycogenolysis/ glycolysis to maintain phosphorylation potential (anaerobic ADP rephosphorylation).
Given the importance of TCA cycle anaplerosis to energy production in skeletal muscle during exercise (Gibala et al. 1997a), this pathway requires careful evaluation in AMPD patients. An ongoing issue in the study of cycle anaplerosis is the source of the TCA cycle intermediates, but recent evidence has questioned the role of the PNC in this process (Gibala et al. 1997b). The study of anaplerosis in patients with high grade AMPD deficiency provides a unique opportunity to determine whether or not PNC function is required for TCA cycle anaplerosis during exercise in humans. In a review of the potential role of AMPD1 in skeletal muscle, it was concluded that, ‘Further study is needed to explain the significance of purine nucleotide catabolism in exercising muscle. Inherited diseases reveal the secret of nature …’ (Mineo & Tarui, 1995). In essence, the use of patients with AMPD1 deficiency to study the role of the PNC in TCA cycle anaplerosis represents a form of human knockout experimentation.
To date, there has only been one investigation comparing skeletal muscle metabolism in healthy controls and AMPD deficient patients during exhaustive exercise (Sinkeler et al. 1987). Given the uncertainty of the role for the PNC in muscle metabolism, and the suggestion that dysfunction of the PNC limits exercise capacity in AMPD deficiency (Sabina et al. 1980), we directly measured the contribution of the PNC to TCA cycle anaplerosis by examining patients with partial and complete deficiency of AMPD activity. A secondary issue addressed in the current study was the evaluation of the possible causes of ‘functional’ myoadenylate deficiency mentioned above.
METHODS
The study was reviewed and received approval from the McMaster Medical Center Ethics Review Committee and conformed to the Declaration of Helsinki guidelines. All subjects gave informed written consent to the procedures.
Subjects/patients
Patients who met criteria for complete (homozygous; n = 3) and partial (heterozygous; n = 4) myoadenylate deaminase (AMPD) deficiency were recruited from the Neuromuscular Clinic at McMaster University Medical Center. The classification was based upon the following criteria: homozygous – absent histochemical stain for AMPD activity plus failure of ammonia to increase on forearm ischaemic testing plus homozygous for the C34T transition mutation in AMPD1 in whole blood (see below); heterozygous – positive histochemical stain for AMPD activity plus ammonia increase on forearm ischaemic testing that was < 10 % of normal control values (with a normal lactate rise) plus heterozygous for the AMPD1 mutation as above, and no other neuromuscular disorder as ruled out by the screening process outlined below (Fishbein et al. 1978, 1990). Forearm ischaemic testing was performed as previously described (Tarnopolsky et al. 1998). We also recruited 12 age and sex matched control subjects (control) from the clinic who were investigated for symptoms of muscle fatigue and/or myalgia, and were subsequently found to have a completely normal work-up for neuromuscular disease including: neurological examination, plasma creatine kinase activity, plasma TSH, B12, complete blood count, electrolytes, electromyography and nerve conduction studies, lactate and ammonia response on forearm ischaemic testing, normal light and electron microscopic evaluation of skeletal muscle, normal AMPD activity on a histochemical assay, and did not have an allele with the C34T mutation. Subject characteristics are presented in Table 1.
Table 1. Subject characteristics.
| Group | Homozygous (n = 3) | Heterozygous (n = 4) | Control (n = 12) | Significance |
|---|---|---|---|---|
| Age (years) | 38 (19) | 43 (8) | 36 (16) | n.s |
| Height (cm) | 169 (7) | 173 (6) | 170 (10) | n.s. |
| Weight (kg) | 69.3 (10.6) | 79.4 (13.6) | 71.4 (10.4) | n.s |
Values are means (S.D.); not significant (n.s., P > 0.05).
Design
The subjects arrived at the laboratory following an overnight fast. Upon arrival, subjects were weighed, and a 20 gauge plastic catheter was inserted into the antecubital vein for blood sampling. A 10 ml blood sample was collected into pre-chilled, heparinized tubes and centrifuged immediately (200 g for 5 min), and plasma lactate concentration was determined using an enzymatic analyser (Yellow Springs Instruments, YSI 23L, Yellow Springs, OH, USA).
Following this, a muscle biopsy of the vastus lateralis was taken using a suction modified Bergstrom needle under sterile conditions. At the same time, a small incision was made in the ipsilateral thigh, ≈10 cm proximal to the original incision. This was then closed with sterile gauze and paper tape and subsequently used for the post-exercise biopsy (see below). The muscle sample (125–200 mg) was blotted and quick-frozen in liquid nitrogen.
The subjects then performed a progressive cycling test to voluntary exhaustion on an ergometer (Lode, Netherlands). Exhaustion was defined as the inability to maintain 60 revolutions per minute for 20 s in spite of vigorous verbal encouragement. The initial workload was set at 45 W for 4 min, followed by 15 W increments every 2 min until voluntary exhaustion. During exercise, the subjects had continuous measurement completed for oxygen uptake ( ), carbon dioxide production (
), carbon dioxide production ( ), respiratory exchange ratio (RER), and minute ventilation (
), respiratory exchange ratio (RER), and minute ventilation ( ) using an open circuit system as previously described (Tarnopolsky et al. 1995). Heart rate was also monitored continuously using a telemetry monitor (Polar Electro, Woodbury, NY, USA). When the subjects were close to exhaustion, the paper tape was removed and the leg was re-cleansed while the subject rode. At voluntary exhaustion, the subject was supported on the bike with the leg extended and held by two other assistants while the leg was quickly re-sterilized and a muscle biopsy completed within 4 s of exercise termination. A post-exercise blood sample was taken and analysed as described above. Pressure was held on the incision site and the subject was assisted off the cycle, while the biopsy was processed as described above. The muscle biopsies were stored at −70 °C for < 2 months before being analysed for AMPD activity, nucleotides, amino acids, phosphocreatine (PCr), creatine (Cr), and several TCA cycle intermediates (fumarate, malate and citrate) as outlined below.
