

Abstract
Ventricular myocytes demonstrate a steeply inwardly rectifying K+ current termed IK1. We investigated the molecular basis for murine IK1 by removing the genes encoding Kir2.1 and Kir2.2. The physiological consequences of the loss of these genes were studied in newborn animals because mice lacking Kir2.1 have a cleft palate and die shortly after birth.
Kir2.1−/− ventricular myocytes lack detectable IK1 in whole-cell recordings in 4 mM external K+. In 60 mM external K+ a small, slower, residual current is observed. Thus Kir2.1 is the major determinant of IK1. Sustained outward K+ currents and Ba2+ currents through L- and T-type channels were not significantly altered by the mutation. A 50 % reduction in IK1 was observed in Kir2.2−/− mice, raising the possibility that Kir2.2 can also contribute to the native IK1.
Kir2.1−/− myocytes showed significantly broader action potentials and more frequent spontaneous action potentials than wild-type myocytes.
In electrocardiograms of Kir2.1−/− neonates, neither ectopic beats nor re-entry arrhythmias were observed. Thus the increased automaticity and prolonged action potential of the mutant ventricular myocytes were not sufficiently severe to disrupt the sinus pacing of the heart. The Kir2.1−/− mice, however, had consistently slower heart rates and this phenotype is likely to arise indirectly from the influence of Kir2.1 outside the heart.
Thus Kir2.1 is the major component of murine IK1 and the Kir2.1−/− mouse provides a model in which the functional consequences of removing IK1 can be studied at both cellular and organismal levels.
Inwardly rectifying potassium channels have been identified in a wide range of tissues and are distinguished by their ability to allow the passage of inward current to a much greater extent than outward current. The attenuated outward flow of K+ through these channels at positive potentials is thought to occur because the internal pore of the channel becomes plugged by Mg2+ and positively charged polyamines (Matsuda et al. 1987; Vandenberg, 1987; Silver & DeCoursey, 1990; Matsuda, 1991; Ficker et al. 1994; Elam & Lansman, 1995; Fakler et al. 1995; Lopatin et al. 1995; Shyng et al. 1996). Cardiac tissue, especially atrial and ventricular myocytes, demonstrates an inward potassium current that is strongly and steeply rectifying and which has been termed IK1. This current is thought to be responsible for maintaining the negative resting potential seen in these cells (Nichols et al. 1996). In addition, because the channels responsible for IK1 close during depolarization, the conductance of cardiac myocytes decreases during the cardiac action potential. The rectification thereby conserves metabolic energy in a tissue that is constantly electrically active (Isomoto et al. 1997). Finally, the outward component of IK1 is thought to contribute to phase 3 repolarization of the cardiac action potential (Ibarra et al. 1991; Varro & Papp, 1992; Carmeliet, 1993; Deal et al. 1996).
Although the physiological properties of IK1 have been extensively studied, its molecular composition is unknown. To date, seven families of genes encoding inward rectifiers have been identified. These Kir channels share a similar proposed subunit structure: a pore-forming loop with a single transmembrane segment on either side. Functional inward rectifier channels are thought to be made up of four subunits (Yang et al. 1995). In some cases, subunits encoded by separate genes can be assembled into a heteromultimeric channel. A notable example of such a channel is the acetylcholine-activated inward rectifier, which is composed of Kir3.1 and Kir3.4 subunits (Krapivinsky et al. 1995). However, not all gene products of the inward rectifier family appear to be able to co-assemble in vitro, and in many cases, including Kir2.1 and Kir2.2, functional channels appear to be formed by four identical subunits (Tinker et al. 1996).
Attempts to pinpoint the molecular basis of IK1 have been hampered by the lack of suitable pharmacological agents. Most Kir channels are Ba2+ sensitive and there are no known toxins that distinguish them. Lately, transcript analysis in cardiac tissues has attempted to identify the inward rectifier responsible for the IK1 current (Dixon & McKinnon, 1994; Barry et al. 1995; Brahmajothi et al. 1996; Dixon et al. 1996). This analysis, in combination with comparisons of the biophysical characteristics of IK1 with those of cloned channels expressed in oocytes, has pointed to the Kir2 family. Within this family, Kir2.1 is the leading candidate because levels of its transcript correlate strongly with the amount of IK1 seen electrophysiologically (Brahmajothi et al. 1996). In the sinoatrial (SA) node where IK1 is absent, there is no Kir2.1 transcript. In the ventricle, where IK1 is present, Kir2.1, when compared to Kir2.2 or Kir2.3, has the highest level of transcript. However, these conclusions are tempered by the poor correlation seen between RNA and protein expression for some cardiac channels (Barry et al. 1995; Xu et al. 1996). Attempts to further pinpoint the molecular identity of IK1 by comparing its single channel conductance with that of Kir2.1, Kir2.2 or Kir2.3 are also problematic. The single channel conductances of Kir2.1, Kir2.2 and Kir2.3 are 22, 34 and 13 pS, respectively, with 150 mm external K+, while the reported predominant single channel conductances of IK1 range from 20 to 30 pS in 150 mm external K+ (Sakmann & Trube, 1984; Kurachi, 1985; Matsuda, 1988; Kubo et al. 1993; Takahashi et al. 1994; Morishige et al. 1994; Isomoto et al. 1997). Recently there has been more definitive evidence linking Kir2.1 to IK1 through genetic manipulation of cardiac cells in vitro. Nakamura (1998), using antisense oligonucleotides, partially suppressed IK1 in adult rat ventricular myocytes and thereby demonstrated that the Kir2.1 gene encodes at least one component of the native IK1.
To further define the roles of the genes encoding Kir2.1 and Kir2.2 in the generation of IK1, we have engineered mice lacking the open reading frames encoding Kir2.1 and Kir2.2 (Zaritsky et al. 2000). This gene knockout strategy has allowed us to study the influence of the deletion of a single cardiac channel gene on the cellular and whole animal levels. In particular, we have examined the impact of the removal of these channels on individual currents as well as on action potential shape in isolated cardiac myocytes. In addition, we have attempted to correlate any changes seen at this level with changes seen in the behaviour of the heart in the intact animal.
METHODS
Mouse genetics
Generation and genotyping of mice lacking either Kir2.1 or Kir2.2 is described elsewhere (Zaritsky et al. 2000). Both strains were in an FVB genetic background and had undergone at least five backcrosses to an FVB strain. Wild-type FVB mice of the same age served as controls, except in ECG studies for which control animals consisted of Kir2.1-/+ and Kir2.1+/+ littermates. All animals were housed and cared for in a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care. For experiments on neonatal dissociated myocytes and explanted tissues, mice were killed by decapitation according to protocols approved by the animal use and care committee of Stanford University.
Transcript analysis
Twenty-five micrograms of total RNA extracted with Trizol (Gibco-BRL) was separated on a formaldehyde gel and transferred to a nylon membrane by capillary action. 32P-labelled probes were generated by random-priming a 389 base pair (bp) fragment from the Kir2.1 open reading frame (ORF) (bp 3-391), an 892 bp fragment from the Kir2.2 ORF (bp 250–1141) or a 432 bp fragment from the Kir2.3 ORF (bp 120–551). Equal loading of total RNA was confirmed by re-probing the blots with a 1.5 kb fragment of the glyceraldehyde phosphate dehydrogenase (GAPDH) open reading frame.