) using an open circuit system as previously described (Tarnopolsky et al. 1995). Heart rate was also monitored continuously using a telemetry monitor (Polar Electro, Woodbury, NY, USA). When the subjects were close to exhaustion, the paper tape was removed and the leg was re-cleansed while the subject rode. At voluntary exhaustion, the subject was supported on the bike with the leg extended and held by two other assistants while the leg was quickly re-sterilized and a muscle biopsy completed within 4 s of exercise termination. A post-exercise blood sample was taken and analysed as described above. Pressure was held on the incision site and the subject was assisted off the cycle, while the biopsy was processed as described above. The muscle biopsies were stored at −70 °C for < 2 months before being analysed for AMPD activity, nucleotides, amino acids, phosphocreatine (PCr), creatine (Cr), and several TCA cycle intermediates (fumarate, malate and citrate) as outlined below.
Genotype analysis (PCR)
Genomic DNA was isolated from whole blood and the region surrounding exon 2 of the AMPD1 gene was amplified using a modification of previously described methods (Norman et al. 1998). Briefly, the Chelex resin-extracted DNA (Jarnopolsky et al. 1998) was amplified using primers (forward-5′-CTTCATACAGCTGAA GAGACA-3′; reverse-5′-GAATCCAGAAAAGCCATGAGC-3′) designed to create an NspI restriction site when the C34T transition mutation was present. Amplification was completed using one cycle of 5 min at 95 °C (hot start), followed by 30 cycles of 95 °C denaturing for 20 s, 56 °C annealing for 20 s and 72 °C extension for 45 s, with a final cycle of 72 °C for 7 min (Progene, Techne (Cambridge) Ltd, Duxford, UK). One-tenth of the PCR amplimer mixture was run on a 2 % agarose gel (100 V for 30 min) to visualize the 214 bp product. After confirmation that the 214 bp product was present, the amplimers were extracted from the mixture using chloroform-ethanol and resuspended in 10 μl of DNase-free water, and the amplimer mixture was digested with 2.0 U of NspI overnight at 37 °C in a buffer supplied by the manufacturer (Gibco BRL, Gaithersburg, MD, USA). About 90 % of the digested product was then run on a 2 % agarose gel (100 V for 30 min). When the C34T transition mutation is present, the NspI endonuclease cuts the 214 bp amplimer into a 191 and 23 bp product. Thus, the homozygous patients (-/-) had a 191 product only (the smaller product is not consistently visible at the bottom of the gel), the heterozygous patients (+/-) had a 191 and a 214 bp product, while the non-affected controls (+/+) had only the 214 bp product.
Muscle analyses
Muscle AMPD enzymatic activity
Portions (10–20 mg wet weight) of muscle biopsies were homogenized (1:19, w:v) in ice cold 100 mm KCl, 10 mm reduced glutathione and 50 mm Imidazole, pH 7.0. An aliquot of the whole homogenate was retained and the remainder was centrifuged (13 000 g for 1 min, at 4 °C) to separate soluble from particulate cell material. The supernatant (soluble cell fraction) was recovered, centrifuged again and retained. AMPD capacity (Vmax, enzyme activity at near-saturating substrate concentration; 15 mm AMP at pH 7.0 and 30 °C) was determined as previously described in detail (Rush et al. 1998). Briefly, a 10 μl aliquot of the whole homogenate or the soluble cell fraction was added to 990 μl of reaction buffer consisting of 150 mm KCl, 15 mm adenosine monophosphate (AMP), and 50 mm Imidazole, pH 7.0, pre-warmed to 30 °C. The reactants were mixed by vortex and the reactions proceeded for 5 min at 30 °C. Reactions were terminated by the addition of 5 volumes of 4.5 % (w/v) perchloric acid. Acid extracts were neutralized (KOH/triethanolamine), and the reaction product, inosine monophosphate (IMP), was determined by reversed-phase high performance liquid chromatography (Tullson et al. 1990). The particulate cell fraction AMPD capacity was calculated by subtraction of the determined soluble cell fraction AMPD capacity from that of the whole homogenate. Under conditions used for the data reported, this assay system produced IMP linearly with respect to both time and volume of extract used.
Muscle nucleotide/nucleoside and base analysis
Portions (5–10 mg wet weight) of muscle biopsies were homogenized in ice cold 4.5 % (w/v) perchloric acid, and the extracts were centrifuged (13 000 g for 7 min at 4 °C) to pellet acid-precipitable material. Supernatants were recovered, and neutralized with 0.5 m tri-n-octylamine in 1,1,2-trichloro-trifluoroethane. Neutralized extracts were centrifuged (13 000 g for 7 min at 4 °C), and the aqueous (upper) phase was recovered for purine analysis. Muscle nucleotides (ATP, ADP, AMP, IMP), nucleosides (adenosine, inosine), and bases (hypoxanthine, xanthine, adenine) were quantified by reversed-phase high performance liquid chromatography, using procedures identical to those previously described (Tullson et al. 1990).