Isolation of myocytes
Ventricular myocytes were isolated from neonatal animals by adapting the methods of Davies et al. (1996). Briefly, hearts were aseptically removed from 1- to 10-h-old pups. The hearts were placed in calcium- and bicarbonate-free Hanks' solution with Hepes buffer containing (mm): 137 NaCl, 20 Hepes, 5.5 glucose, 5.4 KCl, 0.8 MgSO4, 0.44 KH2PO4 and 0.34 NaH2PO4, pH 7.5. The atria were cut away and then each ventricle was placed into 0.5 ml of modified Hanks' buffer containing 100 units ml−1 collagenase type II (Gibco-BRL) and 25.0 mg ml−1 pancreatin (Gibco-BRL). Digestion took place at 37 °C for 30 min on a rotating platform. Each ventricle was then triturated by several passes through a 1 ml pipette tip. The tubes were allowed to sit for 1 min and the supernatant was transferred to a tube containing 0.5 ml of Dulbecco's modified Eagle's medium supplemented with 10 % horse serum and 5 % fetal calf serum. The tubes were then centrifuged at 420 g for 1 min and the supernatant was discarded. The cell pellet was resuspended in medium and plated on laminin-treated (20 μg ml−1, Boehringer Mannheim) glass coverslips. The myocytes were cultured at 37 °C, 5 % CO2. Most electrophysiological recordings were made after 24 h in culture. A series of recordings were also done 3 h after dissociation and it was determined that 24 h in culture did not alter the observed spectrum of currents.
Single cell electrophysiology
Whole-cell patch-clamp recordings were made at room temperature (24 °C) in a perfusion chamber under standard conditions (Nuss & Marban, 1994) using an Axopatch 200B amplifier and pCLAMP software (Axon Instruments, Foster City, CA, USA). The patch pipettes (borosilicate glass, KG-33, Garner Glass Company, Claremont, CA, USA) were pulled and fire-polished to resistances of 2–4 MΩ when filled with an internal recording solution. The series resistance was compensated (70 %) and cell capacitance was determined by analog measurement with the patch-clamp amplifier. Currents were filtered with a cut-off frequency of 10 kHz and sampled at 5 kHz.
During recordings of inwardly rectifying potassium currents, the extracellular solution was composed of (mm): 136 NaCl, 10 glucose, 10 Hepes, 4 KCl, 2 MgCl2 and 1.0 CaCl2 (pH 7.4 with NaOH). When 60 mm KCl was used, the NaCl concentration was adjusted to 80 mm. Electrodes were filled with (mm): 135 KCl, 10 EGTA, 10 Hepes, 5 Mg-ATP and 5 glucose (pH 7.3 with KOH).
Ba2+ currents carried through Ca2+ channels were recorded in a solution containing (mm): 127 NaCl, 20 CsCl2, 10 Hepes, 10 BaCl2 and 1 MgCl2 (pH 7.4 with NaOH). The pipette-filling solution was composed of (mm): 190 N-methyl-d-glucamine (NMDG), 40 Hepes, 12 phosphocreatine, 5 EGTA, 3 Mg-ATP, 0.4 MgCl2; and 200 μm GTP (pH 7.2 with H2SO4). TTX (1 μm; Calbiochem, San Diego, CA, USA) was included in the external solution in order to block Na+ channels. When noted, 1 μm nifedipine (Sigma) was used to block L-type Ca2+ channels.
Voltage-activated K+ currents were recorded in a Na+-free external solution containing (mm): 136 NMDG, 15 glucose, 10 Hepes, 4 KCl, 2 MgCl2 and 0.1 CaCl2 (pH 7.4 with HCl). Cd+ at 500 μm was included to block Ca2+ currents. The internal solution was identical to that used for inwardly rectifying currents.
To record action potentials, whole-cell recordings were established in voltage-clamp mode and, after compensation for pipette capacitance, the amplifier was switched to current-clamp mode. External and internal pipette solutions were identical to those used to record inwardly rectifying currents. To examine individual action potentials, a small amount of current was injected to hyperpolarize the cell to −80 mV and short depolarizing current pulses were used to trigger action potentials. For recordings of spontaneous activity, no current was injected. Instead, current-clamp recordings captured any spontaneously generated action potentials over a 30 s interval.
Myocyte automaticity
Myocyte cultures were removed from the incubator, placed on the heated (37 °C) stage of an inverted microscope and inspected immediately. Individual myocytes were examined for 3 s and scored as beating or quiescent. Approximately 60 cells per dish were scored and the results were tabulated to determine the fraction of beating cells per dish. The observer was blind to the phenotype of the animal from which the cells were isolated.
Electrocardiograms
Surface electrocardiograms (ECGs) were recorded from 1- to 10-h-old pups using the PhysioTEL telemetry system (Data Sciences International, Saint Paul, MN, USA). Pups were housed in a 28 °C incubator with supplemental O2 (1 l min−1). The control population contained both wild-type and heterozygous littermates of Kir2.1−/− mice. Needle electrodes were placed on the skin of the left and right shoulders. Data were digitized at 2 kHz and stored and analysed with MacLab software (ADInstruments, Mountain View, CA, USA). For interval measurements the following conventions were used: the RR interval was measured from the peak of one R wave to the peak of the subsequent R wave, the PR interval was measured from the peak of the P wave to the onset of the Q wave, the QRS interval was measured from the onset of the Q wave to the end of the S wave and the QT interval was measured from the onset of the Q wave to the maximum deflection of the T wave. This QT interval was used because the determination of the T wave endpoint was often obscured by a shifting baseline. Within a 3 min recording, little beat-to-beat variation was observed and so a representative beat was selected for measurement from each animal.
Data analysis
Voltage-clamp and current-clamp data were compiled and analysed using Clampfit (Axon Instruments) and Prism (GraphPad Software, San Diego, CA, USA). Single cell current amplitudes were normalized for differences in cell size by whole-cell membrane capacitance. All data are presented as means and standard error of the mean (mean ±s.e.m.). Differences between means of two groups were tested using Student's unpaired t test. Statistical significance was taken at P < 0.05.
RESULTS
Generation and phenotype of mice lacking the Kir2.1 or Kir2.2 gene
Elsewhere we describe the generation and gross phenotype of transgenic mice in which the genes encoding Kir2.1 and Kir2.2 had been disrupted (Zaritsky et al. 2000). In the construction of the targeting vectors, we removed the entire open reading frame encoding that gene. This ensured that in the knockout animals, a truncated version of the protein would not be produced. Such a truncated protein might have had some residual function or interfered with another Kir channel, and such an action would have complicated the analysis of the knockout animal. Homozygous Kir2.2−/− mice are viable as adults and are fertile. They appear grossly normal and a histological analysis of their hearts and brains has revealed no abnormalities. In contrast, all of the homozygous Kir2.1−/− animals die shortly after birth from a complete cleft of the secondary palate. Therefore, in the present study we restricted our examination to neonatal animals 1–10 h after birth.