TCA intermediates, phosphocreatine and creatine
A 3–8 mg portion of lyophylized muscle was extracted into 0.5 m perchloric acid (containing 1 mm EDTA), neutralized with 2.2 m KHCO3, and assayed for phosphocreatine, creatine, citrate, malate and fumarate using enzymatic methods (Passoneau & Lowry, 1993) adapted for fluorometry (Hitachi F-2000 fluorescence spectrophotometer, Hitachi Instruments). To correct for differences in non-muscle elements between samples, muscle metabolites were corrected to the highest total creatine value obtained within two biopsy samples for a given subject.
Muscle amino acid analysis
A 2–4 mg portion (dry weight) of the biopsy was glass homogenized for 1 min in 100 μl of deionized (Milli Q) water and was then centrifuged for 3 min at 1200 g. The supernatant was used for free amino acid analysis by HPLC (Waters Corp., Milford, MA, USA) according to a modified method (Graham et al. 1995), first described by Heinrikson & Meredith (1984).
Statistics
Descriptive data between the groups were analysed using a one-way analysis of variance (ANOVA). The muscle data were analysed using two-way ANOVA with a between (groups, 3 levels) and within (pre/post) design. Pair-wise comparisons were made using a Tukey post hoc test when significant F ratios were obtained. All data in tables and figures are presented as means (± s.d.). A value of P < 0.05 was considered to be statistically significant. All data were analysed using a commercially available statistical program (Statistica v. 5.1, StatSoft, Inc., Tulsa, OK, USA).
RESULTS
Cycle ergometry
There were no differences in peak , , , RER, peak power (watts), heart rate, or time to fatigue between the groups (Table 2). Plasma lactate increased similarly for all groups in response to the ergometry testing (P < 0.001; Table 2).
Table 2. Cycle ergometry testing characteristics.
| Group | ||||
|---|---|---|---|---|
| Homozygous | Heterozygous | Control | Significance | |
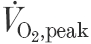 (l min−1) (l min−1) |
1.3 (0.5) | 1.6 (0.6) | 1.7 (0.5) | n.s |
| RER | 1.09 (0.10) | 1.17 (0.12) | 1.13 (0.12) | n.s |
| Peak power (W) | 110 (23) | 118 (69) | 130 (49) | n.s |
| Heart rate (beats min−1) | 137 (23) | 157 (13) | 158 (27) | n.s |
| Time to fatigue (s) | 680 (39) | 868 (572) | 925 (362) | n.s |
| Lactate (mmol l−1): pre | 0.69 (0.23) | 0.67 (0.28) | 0.63 (0.24) | n.s |
| post | 1.87 (0.60)* | 3.20 (1.11)* | 3.40 (1.27)* | n.s |
Values are means (S.D.). RER, respiratory exchange ratio. Values of , peak power, RER and heart rate were taken at voluntary exhaustion. pre, pre-exercise; post, post-exercise.
Significant increase with exercise (P < 0.001). n.s., not significant (P > 0.05).
Muscle AMPD activity
The enzyme activity for the homozygous group was significantly lower than both the heterozygous and control groups (1.43 vs. 59.31 vs. 154.30 μmol (g wet wt)−1 min−1, respectively, P < 0.001). The activity for the homozygous group was 0.9 % of that for the control group and the heterozygous group showed 38 % of control activity (Table 3). The ratio of soluble cell fraction:total AMPD activity was 0.78 (s.d., 0.05), 0.66 (0.06) and 0.69 (0.05) for the homozygous, heterozygous and control groups, respectively.
Table 3. Muscle AMPD activity, nucleotides, nucleosides, bases and energy charge.
| Group | |||||||
|---|---|---|---|---|---|---|---|
| Homozygous | Heterozygous | Control | |||||
| AMPD activity | Total | 1.43 | (0.34) | 59.31 | (8.26) | 154.30 | (34.36)a |
| (μmol (g wet wt)−1 min−1) | Soluble | 1.14 | (0.24) | 39.10 | (10.52) | 107.47 | (28.72)a |
| ATP (μmol (g wet wt)−1) | pre | 5.2 | (0.6) | 4.3 | (0.7) | 5.0 | (0.6) |
| Post | 5.2 | (1.0) | 3.5 | (0.8) b | 3.9 | (0.5)b | |
| ADP (μmol (g wet wt)−1) | pre | 0.82 | (0.11) | 0.77 | (0.09) | 0.86 | (0.07) |
| post | 0.92 | (0.14) | 0.76 | (0.12) | 0.77 | (0.11) | |
| AMP (μmol (g wet wt)−1) | pre | 0.03 | (0.02) | 0.06 | (0.03) | 0.05 | (0.07) |
| post | 0.19 | (0.20)c | 0.03 | (0.03) | 0.05 | (0.04) | |
| Energy charge | pre | 0.93 | (0.00) | 0.91 | (0.01) | 0.92 | (0.01) |
| post | 0.89 | (0.06) | 0.90 | (0.02) | 0.91 | (0.02) | |
| IMP (μmol (g wet wt)−1) | pre | 0.01 | (0.01) | 0.07 | (0.03) | 0.07 | (0.04) |
| Post | 0.01 | (0.00) | 0.24 | (0.14) | 0.36 | (0.21)d | |
| Xanthine (μmol (g wet wt)−1) | pre | 0.06 | (0.01) | 0.06 | (0.02) | 0.06 | (0.02) |
| post | 0.06 | (0.01) | 0.09 | (0.05) | 0.09 | (0.04) | |
| Adenosine (μmol (g wet wt)−1) | pre | 0.004 | (0.002) | 0.005 | (0.005) | 0.008 | (0.007) |
| post | 0.005 | (0.006) | 0.003 | (0.002) | 0.006 | (0.006) | |
| Inosine (μmol (g wet wt)−1) | pre | 0.004 | (0.001) | 0.008 | (0.001) | 0.012 | (0.011) |
| post | 0.030 | (0.024)e | 0.113 | (0.110)e | 0.096 | (0.06)e | |
| Hypoxanthine (μmol (g wet wt)−1) | pre | 0.023 | (0.013) | 0.022 | (0.013) | 0.025 | (0.020) |
| Post | 0.043 | (0.040)d | 0.077 | (0.079)d | 0.050 | (0.023)d | |
| Uric acid (μmol (g wet wt)−1) | pre | 0.02 | (0.00) | 0.0 | (0.01) | 0.02 | (0.02) |
| post | 0.04 | (0.01)e | 0.05 | (0.05)e | 0.04 | (0.01)e | |
Values are means (S.D.). pre, pre-exercise; post, post-exercise.
Each group's enzyme activity was significantly different from each other (P < 0.001);
significant decrease with exercise (P < 0.01);
significant increase with exercise (P < 0.05);
significant increase with exercise (P < 0.05);
significant increase with exercise (P < 0.01).
Muscle nucleotide/nucleoside and base analysis
Muscle ATP concentration was maintained for the homozygous group before and after exercise, yet showed a significant decrease for the heterozygous and control groups (P < 0.01). ADP concentration and cellular energy charge ([ATP]+½[ADP]/[ATP]+[ADP]+[AMP]) were unchanged before and after exercise and were not different between groups. There was a trend towards a drop in energy charge with exercise for all groups (P = 0.052). AMP concentration was not different before or after exercise for the heterozygous and control groups, yet it increased significantly due to exercise for the homozygous group (P < 0.05). IMP concentration was unchanged with exercise for the homozygous and heterozygous group, yet increased significantly for the control group (P < 0.05). There was a trend for IMP to increase for the heterozygous group (P = 0.072). Xanthine and adenosine concentrations did not change with exercise for any of the groups, whereas inosine (P < 0.01), hypoxanthine (P < 0.05) and uric acid concentrations (P < 0.01) increased similarly for all groups (Table 3).
TCA intermediates, phosphocreatine and creatine
The concentration of each of the three TCA cycle intermediates (Table 4) and the sum of the three TCAi intermediates (ΣTCAi; Fig. 1) increased similarly for each of the groups following exercise (P < 0.001). Resting phosphocreatine (PCr) and creatine (Cr) concentrations were similar between groups. PCr decreased significantly and similarly for all groups (P < 0.001), with a reciprocal increase in Cr (P < 0.001) (Table 4).
Table 4. Concentrations of muscle TCA cycle intermediates, phosphocreatine and creatine (mmol (kg dry wt)−1).
| Group | ||||
|---|---|---|---|---|
| Homozygous | Heterozygous | Control | ||
| Fumarate | pre | 0.05 (0.00) | 0.08 (0.03) | 0.06 (0.02) |
| post | 0.42 (0.04)* | 0.50 (0.13)* | 0.46 (0.13)* | |
| Malate | pre | 0.49 (0.11) | 0.57 (0.18) | 0.51 (0.16) |
| post | 2.90 (0.98)* | 2.29 (0.54)* | 2.24 (0.42)* | |
| Citrate | pre | 0.26 (0.09) | 0.31 (0.14) | 0.31 (0.16) |
| post | 0.63 (0.27)* | 0.64 (0.25)* | 0.65 (0.31)* | |
| Phosphocreatine | pre | 85.9 (8.8) | 79.3 (9.9) | 88.3 (8.7) |
| pos | 61.6 (14.9)† | 58.7 (8.8)† | 51.8 (10.6)† | |
| Creatine | pre | 42.3 (15.2) | 45.0 (7.9) | 37.5 (5.2) |
| post | 66.5 (17.5)* | 59.3 (13.6)* | 63.5 (17.0)* | |
Values are means (S.D.). pre, pre-exercise; post, post-exercise.
Significant increase with exercise (P < 0.001);
significant decrease with exercise (P < 0.001)
Figure 1. Sum of concentration of intramuscular TCA cycle intermediates at rest and after exercise.

Values represent the sum of citrate + malate + fumarate concentrations, expressed as means ± s.d. * Significantly different (P < 0.001) vs. within same age group.
Muscle amino acids
There were no between-group differences in any of the nine essential amino acid concentrations (histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan and valine), nor was there a significant effect of exercise upon these amino acids (data not shown). For the non-essential amino acids there were no between-group nor exercise effects for alanine, asparagine, serine, ornithine, tyrosine, proline, aspartate, glycine and arginine. Muscle glutamate decreased significantly and similarly for all groups as a result of the exercise (Table 5). There was a significantly higher taurine concentration for the homozygous compared with heterozygous and control groups (main effect for group, P < 0.05; Table 5). A similar difference was found for glutamine concentration with the homozygous group showing higher overall concentrations as compared with heterozygous and control groups (main effect for group, P < 0.05, Table 5).