Transcript analysis of cardiac tissue derived from Kir2.1−/−, Kir2.2−/− and wild-type animals
The Kir2.1 and Kir2.2 proteins share a very high level of sequence homology and their patterns of expression overlap in several tissues. In the mouse heart both proteins are expressed in the atria and ventricles. To interpret the cardiac phenotype of the Kir2.1 knockout it was necessary to know if a compensatory up-regulation of Kir2.2 or Kir2.3 transcription had occurred. A similar potential existed for up-regulation of Kir2.1 or Kir2.3 in the Kir2.2 knockout. To address these possibilities, we analysed total RNA isolated from the cardiac tissue of neonatal Kir2.1−/−, Kir2.2−/− and wild-type animals (Fig. 1). Kir2.1 transcript, as expected, was the most abundant member of this family in the heart, even at this neonatal stage. Kir2.2 transcripts were present as well, but little Kir2.3 was observed in wild-type animals. The transcripts for the disrupted genes were completely undetectable, which was to be expected once the open reading frame was removed. Neither Kir2.1 nor Kir2.2 transcripts were up-regulated in response to the deletion of one another. The level of Kir2.3 transcript was increased in the Kir2.1 knockout hearts (Fig. 1), but this increase lacks detectable physiological significance (see below).
Figure 1. Transcript analysis of cardiac tissue.
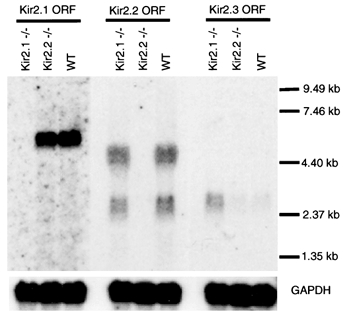
Each lane was loaded with 25 μg of total neonatal ventricular RNA from wild-type (WT), Kir2.1−/− or Kir2.2−/− mice and probed with a portion of the open reading frame of Kir2.1, Kir2.2 or Kir2.3. Kir2.1 and 2.3 are each encoded by a single transcript while Kir2.2 is encoded by two. Equal loading of RNA was confirmed by re-probing the blots with a fragment of the GAPDH open reading frame.
Inward K+ currents in wild-type, Kir2.1−/− and Kir2.2−/− neonatal ventricular myocytes
To determine if either Kir2.1 or Kir2.2 encoded the subunits of the channel conducting IK1, whole-cell voltage-clamp recordings were made from dissociated neonatal ventricular myocytes. Representative inward K+ currents in 4 mm extracellular K+ were recorded from individual ventricular myocytes isolated from wild-type and Kir2.1−/− mice (Fig. 2A and B). Currents were evoked by voltage steps to −70 to −130 mV (in 15 mV increments) from a holding potential of −40 mV. Leak currents were subtracted using currents evoked by small hyperpolarizing pulses (P/4 method), as has previously been employed for the analysis of this current (Masuda & Sperelakis, 1993). In general, the evoked IK1 was large and the leak currents minor and in consequence the subtraction had little influence on the amplitude or shape of the current. Using this protocol, the currents recorded from wild-type myocytes closely resembled those that have been previously described in neonatal rat myocytes (Masuda & Sperelakis, 1993). Notably, IK1 currents were not detected in myocytes isolated from Kir2.1−/− animals though they were prominent in similarly aged wild-type cells. The mean ±s.e.m. steady state inward currents at −130 mV (normalized to cellular capacitance) were −10.0 ± 1.0 pA pF−1 (n = 11) in wild-type myocytes and 0.4 ± 0.5 pA pF−1 (n = 8) in Kir2.1−/− myocytes; these values are significantly different (P < 0.001) (Fig. 2C).
Figure 2. Inwardly rectifying K+ currents in 4 mm external K+.
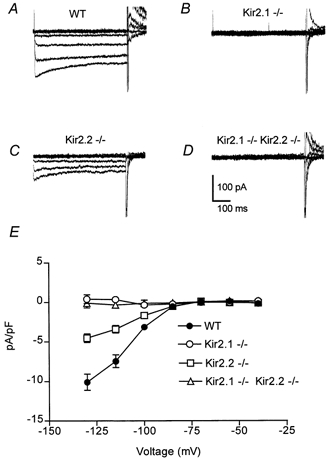
A-D, inwardly rectifying currents were recorded in 4 mm external K+ from voltage-clamped neonatal myocytes isolated from wild-type (WT), Kir2.1−/−, Kir2.2−/− and Kir2.1−/−-Kir.2.2−/− mice. For each family of current traces, the voltage was stepped from a holding potential of −40 mV to potentials of −70 to −130 mV (in 15 mV increments). Leak currents were subtracted using currents evoked by small hyperpolarizing pulses (P/4 method). Cell capacitances were 14, 18, 19 and 19 pF, respectively. E, steady state currents (means ±s.e.m.) plotted as a function of voltage in wild-type (n = 11), Kir2.1−/− (n = 8), Kir2.2−/− (n = 11) and Kir2.1−/−-Kir.2.2−/− (n = 4) myocytes. Current amplitudes were measured at the end of the 500 ms voltage-clamp step and were normalized to cell capacitance to control for cell surface area.
Inward K+ currents can be enhanced by raising the extracellular K+ concentration to increase the available charge carrier. To look more stringently for any remaining IK1 in the Kir2.1−/− animal, we repeated the voltage-clamp measurements in 60 mm[K+]o. Currents were evoked in wild-type and Kir2.1−/− myocytes by voltage steps to −10 to −70 mV (in 15 mV increments) from a holding potential of −10 mV (Fig. 3A and B). For leak subtraction small depolarizing pulses were used because hyperpolarizing pulses would have evoked IK1. The increase in external K+ concentration did uncover a small but observable steady state inward current in Kir2.1−/− myocytes (mean ±s.e.m. at −70 mV, −11.7 ± 1.2 pA pF−1, n = 8), which represents 16 % of the amplitude of the wild-type current (mean ±s.e.m. at −70 mV, −71.5 ± 2.9 pA pF−1, n = 4) (Fig. 3E).
Figure 3. Inwardly rectifying K+ currents in 60 mm external K+.
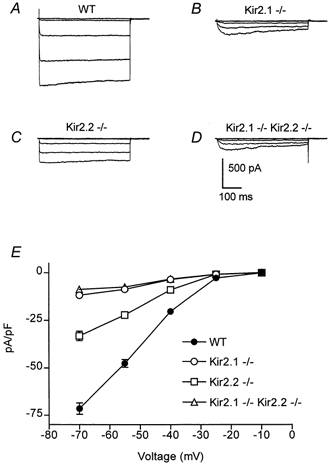
A–D, inwardly rectifying currents were recorded in 60 mm external K+ from voltage-clamped neonatal myocytes isolated from wild-type, Kir2.1−/−, Kir2.2−/− and Kir2.1−/−-Kir.2.2−/− mice. For each family of current traces, the voltage was stepped from a holding potential of −10 mV to potentials of −10 to −70 mV (in 15 mV increments). Leak currents were subtracted using currents evoked by small depolarizing pulses (P/4 method). Cell capacitances were 15, 14, 11 and 15 pF, respectively. E, steady state currents (means ±s.e.m.) were plotted as a function of voltage in wild-type (n = 4), Kir2.1−/− (n = 8), Kir2.2−/− (n = 11) and Kir2.1−/−-Kir.2.2−/− (n = 9) myocytes. Current amplitudes were measured at the end of the 500 ms voltage-clamp step and were normalized to cell capacitance to control for cell surface area.