Table 5. Selected muscle amino acid concentrations (mmol (kg dry wt)−1).
| Group | ||||
|---|---|---|---|---|
| Homozygous | Heterozygous | Control | ||
| Alanine | pre | 13.7 (5.4) | 12.9 (2.4) | 11.8 (3.2) |
| post | 16.7 (2.4) | 14.6 (5.2) | 13.8 (2.8) | |
| Aspartate | pre | 0.67 (0.32) | 0.78 (0.47) | 0.98 (0.35) |
| post | 0.79 (0.21) | 0.84 (0.23) | 0.94 (0.46) | |
| Taurine | pre | 81.8 (10.1)* | 54.0 (9.9) | 42.9 (10.8) |
| pos | 59.1 (14.4)* | 51.5 (15.3) | 43.1 (10.9) | |
| Glutamine | pre | 81.8 (10.1)* | 49.7 (13.7) | 53.5 (11.2) |
| post | 63.9 (5.7)* | 43.6 (9.4) | 51.7 (9.8) | |
| Glutamate | pre | 16.4 (5.1) | 12.1 (2.1) | 12.5 (3.2) |
| post | 8.1 (3.8)† | 9.9 (2.7)† | 8.4 (3.7)† | |
Values are means (S.D.). pre, pre-exercise; post, post-exercise.
Significantly higher for homozygous (P < 0.05);
significantly lower after exercise (P < 0.05).
DISCUSSION
A major finding in the current study was the ability of the muscle to maintain TCA cycle intermediate (TCAi) concentrations and cellular energy charge during exercise in spite of AMPD activity being less than 1 % of control values. This finding demonstrates the utility of using an inborn error of metabolism to unravel a fundamental question in muscle physiology, namely, that the PNC is not required for TCA cycle anaplerosis. From a clinical perspective, the main finding in this study is that patients with less than 1 % residual AMPD activity were not markedly different from subjects with normal AMPD activity in their tolerance for progressive cycle ergometry exercise.
One conceivable consequence of AMPD deficiency would be an impairment of the PNC with a resultant attenuation of TCAi anaplerosis (Sabina et al. 1980; Flanagan et al. 1986). It has previously been demonstrated that the total muscle TCAi content increased severalfold during the initial minutes of moderate to intense contraction, but the TCAi gradually declined when the bout was prolonged (Sahlin & Broberg, 1990; Gibala et al. 1997b). The PNC results in the re-amination of IMP to AMP and is catalysed by adenylosuccinate synthetase and adenylosuccinate lyase, respectively: IMP + aspartate + GTP / adenylosuccinate + GDP + Pi/ AMP + fumarate. Thus, fumarate production and TCAi anaplerosis could depend upon AMPD and PNC activity. Using 5-amino-4-imidazolecarboxamide ribose to inhibit the adenylosuccinate lyase reaction, investigators have reported that energy production did not meet energy demand in exercising rodents (Flanagan et al. 1986). This group also reported that lactate production was unimpaired and concluded that inhibition of the PNC must have caused aerobic energy impairment (Flanagan et al. 1986). Another group used the adenylosuccinate synthetase blocker, hadacidin, to study in situ rodent muscle exposed to various frequencies of tetanic stimulation and found no impairment of contractile force, nor any effects upon PCr hydrolysis, or lactate production (Meyer & Terjung, 1980). A case report in a patient with biochemically confirmed AMPD deficiency found a severe decrease in ATP and an increase in adenosine concentration following exercise, and a very prolonged ATP resynthesis rate (≈50 % normal by 3 h of recovery) (Sabina et al. 1980). This group hypothesized that this observation was due to a ‘Disruption of the purine nucleotide cycle’ (Sabina et al. 1980). It is difficult to ascribe this profound drop in ATP content and such an impairment of ADP rephosphorylation to AMPD deficiency, as neither the current study nor another study of ATP/ADP content (Sinkeler et al. 1987) found significant ATP depletion following exercise. It could be that the patient in this single case report (Sabina et al. 1980) had other confounding factors that led to this profound disruption in cellular energy status and would be considered to have coincidental AMPD deficiency.
The results of the current study have demonstrated that near total AMPD enzyme deficiency did not result in an impairment of exercise-induced anaplerosis of the TCAi pool. The magnitude of the increase in the TCAi pool and the strength of the statistical analysis very strongly support that this is a real finding and not a false positive result. This finding is consistent with the concept that the PNC is not quantitatively important in exercise-mediated TCAi anaplerosis, and supports the previously demonstrated importance of the alanine aminotransferase (ALT) reaction for TCAi anaplerosis (Sahlin et al. 1990; Gibala et al. 1997b). It could be argued that changes in three of the eight TCA cycle intermediates may not be representative of the total TCAi pool. However, we (Gibala et al. 1997b) have found that the sum of citrate, malate and fumarate accounts for approximately 70 % of total pool size, and they increase in a direction similar to that of other TCA cycle intermediates at the start of exercise (except for 2-oxoglutarate, which decreases or does not change markedly). From a fatigue perspective, a depletion of TCAi cannot account for contractile failure with this type of exercise, for the TCAi pool remained above resting levels following exercise in all groups at exhaustion in the current study.
The amino acid data from the current study demonstrated a decrease in glutamate concentration with exercise that was similar to that reported by Gibala et al. (1997b). Although we did not find a statistically significant increase in alanine concentration in the current study (up 20 %), it was directionally similar to that reported in recreationally exercise-trained males (up 54 %) (Gibala et al. 1997b). The amino acid data support the hypothesis that the ALT reaction is important for TCAi expansion during exercise in healthy young controls (Gibala et al. 1997b), and in patients with partial and complete muscle AMPD enzyme deficiency. The increase in the content of glutamine and taurine in the patients with complete AMPD deficiency is difficult to explain. Given that glutamine and taurine did not change before or after exercise, it is not likely that these changes are of any significance to energy transduction under high intensity exercise conditions.
In theory, an increase in phosphocreatine (PCr) hydrolysis may increase in response to an impairment of one of the energy generating pathways. Such is the case with mitochondrial cytopathies, where impaired aerobic energy transduction is accompanied by increased glycolytic/glycogenolytic flux (increased lactate) and an increase in PCr hydrolysis (Tarnopolsky, et al. 1998; Tarnopolsky & Parise, 1999). Furthermore, an enhanced PCr hydrolysis has been described in patients with McArdle's disease during exercise (Sahlin et al. 1995). Together, these results suggest that high grade aerobic and anaerobic metabolic defects are partially compensated for by an enhanced PCr hydrolysis. In the current study, there were no between-group differences in PCr hydrolysis in response to exercise, nor were the basal concentrations reduced in the AMPD deficient patients. Although it is possible the shorter exercise intensity on the ergometer for homozygous patients could have masked a greater PCr hydrolysis for this group, our findings are consistent with an earlier study comparing five AMPD deficient patients to ten control subjects during ischaemic, isometric exercise (Sinkeler et al. 1987). The lack of an enhanced PCr hydrolysis (in combination with normal lactate production) during cycling exercise in AMPD deficiency suggests that AMPD deficiency does not result in an upregulation of compensatory anaerobic energy transduction pathways during this kind of exercise.
A recent paper has demonstrated a reduction in the force of contraction of mechanically skinned rat skeletal muscle with 0.4 and 3 mm adenosine (Blazev & Lamb, 1999). This group has suggested that an increase in adenosine concentration could account for the fatigue experienced by patients with AMPD deficiency (Blazev & Lamb, 1999). This group (Blazev & Lamb, 1999) quotes a case report where one patient showed a post-exercise increase in adenosine (Sabina et al. 1980) as evidence for the potential role in fatigue. As we have argued above, the single case report does not have muscle biochemical findings that are consistent with other larger case series (Sinkeler et al. 1987), nor with the results of the current study. Given that we did not observe a significant increase in adenosine in the AMPD deficient subjects from the current study, we do not feel that adenosine causes muscle fatigue during this type of exercise.
The biochemical data reported in the current study in the patients with high grade AMPD deficiency support the conclusions that AMPD deficiency is a ‘harmless genetic variant’ (Verzijl et al. 1998). This group (Verzijl et al. 1998), and others (Norman et al. 1995, 1998), reported that complete AMPD genetic deficiency occurs in about 2 % of an otherwise asymptomatic group of subjects. Furthermore, the allelic frequency (heterozygous state) is between 13.7 and 18 % in asymptomatic healthy controls in the Netherlands (Verzijl et al. 1998) and the United States/Sweden (Norman et al. 1995, 1998). From a phylogenetic perspective, it would be difficult to believe that a genetic mutation that led to significant muscle dysfunction would continue to have such a high allelic frequency in out-bred populations.
Some have suggested that AMPD deficiency is found with a higher frequency in patients referred for exercise-induced myalgias (Kelemen et al. 1982; Gross, 1998). This observation was called into question based upon the results of Verzijl et al. (1998) who found similar homozygous frequency rates for patients with exertional myalgia (1 %, n = 1836 patients) and neuromuscular complaints (1.8 %, n = 2388 patients). One possible explanation for the discrepancy in the results could be the fact that the diagnosis of AMPD deficiency was based upon forearm ischaemic testing and histochemical AMPD activity where 8.3 % of the patients with exertional myalgia were completely deficient (Kelemen et al. 1982). We have found that the combination of the forearm ischaemic test and the AMPD histochemical staining technique may result in a false positive diagnosis of ‘primary’ AMPD deficiency (Tarnopolsky et al. 1999). For example, in the current study we had identified six patients who could be classified as having primary AMPD deficiency using solely forearm ischaemic testing and muscle histochemistry for AMPD activity, yet only three had absent biochemical enzyme activity, one had a partial reduction in activity (≈50 %) and two had normal enzyme activity. The genetic analysis was 100 % concordant with the direct biochemical assay used in the current study. Therefore, we feel that either direct biochemical enzyme activity determination and/or genetic analysis are required to classify patients as having ‘primary’ AMPD deficiency. The use of correlative associations between AMPD deficiency and symptoms has led to an ongoing debate about the pathogenicity of the enzyme deficiency (Fishbein, 1999). For this reason, we chose to examine the physiological and muscle metabolic responses to high intensity exercise after rigorously assessing the AMPD enzyme activity of each subject through direct biochemical assay, to objectively determine whether AMPD enzymatic deficiency had a negative impact upon muscle metabolism/fatigue. We do concede that with the variation in exercise duration, and hence intensity, and the small number of subjects, we may not have detected a negative effect of AMPD deficiency on performance (type II error). It is probable that if more than three homozygous subjects had been tested then a statistically significant difference in performance may have been revealed, albeit unrelated to impaired TCAi depletion. However, the biochemical data from the current study and from a previous study using isometric exercise (Sinkeler et al. 1987) provide strong support against a role for the pathways thought to be altered by AMPD deficiency that could alter exercise performance. It is possible, however, that the similar decrease in PCr hydrolysis and somewhat shorter exercise duration for the homozygous subjects, as compared with control subjects, could indicate a greater PCr hydrolysis per unit of time in the presence of AMPD deficiency. Future studies should evaluate exercise performance in a large number of patients with genetically or biochemically proven AMPD mutations/deficiency to rule out a more subtle effect of the defect. If one is found, it must be through a pathway unrelated to TCAi anaplerosis. A final finding in the current study is that the patients who met Fishbein's criteria for functional AMPD deficiency (Fishbein et al. 1990) did not appear to have a Km mutant but instead had enzyme activities (< 50 %) consistent with their heterozygous state for the AMPD allele. If the heterozygous subjects had a Km mutant that was not activated in vivo, we would have expected to see an attenuation of IMP accumulation and/or an increase in AMP accumulation following exercise. These findings do not rule out that some patients who meet the criteria for functional AMPD deficiency may have a Km mutant enzyme or an impairment of ammonia efflux (Fishbein et al. 1990). However, the high frequency of the mutant allele would suggest that the most likely scenario would be heterozygosity for the AMPD1 mutant allele. We also considered that an abnormality of the soluble:total AMPD enzyme activity ratio could possibly explain some cases of functional AMPD deficiency, for one study found three regions in the AMPD1 peptide that regulated protein binding (Hisatome et al. 1998). However, our data have shown that the ratio of soluble:total AMPD activity was similar in all groups (≈0.70). This indicated that protein binding is not affected by the AMPD1 mutation, yet does not rule out the remote possibility that some cases of functional AMPD deficiency are due to a mutation in one of the three regions that are important in protein binding (Hisatome et al. 1998).