Because this residual inwardly rectifying current could be due to another member of the Kir family, we next examined myocytes isolated from Kir2.2−/− animals. Inward K+ currents were recorded from Kir2.2−/− cells as above. In 4 mm external K+ there was a 50 % reduction in the steady state inward current density with a mean of −4.5 ± 0.5 pA pF−1 (n = 11) at −130 mV, which was significantly different from both Kir2.1−/− myocytes and wild-type myocytes (P < 0.001) (Fig. 2C and E). When these recordings were repeated using 60 mm external K+ (Fig. 3C and E), the inward currents were again significantly smaller than wild-type (mean ±s.e.m. at −70 mV, −33.1 ± 2.5 pA pF−1, n = 11; P < 0.001). Thus, though the dramatic effect of the loss of Kir2.1 would suggest that IK1 is primarily conducted through channels encoded by that gene, the phenotype of the Kir2.2 knockouts indicates that Kir2.2 may also play a role in the generation of this current. Indeed, the two genes together would appear to account for substantially more than 100 % of the current and the possible explanations of this observation are discussed below.
To further explore the possibility of Kir2.2 contributing to the small inwardly rectifying current seen in 60 mm external K+ in the absence of Kir2.1, we examined myocytes from animals lacking both genes. In 4 mm external K+, myocytes isolated from Kir2.1−/−-Kir2.2−/− animals showed no inward rectification with a steady state inward density mean of −0.1 ± 0.6 pA pF−1 (n = 4) at −130 mV (Fig. 2D and E). In 60 mm external K+, however, inward K+ currents recorded from Kir2.1−/−- Kir2.2−/− myocytes were very similar in amplitude to those from Kir2.1−/− (−8.7 ± 0.9 pA pF−1 at −70 mV, n = 9) (Fig. 3D and E). Thus, the inward K+ current of neonatal ventricular myocytes appears to consist of two component currents. The major current requires Kir2.1 and its amplitude may be partially dependant on Kir2.2. The minor component that is only detectable in 60 mm external K+ is independent of these genes.
Ca2+ currents and outward K+ currents in wild-type and Kir2.1−/− ventricular myocytes
Before studying the functional consequences of removing IK1, we characterized more extensively Kir2.1−/− myocytes to address the possibility that some of the other major ionic currents had also been altered (Barry et al. 1998). Such changes might be due to a direct interaction of the channels or an indirect compensatory response of the Kir2.1−/− myocytes. In particular, Ca2+ and outward K+ currents were compared in wild-type and Kir2.1−/− ventricular myocytes.
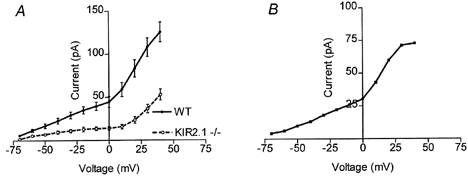
Currents through L- and T-type Ca2+ channels from wild-type and Kir2.1−/− myocytes were measured in response to voltage steps from −80 to +50 mV (in 15 mV increments) from a −80 mV holding potential (Fig. 4A and B). The external solution contained 10 mm Ba2+ in order to increase the amplitude of the currents, and 1 μm TTX to block Na+ channels. Outward K+ currents were blocked by 20 mm Cs+ in the internal solution. Figure 4C shows the current density as a function of the depolarizing voltage in wild-type and Kir2.1−/− myocytes. In contrast to the marked reduction of inward K+ currents, there were no significant differences in the inward current amplitude through Ca2+ channels between wild-type and Kir2.1−/− myocytes using the −80 mV holding potential (mean at −10 mV, −36.9 ± 5.1 pA pF−1 (n = 9); −36.4 ± 5.4 pA pF−1 (n = 9), respectively). The addition of 1 μm nifedipine to reduce L-type currents also failed to reveal any significant difference between the two cell populations (mean at −10 mV, wild-type, −17.9 ± 2.1 pA pF−1, n = 9; Kir2.1−/−, −17.6 ± 1.5 pA pF−1, n = 9; Fig. 4C). We repeated the above protocol with a −40 mV holding potential in order to inactivate the T-type channels and isolate the L-type current. Once again there was no significant difference in the amount of inward current between wild-type and Kir2.1−/− cells (data not shown).
Figure 4. Inward Ba2+ currents in wild-type and Kir2.1−/− neonatal ventricular myocytes.
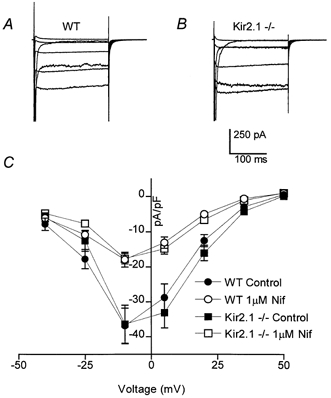
A and B, Ba2+ currents through T- and L-type Ca2+ channels were recorded in the presence of external TTX and internal Cs+ from voltage-clamped neonatal myocytes isolated from wild-type and Kir2.1−/− mice. For each family of current traces, the voltage was stepped from a holding potential of −80 mV to potentials of −40 to +50 mV (in 15 mV increments). Cell capacitances were 17 and 16 pF, respectively. C, steady state currents (means ±s.e.m.) were plotted as a function of voltage in wild-type (n = 9) and Kir2.1−/− (n = 9) myocytes, with and without the addition of 1 μm nifedipine to reduce L-type currents. Currents were measured 50 ms into the voltage pulse and were normalized to cell capacitance to control for cell surface area.
Outward potassium currents from wild-type and Kir2.1−/− myocytes were examined using a −40 mV holding potential with steps to potentials of −40 mV to +80 mV (in 20 mV increments) (Fig. 5A and B). The Na+-free external solution contained 500 μm Cd2+ to block Ca2+ currents. Steady state current density was measured at the end of the 500 ms voltage clamp step and plotted as a function of the depolarizing voltage in wild-type and Kir2.1−/− myocytes (Fig. 5C). As with the currents through the Ca2+ channels, there did not appear to be any difference in the steady state outward K+ currents between wild-type (at +80 mV, mean 8.7 ± 0.7 pA pF−1, n = 6) and Kir2.1−/− myocytes (mean 9.1 ± 1.1 pA pF−1, n = 6).
Figure 5. Outward K+ currents in wild-type and Kir2.1−/− neonatal ventricular myocytes.
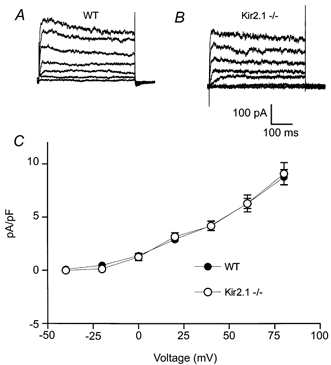
A and B, outward K+ currents were recorded from voltage-clamped myocytes isolated from wild-type and Kir2.1−/− mice. For each family of current traces, the voltage was stepped from a holding potential of −40 mV to potentials of −40 to +80 mV (in 20 mV increments). The Na+-free external solution contained 500 μm Cd2+ to block Ca2+ currents. Cell capacitances were 22 and 23 pF, respectively. C, steady state currents (means ±s.e.m.) plotted as a function of the depolarizing voltage in wild-type (n = 6) and Kir2.1−/− (n = 6) myocytes. Currents were measured at the end of the 500 ms voltage-clamp step and were normalized to cell capacitance to control for cell surface area.
Action potential duration in wild-type and Kir2.1−/− ventricular myocytes
The functional consequences of removing IK1 were investigated by examining action potential waveforms from Kir2.1−/− myocytes in current-clamp mode. Myocytes were held at approximately −80 mV by the constant injection of a small hyperpolarizing current and single action potentials were triggered by brief injections of depolarizing current (Fig. 6A and B). Although the action potential amplitudes did not differ on average between the wild-type and Kir2.1−/− cells, the action potentials recorded from Kir2.1−/− myocytes were significantly broader than those recorded from wild-type myocytes (Fig. 6C). The loss of Kir2.1 doubled the 90 % to 10 % decay time from 99 ± 10 ms (n = 17, wild-type) to 200 ± 23 ms (n = 11, Kir2.1−/−); these values are significantly different at the P < 0.001 level. Similarly, action potential half-widths differed significantly (P < 0.001), increasing from 27 ± 3 ms (n = 11, wild-type) to 75 ± 13 ms (n = 11, Kir2.1−/−).
Figure 6. Triggered action potentials in wild-type and Kir2.1−/− neonatal ventricular myocytes.
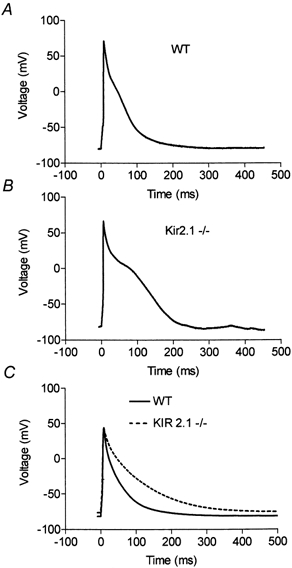
A and B, representative triggered action potentials recorded from current-clamped myocytes isolated from wild-type and Kir2.1−/− mice. The myocytes were held at approximately −80 mV by the injection of a small hyperpolarizing current and action potentials were triggered by brief injections of depolarizing current. C, superimposed average action potentials from wild-type (n = 17) and knockout (n = 11) myocytes. Though the holding potentials of cells occasionally varied by up to 5 mV, the broadening of the action potential correlated consistently with the genotype of the cell and not with the minor variations in holding potential.
Because the Kir2.1 current and ventricular IK1 are strongly rectifying, this current is predicted to be influential only late in the falling phase of the action potential, when the membrane potential is close to EK (Ibarra et al. 1991; Varro & Papp, 1992; Carmeliet, 1993; Deal et al. 1996). The action potentials in the Kir2.1−/− mice, however, appeared to be broader throughout the falling phase; the rate of repolarization appeared slower than wild-type even at very depolarized potentials. To examine this more closely, the net current at each time point during repolarization was determined by taking the derivative of individual action potentials and multiplying by the capacitance of the cell. The resultant net currents were averaged for several cells and plotted versus membrane potential (Fig. 7A). As expected from the slowing of the falling phase, the net currents measured in the knockout animals were significantly smaller than those in wild-type cells at voltages from +40 to −50 mV (P < 0.01). The increased duration of the action potential in the Kir2.1−/− animals was caused, therefore, by slower repolarization throughout the action potential and was not confined to the final phase. Thus the physiology of the myocytes had been altered in a voltage range where the inward rectifier current is not conventionally thought to act. This finding is discussed below.
Figure 7. Net current during the falling phase of the action potential as a function of voltage.
A, individual triggered action potential traces like those in Fig. 6 were differentiated with respect to time and multiplied by the capacitance of each cell to calculate the net current flowing at each time point of the falling phase of the action potential. The resultant net currents were determined for each membrane potential and average values ±s.e.m. are shown for wild-type (n = 16) and knockout (n = 10) myocytes. B, the difference current was calculated by subtracting the curves in part A to reveal the net current change resulting from the loss of Kir2.1.
Spontaneous activity of wild-type and Kir2.1−/− ventricular myocytes
In addition to individual triggered action potentials, we also examined the spontaneous electrical activity of isolated Kir2.1−/− myocytes by establishing a whole-cell configuration and immediately thereafter recording in current-clamp mode with no current application. Likely damaged cells with resting potentials above −70 mV were excluded from the analysis. Over 75 % of wild-type cells were quiescent (n = 25) and their average resting potential immediately after attaining the whole-cell configuration was −72 mV. In contrast, only 9 % of Kir2.1−/− myocytes (n = 22) were quiescent. Instead, most of the knockout myocytes showed spontaneous rhythmic action potentials. Because of this activity, it was not possible to measure a true resting potential and the minimum potential for the interval between spikes was measured instead (−73 mV, n = 22). A cumulative probability histogram (Fig. 8A) reveals the complete distribution of beat frequencies in both populations and thereby illustrates the increased probability and frequency of spontaneous activity in the Kir2.1−/− cells (P < 0.01 using the Kolmogorov-Smirnov two sample test). For example, 95 % of the wild-type cells were either quiescent or showed less than 0.75 beats min−1. In contrast only 60 % of the Kir2.1−/− myocytes were in this category. Overall, the mean frequency of action potentials in the active knockout cells was 0.8 Hz. The six wild-type cells that evinced spontaneous activity had a mean beat frequency of 0.5 Hz. Lest the increased activity were due to the formation of the whole-cell recording, the non-physiological temperature of 23 °C, or a bias in cell selection, the spontaneous activity was also assessed by visual examination of myocytes at 37 °C. For these experiments individual myocytes were observed for 3 s and were scored as beating or quiescent. The observer was blind to the phenotype of the animal from which the cells were isolated and a systematic and uniform protocol was applied to each dish of dissociated cells (see Methods). In order to insure uniformity of analysis from dish to dish, all of the individual myocytes in a high power field were observed. Approximately six to seven fields were observed in sequence, which resulted in the scoring of roughly 60 cells per dish. Again, the Kir2.1−/− myocytes showed a significantly (P < 0.001) greater level of spontaneous activity with 73 ± 2 % (n = 12 dishes) of the mutant myocytes judged to be beating versus 48 ± 2 % (n = 12 dishes) of the control myocytes (Fig. 8B).
Figure 8. Spontaneous activity of wild-type and Kir2.1−/− myocytes.