In summary, a high grade deficiency of AMPD activity does not result in an apparent impairment of TCAi anaplerosis, exercise capacity, phosphocreatine hydrolysis, or cellular energy charge during exhaustive cycling exercise.
Acknowledgments
This study was supported by NSERC Canada and the Department of Physical Medicine and Rehabilitation of Hamilton Health Sciences Corporation. Ms Joan Martin was invaluable in data collection and manuscript preparation. Some of these data were presented in abstract form at the Federation of the American Society for Experimental Biology Meeting, Washington, 1999.
References
- Blazev R, Lamb GD. Adenosine inhibits depolarization-induced Ca2+ release in mammalian skeletal muscle. Muscle and Nerve. 1999;22:1674–1683. doi: 10.1002/(sici)1097-4598(199912)22:12<1674::aid-mus9>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Fishbein WN. Primary, secondary, and coincidental types of myoadenylate deaminase deficiency (letter) Annals of Neurology. 1999;45:547–548. doi: 10.1002/1531-8249(199904)45:4<547::aid-ana25>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Fishbein WN, Armbrustmacher VW, Griffin JL. Myoadenylate deaminase deficiency: a new disease of muscle. Science. 1978;200:545–548. doi: 10.1126/science.644316. [DOI] [PubMed] [Google Scholar]
- Fishbein WN, Foellmer JW, Davis JI. Medical implications of the lactate and ammonia relationship in anaerobic exercise. International Journal of Sports Medicine. 1990;11(2):S91–S100. doi: 10.1055/s-2007-1024860. [DOI] [PubMed] [Google Scholar]
- Flanagan WF, Holmes EW, Sabina RL, Swain JL. Importance of purine nucleotide cycle to energy production in skeletal muscle. American Journal of Physiology. 1986;251:C795–802. doi: 10.1152/ajpcell.1986.251.5.C795. [DOI] [PubMed] [Google Scholar]
- Gibala MJ, Maclean DA, Graham TE, Saltin B. Anaplerotic processes in human skeletal muscle during brief dynamic exercise. Journal of Physiology. 1997a;502:703–713. doi: 10.1111/j.1469-7793.1997.703bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibala MJ, Tarnopolsky MA, Graham TE. Tricarboxylic acid cycle intermediates in human muscle at rest and during prolonged cycling. American Journal of Physiology. 1997b;272:E239–244. doi: 10.1152/ajpendo.1997.272.2.E239. [DOI] [PubMed] [Google Scholar]
- Graham TE, Turcotte LP, Kiens B, Richter EA. Training and muscle ammonia and amino acid metabolism in humans during prolonged exercise. Journal of Applied Physiology. 1995;78:725–735. doi: 10.1152/jappl.1995.78.2.725. [DOI] [PubMed] [Google Scholar]
- Gross M. Clinical heterogeneity and molecular mechanisms in inborn muscle AMP deaminase deficiency. Journal of Inherited Metabolic Diseases. 1997;20:186–192. doi: 10.1023/a:1005352605421. [DOI] [PubMed] [Google Scholar]
- Heinrikson RL, Meredith SC. Amino acid analysis by reverse phase high-performance liquid chromatography: precolumn derivatization with phenylisothiocyanate. Analytical Biochemistry. 1984;36:65–74. doi: 10.1016/0003-2697(84)90307-5. [DOI] [PubMed] [Google Scholar]
- Hisatome I, Morisake T, Kamma H, Sugama T, Morisaki H, Ohtahara A, Holmes EW. Control of AMP deaminase 1 binding to myosin heavy chain. American Journal of Physiology. 1998;275:C870–881. doi: 10.1152/ajpcell.1998.275.3.C870. [DOI] [PubMed] [Google Scholar]
- Kelemen J, Rice DR, Bradley WG, Munsat TL, Dimauro S, Hogan EL. Familial myoadenylate deaminase deficiency and exertional myalgia. Neurology. 1982;32:857–863. doi: 10.1212/wnl.32.8.857. [DOI] [PubMed] [Google Scholar]
- Meyer RA, Terjung RL. AMP deamination and IMP reamination in working skeletal muscle. American Journal of Physiology. 1980;239:C32–38. doi: 10.1152/ajpcell.1980.239.1.C32. [DOI] [PubMed] [Google Scholar]
- Mineo I, Tarui S. Myogenic hyperuricemia: What can we learn from metabolic myopathies. Muscle and Nerve. 1995;(3):S75–S81. doi: 10.1002/mus.880181416. [DOI] [PubMed] [Google Scholar]
- Morisaki T, Gross M, Morisaki H, Pongratz D, Zollner N, Holmes EW. Molecular basis of AMP deaminase deficiency in skeletal muscle. Proceedings of the National Academy of Sciences of the USA. 1992;89:6457–6461. doi: 10.1073/pnas.89.14.6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman B, Glenmark B, Jansson E. Muscle AMP deaminase deficiency in 2 % of a healthy population. Muscle and Nerve. 1995;18:239–241. doi: 10.1002/mus.880180216. [DOI] [PubMed] [Google Scholar]