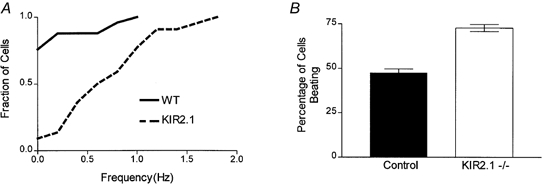
A, cumulative probability histogram of the beat frequency of spontaneously active wild-type (n = 20) and Kir2.1−/− (n = 19) ventricular myocytes. The frequency of the beats was calculated by determining the number of spontaneously generated action potentials seen in a 30 s period of current clamp recordings (I = 0). B, individual myocytes at 37 °C were observed for 3 s and were scored as beating or quiescent. Approximately 60 cells were examined per dish from wild-type (n = 12) and Kir2.1−/− (n = 12) animals. The observer was blind to the phenotype of the mouse from which the cells were isolated.
ECG recordings of wild-type and Kir2.1−/− animals
Finally, the analysis was extended to the in vivo properties of the heart by performing surface ECGs on neonatal Kir2.1−/− animals and their control littermates (which included both wild-type mice and animals heterozygous for the deleted gene). We hypothesized that the prolonged action potential duration and increased spontaneous activity seen in the Kir2.1−/− myocytes would be manifested as changes in the ECG waveforms recorded from Kir2.1−/− mice. In particular, we wished to determine if the increased automaticity of the ventricular myocytes that we had observed in culture would be reflected in arrhythmias in which ventricular activity disrupted the normal pacing by the SA node. In addition, it was possible that the broadening of individual action potentials seen in culture might correspond to a delayed repolarization of the myocardium of the intact heart. ECGs, however, revealed a sinus-paced rhythm in all mice lacking Kir2.1 as well as their control littermates (Fig. 9A and B). In roughly 500 min of recordings from each group we did not observe any examples of ectopic beats that might arise from spontaneous activity in the ventricles; each QRS complex was preceded by a P-wave. Analysis of the ECGs revealed a marked bradycardia in the Kir2.1−/− animals; the interval from one beat to the next was 58 % longer (P < 0.001) in the homozygous mutants than in their control littermates (Table 1). In addition the PR, QRS and QT intervals were significantly longer in the Kir2.1−/− animals (Table 1). Because the loss of Kir2.2 also caused a partial reduction in IK1, we performed a similar analysis of Kir2.2−/− neonates. In these animals, a regular sinus rhythm was again observed but the bradycardia was not present. Thus, despite the ablation of IK1 in the Kir2.1−/− myocytes and the dramatic influence of this mutant on individual action potentials and spontaneous activity of dissociated myocytes, the consequences for the intact neonatal heart were surprisingly and paradoxically mild.
Figure 9. Examples of control and Kir2.1−/− ECGs.
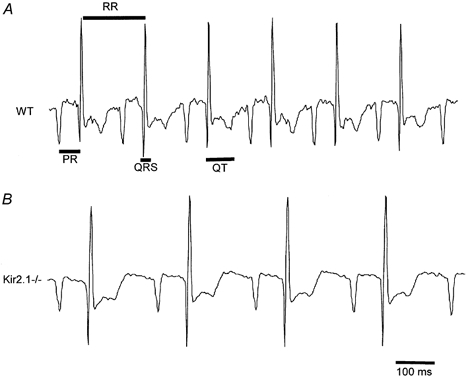
Representative ECGs from a wild-type (A) and a Kir2.1−/− (B) pup. The mutants are consistently bradycardic but have a normal sinus rhythm. Time intervals, analysed quantitatively in Table 1 and described in Methods, are indicated on the wild-type trace.
Table 1.
Mean ECG intervals (in ms) of control and mutant strains
| Control (n = 33) | Kir2.1−/− (n = 19) | Kir2.2−/− (n = 34) | |
|---|---|---|---|
| RR | 156 ± 15 | 248 ± 31 | 144 ± 15 |
| PR | 44 ± 5 | 65 ± 14 | 43 ± 5 |
| QRS | 15 ± 1 | 18 ± 2 | 13 ± 1 |
| QT | 50 ± 6 | 74 ± 9 | 44 ± 7 |
Data are means ± S.D.
DISCUSSION
We have attempted to identify the molecular basis for murine ventricular IK1-conducting channels by studying lines of mice in which the genes encoding Kir2.1 or Kir2.2 have been removed by gene targeting techniques (Zaritsky et al. 2000). To date, the identification of the molecular composition of the cardiac IK1 has been complicated by the multitude of genes for inwardly rectifying K+ channels that have similar biophysical and pharmacological properties. Moreover, differences between in vivo properties and those of expressed, cloned channels may be attributable to missing subunits, differential glycosylation, or other influences of the cellular milieu. With the genetic approach, however, we have revealed a requirement for an individual gene.
The Kir2.1 gene is essential for the expression of IK1 in mouse neonatal ventricular myocytes. In physiological extracellular K+, myocytes from the Kir2.1 knockout show no detectable inwardly rectifying current, and in 60 mm K+ the remaining current is seven times smaller than in wild-type. This remainder cannot be attributed to Kir2.2, because a current of the same magnitude is seen in the double knockout. One possibility is that Kir2.3, which appears to be expressed at low levels in the mouse neonatal heart (Fig. 1), or the newly described Kir2.4 (Topert et al. 1998), can give rise to this minor component. Alternatively, this may represent an ATP- or G protein-activated current that is distinct from IK1 and may not be activated by our recording conditions. The slower kinetics of this residual current suggest that it may be quite distinct from IK1 and may perhaps be encoded by an erg family member.
The finding that IK1 depends critically on Kir2.1 is consistent with a report of Kir2.1 antisense oligonucleotides partially reducing IK1 in cultured adult rat ventricular myocytes (Nakamura et al. 1998). This reduction was ascribed to the suppression of a 21 pS channel. Though the incomplete suppression of the current in the previous study could arise from the different species that was used, the antisense approach probably did not completely prevent synthesis of the Kir2.1 protein. It is also possible that other inward rectifiers contribute more prominently to the production of IK1 after the neonatal period. Several studies (Huynh et al. 1992; Wahler, 1992; Masuda & Sperelakis, 1993) have noted a postnatal increase in IK1, but it remains unclear as to whether this increase is due to the appearance of a distinct species of channel, or an increase in the number or open probability of the same type of channel.
Surprisingly, though Kir2.1 appeared to be sufficient to account for all of IK1 in neonatal ventricular myocytes with the exception of the kinetically distinct residual current, recordings from the Kir2.2 knockout myocytes show a 50 % reduction of the current in both normal and high extracellular K+. This result poses an interesting question: why do the two genes together appear to account for 150 % of the amplitude of the current. A straightforward compensatory change in the regulation of the channels cannot provide a simple explanation: in such a case, the loss of one gene would be expected to increase the expression of the other and so the combined phenotypes would be observed to account for less than 100 % of IK1. The observed phenotype would suggest instead that the loss of one gene led to a counterproductive loss of expression of the second gene and such an effect cannot be excluded. Though no change in the amount of Kir2.1 transcript was observed in the Kir2.2 knockout hearts, a regulatory change at the level of protein expression or channel function remains possible. Moreover, this regulation could arise indirectly outside of the heart as a consequence of a Kir2.2 phenotype that influenced hormonal levels or neuronal inputs.
An additional explanation that is commonly evoked in genetic observations of this sort is an interaction of the protein products of the two genes. In the present case, the data could be accounted for if Kir2.1 and Kir2.2 subunits normally co-assemble to form heteromultimeric channels. Kir2.1 would be essential either for the function of the channel or its translocation to and maintenance on the surface membrane. Kir2.2 would be less essential and thus, in the absence of Kir2.2, the remaining Kir2.1 subunits would form channels that could give rise to a current 50 % smaller than wild-type. The reduction could arise either from fewer channels being expressed on the membrane or from a reduction in the current carried by each channel.
Is co-assembly the correct explanation of the phenotype? If so, then it is noteworthy that Kir2.1, Kir2.2 and Kir2.3 are co-expressed in several regions of the brain (Horio et al. 1996; Karschin et al. 1996), and thus potentially there are heteromultimers of these channels in the brain as well. Yet, though some portions of these channels may be able to interact (Fink et al. 1996), the majority of evidence from heterologous expression systems has indicated that subunit co-assembly among Kir2 family members does not occur. Tinker et al. (1996) showed that tagged Kir2.1 and Kir2.2 proteins expressed in HEK293 cells did not significantly co-precipitate. Also, though a dominant negative Kir2.1 construct prevented the expression of a functional Kir2.1 current in oocytes, it did not affect Kir2.2 or Kir2.3 currents. Thus, if Kir2.2 co-assembly with Kir2.1 channels is indeed the correct explanation of the phenotype, it would require subunit interactions to occur that do not appear in these heterologous systems. Therefore, though a heteromultimeric Kir2.1-Kir2.2 channel could explain the phenotypes observed in these studies, this hypothesis must be tested by biochemical studies of the native channels in heart.
Because the IK1 current depends so heavily on expression of Kir2.1, the Kir2.1 knockout mouse has provided a model in which to study the effects of removing IK1 on cardiac electrophysiology. Though it is not possible completely to rule out secondary changes in other channels, no other inward rectifier channel appeared to have been up-regulated to replace Kir2.1 and measurements of steady state K+ currents as well as Ca2+ currents indicated that the densities of these currents also were unchanged. Though the magnitude of the transient outward current (ITO) was not systematically studied, we observed that it varied considerably from cell to cell, perhaps because of the heterogeneity of the ventricle. The cellular phenotypes, however, were observed very consistently, regardless of the magnitude of ITO, and thus secondary changes in ITO are also unlikely to have confounded the analysis. Previous studies of the role of IK1 necessarily depended on block by Ba2+, which is both voltage and time dependent, and Ba2+ is likely to block other currents as well (Ibarra et al. 1991; Carmeliet, 1993; Deal et al. 1996; Ishihara, 1997; Ishihara & Ehara, 1998). Nonetheless, both pharmacological studies and modelling have indicated an important role for IK1 in ventricular myocytes, particularly in the final repolarization of the action potential. In the rabbit heart, the predominance of IK1 in ventricular cells is thought to be a major determinant of the physiological differences of atrial and ventricular cells (Giles & Imaizumi, 1988). The Kir2.1 knockout animals permitted an independent examination of the impact of this current. The significance of the current was determined for the single myocyte, its action potential and intrinsic automaticity, and also for the cardiac physiology of the intact organism. At each level of analysis, the disruption of Kir2.1 had a major impact.
The duration of the action potential of Kir2.1 knockout myocytes was substantially longer than in wild-type cells. Previous work had suggested that IK1 helped to repolarize the action potential (Ibarra et al. 1991; Varro & Papp, 1992; Carmeliet, 1993; Deal et al. 1996) and in particular, action potentials elicited in the presence of Ba2+ had significantly slowed phase 3 repolarization, as would be anticipated for a strongly rectifying channel. However, in the Kir2.1 knockout myocytes, the falling phase was slower throughout. As can be seen from the analysis of dV/dt during the repolarization (Fig. 7), even during the positive overshoot of the action potential, considerably less net outward current was occurring in the Kir2.1 knockout animal. This result is surprising: if the inward rectification of IK1 is as steep and rapid as is thought (Hille, 1992), very little if any outward current would be expected to flow through IK1 channels at voltages above −10 mV. One possibility is that the disruption of IK1 causes a change in the initial phase of the upstroke of the action potential and, as a consequence, there is a change in the degree of activation or inactivation of another outward current, perhaps ITO. If the net effect were a decrease in this outward current during repolarization, such a change could explain the slowing of repolarization. Alternatively, if the rectification of IK1 in neonatal murine myocytes is slower or less complete than has been thought, IK1 itself could directly account for the brevity of the action potential in these cells. At present, the broadening of the early phases of repolarization in the Kir2.1−/− animals remains an enigmatic but significant finding for the role of IK1 and a challenge for further modelling of the ventricular action potential.
Another prominent difference between the behaviour of the knockout cells and their wild-type counterparts was the degree of spontaneous activity. During current clamp recordings, nearly all of the wild-type cells were quiescent while over 70 % of the knockout cells showed activity in the form of spontaneously generated action potentials. This difference in activity was also seen when the same populations were observed under more physiological conditions: by looking optically for contractions at 37 °C in tissue culture media. The tendency of IK1 to resist automaticity in ventricular myocytes has been predicted by models of myocyte activity and observed in pharmacological experiments (Noble, 1986; Hirano & Hiraoka, 1988; Valenzuela & Vassalle, 1991; Nichols et al. 1996). Without IK1, the resting potential of the ventricular myocytes is less firmly clamped at EK and, once the voltage-activated K+ channels have closed, a depolarizing current such as IH is left relatively unopposed in returning the cells to threshold. Thus, our observations of increased automaticity are in excellent agreement with the expected role of IK1.
The significance of the inward rectifier for the establishment of the resting potential itself was more difficult to ascertain. The whole-cell patch-clamp technique is not ideal for resting potential measurements because of the exchange of the electrode solution with the normal cytosol of the myocyte. A more substantial complication was presented by the increased beating frequency seen in the knockout cells, which required that we examine the minimum diastolic potentials rather than a true resting potential. The minimum diastolic potentials of the Kir2.1−/− myocytes were generally below −70 mV, which was very similar to the resting potentials observed in quiescent wild-type myocytes. It appears likely, therefore, that the conductance of the voltage-activated K+ channels is sufficiently high immediately after the action potential that the membrane potential is brought to EK despite the disruption of IK1. As the voltage-dependent K+ channels deactivate, however, the absence of the normal IK1 presumably allowed the mutant cells to be brought rapidly to threshold.
The substantial changes seen at the single cell level in the knockout mice raised the possibility that arrhythmias might be observed in the intact heart. If the increased automaticity of the ventricular cells, for example, were sufficient to usurp the normal pacemaking role of the SA node, ectopic beats would arise or, indeed, the entire heart might be paced from the ventricles. Alternatively, the slowing of the action potential could induce a separate set of arrhythmias. However, in recordings from the knockout animals, a sinus-paced rhythm was consistently observed and neither ectopic beats nor re-entry arrhythmias were detected. Thus the increased automaticity was not so great as to interfere with normal atrial pacing of the murine heart under these conditions. It may well be, however, that other conditions that would predispose the heart to ectopic activity, such as damage to the myocardium or conduction pathways, might evoke considerably greater problems in animals lacking IK1. Indeed, though the present study was confined to neonatal animals, older animals with larger hearts and slower atrial pacing might have had a more apparent phenotype in the ECG. Nonetheless, the failure of the loss of IK1 to cause a drastic change in ventricular physiology runs counter to the view that this current has a critical role in preventing automaticity in the ventricles and counter to our observations of increased automaticity in isolated myocytes. One possible explanation may be that in the rapidly paced hearts of neonatal mice, voltage-dependent K+ channels are repeatedly being activated and may provide a sufficient level of hyperpolarization to dampen ventricular automaticity, even in the absence of Kir2.1.