- Norman B, Mahnke-Zizelman DK, Vallis A, Sabina RL. Genetic and other determinants of AMP deaminase activity in healthy adult skeletal muscle. Journal of Applied Physiology. 1998;85:1273–1278. doi: 10.1152/jappl.1998.85.4.1273. [DOI] [PubMed] [Google Scholar]
- Passoneau JA, Lowry OH. Enzymatic Analysis: A Practical Guide. Totawa, NJ, USA: Humana Press; 1993. [Google Scholar]
- Rush JW E, Tullson PC, Terjung RL. Molecular and kinetic alterations of muscle AMP deaminase during chronic creatine depletion. American Journal of Physiology. 1998;274:C465–471. doi: 10.1152/ajpcell.1998.274.2.C465. [DOI] [PubMed] [Google Scholar]
- Sabina RL, Fishbein WN, Pezeshkpour G, Clarke PR H, Holmes EW. Molecular analysis of the myoadenylate deaminase deficiencies. Neurology. 1992;42:170–179. doi: 10.1212/wnl.42.1.170. [DOI] [PubMed] [Google Scholar]
- Sabina RL, Swain JL, Patten BM, Ashizawa T, O'brien WE, Holmes EW. Disruption of the purine nucleotide cycle: A potential explanation for muscle dysfunction in myoadenylate deaminase deficiency. Journal of Clinical Investigation. 1980;66:1419–1423. doi: 10.1172/JCI109995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlin K, Broberg S. Adenine nucleotide depletion in human muscle during exercise: Causality and significance of AMP deamination. International Journal of Sports Medicine. 1990;11(2):S62–S67. doi: 10.1055/s-2007-1024856. [DOI] [PubMed] [Google Scholar]
- Sahlin K, Jorfeldt L, Henriksson KG, Lewis SF, Haller RG. Tricarboxylic acid cycle intermediates during incremental exercise in healthy subjects and in patients with McArdle's disease. Clinical Science. 1995;88:687–693. doi: 10.1042/cs0880687. [DOI] [PubMed] [Google Scholar]
- Sahlin K, Katz A, Broberg S. Tricarboxylic acid cycle intermediates in human muscle during prolonged exercise. American Journal of Physiology. 1990;259:C834–841. doi: 10.1152/ajpcell.1990.259.5.C834. [DOI] [PubMed] [Google Scholar]
- Shumate JB, Katnik R, Ruiz M, Kaiser K, Frieden C, Brooke MH, Carroll JE. Myoadenylate deaminase deficiency. Muscle and Nerve. 1979;2:2113–2116. doi: 10.1002/mus.880020309. [DOI] [PubMed] [Google Scholar]
- Sinkeler SP, Binkhorst RA, Joosten EM, Wevers RA, Coerwinkei MM, Oei TL. AMP deaminase deficiency: study of the human skeletal muscle purine metabolism during ischemic isometric exercise. Clinical Science. 1987;72:475–482. doi: 10.1042/cs0720475. [DOI] [PubMed] [Google Scholar]
- Tarnopolsky MA, Atkinson SA, Phillips SM, Macdougall JD. Carbohydrate loading and metabolism during exercise in males and females. Journal of Applied Physiology. 1995;78:1360–1368. doi: 10.1152/jappl.1995.78.4.1360. [DOI] [PubMed] [Google Scholar]
- Tarnopolsky MA, Macguire J, Myint T, Applegarth D, Robinson BH. Clinical, physiological, and histological features in a kindred with the T3217C MELAS mutation. Muscle and Nerve. 1998;21:25–33. doi: 10.1002/(sici)1097-4598(199801)21:1<25::aid-mus4>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Tarnopolsky MA, Parise G. Direct measurement of high energy phosphate compounds in patients with neuromuscular disease. Muscle and Nerve. 1999;22:1228–1233. doi: 10.1002/(sici)1097-4598(199909)22:9<1228::aid-mus9>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Tarnopolsky MA, Rush JW E, Stevens L, Macguire J. The clinical utility of the forearm ischemic test lactate/ammonia response in the diagnosis of neuromuscular disorders: Insights into mitochondrial cytopathies and myoadenylate deaminase deficiency. Neurology. 1999;52(2):A461. [Google Scholar]
- Tullson PC, Whitlock DM, Terjung RL. Adenine nucleotide degradation in slow-twitch red muscle. American Journal of Physiology. 1990;258:C258–265. doi: 10.1152/ajpcell.1990.258.2.C258. [DOI] [PubMed] [Google Scholar]
- Verzijl HTFM, van Engelen BGM, Luyten JAFM, Steenbergen GCH, van den Heuvel LPWJ, Ter Laak HJ, Padberg GW, Wevers RA. Genetic characteristics of myoadenylate deaminase deficiency. Annals of Neurology. 1998;44:140–143. doi: 10.1002/ana.410440124. [DOI] [PubMed] [Google Scholar]