The QT interval in mice lacking Kir2.1 was consistently lengthened relative to control. While it is tempting to attribute this effect to the increased action potential duration seen in the myocytes of those animals, this conclusion is not as yet warranted; the mutant animals exhibited a consistent and pronounced bradycardia and a lengthening of the QT interval is a well-established consequence of bradycardia in general (London et al. 1998; Walker & Pugsley, 1998). In preliminary experiments in which the heart rate of wild-type animals was slowed to the knockout rate with propranolol, a β-blocker, the QT intervals between the two groups were not significantly different (data not shown). Thus, the broadening of individual triggered action potentials may not be paralleled by comparable broadening in a rapidly paced myocyte in vivo, or broadening may not translate into a gross change in the repolarization of the ventricle.
The bradycardia of mice lacking Kir2.1 is in and of itself an intriguing finding: clearly the increased beat frequency of the ventricular myocytes was not matched by a similar increase in SA nodal cells and this is consistent with observations that neither IK1 nor Kir2.1 transcript is characteristic of nodal cells (Noma et al. 1984; Dixon & McKinnon, 1994; Brahmajothi et al. 1996). We suspect that the bradycardia does not originate in the heart. Instead, the bradycardia could be the result of altered vascular tone (Zaritsky et al. 2000), sensory responses to cardiovascular parameters, function of CNS nuclei that regulate cardiac reflexes, or function of the autonomic nervous system itself. Kir2.1 is expressed in the central nervous system (Horio et al. 1996; Karschin et al. 1996) and has been seen in autonomic ganglia as well (Topert et al. 1998). The influence of Kir2.1 on the regulation of heart rate will be particularly important to take into account if clinical attempts are made to target Kir2.1 in cardiac tissue.
In summary, the Kir2.1 knockout mouse has provided clear evidence that the Kir2.1 gene is essential for IK1 in neonatal ventricular myocytes. The knockout myocytes evince significant alterations in their firing rate and in the waveform of the action potential. In the intact organism, the cardiovascular phenotype includes a prominent bradycardia. These mice, as well as the Kir2.2 knockouts, should provide continuing opportunities to reinvestigate the significance of IK1 in the heart and in the physiology and development of other organ systems as well.
Acknowledgments
The authors are grateful to Drs Brian Kobilka, Daniel Bernstein and Kavin Desai for instruction and assistance with mouse electrocardiograms, to Drs Richard Tsien, Richard Aldrich, Iain Robinson and Bryan Stewart for many helpful discussions, and to Irene Inman and Brendan McCullough for technical support. NIH grant GM42376 supported this work and J.J.Z. was supported by an MSTP training grant.
References
- Barry DM, Trimmer JS, Merlie JP, Nerbonne JM. Differential expression of voltage-gated K+ channel subunits in adult rat heart. Relation to functional K+ channels. Circulation Research. 1995;77:361–369. doi: 10.1161/01.res.77.2.361. [DOI] [PubMed] [Google Scholar]
- Barry DM, Xu H, Schuessler RB, Nerbonne JM. Functional knockout of the transient outward current, long-QT syndrome, and cardiac remodeling in mice expressing a dominant-negative Kv4 α subunit. Circulation Research. 1998;83:560–567. doi: 10.1161/01.res.83.5.560. [DOI] [PubMed] [Google Scholar]
- Brahmajothi MV, Morales MJ, Liu S, Rasmusson RL, Campbell DL, Strauss HC. In situ hybridization reveals extensive diversity of K+ channel mRNA in isolated ferret cardiac myocytes. Circulation Research. 1996;78:1083–1089. doi: 10.1161/01.res.78.6.1083. [DOI] [PubMed] [Google Scholar]
- Carmeliet E. K+ channels and control of ventricular repolarization in the heart. Fundamentals of Clinical Pharmacology. 1993;7:19–28. doi: 10.1111/j.1472-8206.1993.tb00214.x. [DOI] [PubMed] [Google Scholar]
- Davies MP, An RH, Doevendans P, Kubalak S, Chien KR, Kass RS. Developmental changes in ionic channel activity in the embryonic murine heart. Circulation Research. 1996;78:15–25. doi: 10.1161/01.res.78.1.15. [DOI] [PubMed] [Google Scholar]
- Deal KK, England SK, Tamkun MM. Molecular physiology of cardiac potassium channels. Physiological Reviews. 1996;76:49–67. doi: 10.1152/physrev.1996.76.1.49. [DOI] [PubMed] [Google Scholar]
- Dixon JE, Mckinnon D. Quantitative analysis of potassium channel mRNA expression in atrial and ventricular muscle of rats. Circulation Research. 1994;75:252–260. doi: 10.1161/01.res.75.2.252. [DOI] [PubMed] [Google Scholar]
- Dixon JE, Shi W, Wang HS, Mcdonald C, Yu H, Wymore RS, Cohen IS, Mckinnon D. Role of the Kv4. 3 K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circulation Research. 1996;79:659–668. doi: 10.1161/01.res.79.4.659. [DOI] [PubMed] [Google Scholar]
- Elam TR, Lansman JB. The role of Mg2+ in the inactivation of inwardly rectifying K+ channels in aortic endothelial cells. Journal of General Physiology. 1995;105:463–484. doi: 10.1085/jgp.105.4.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakler B, Brandle U, Glowatzki E, Weidemann S, Zenner HP, Ruppersberg JP. Strong voltage-dependent inward rectification of inward rectifier K+ channels is caused by intracellular spermine. Cell. 1995;80:149–154. doi: 10.1016/0092-8674(95)90459-x. [DOI] [PubMed] [Google Scholar]
- Ficker E, Taglialatela M, Wible BA, Henley CM, Brown AM. Spermine and spermidine as gating molecules for inward rectifier K+ channels. Science. 1994;266:1068–1072. doi: 10.1126/science.7973666. [DOI] [PubMed] [Google Scholar]
- Fink M, Duprat F, Heurteaux C, Lesage F, Romey G, Barhanin J, Lazdunski M. Dominant negative chimeras provide evidence for homo and heteromultimeric assembly of inward rectifier K+ channel proteins via their N-terminal end. FEBS Letters. 1996;378:64–68. doi: 10.1016/0014-5793(95)01388-1. [DOI] [PubMed] [Google Scholar]
- Giles WR, Imaizumi Y. Comparison of potassium currents in rabbit atrial and ventricular cells. Journal of Physiology. 1988;405:123–145. doi: 10.1113/jphysiol.1988.sp017325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. 2. Sunderland, MA, USA: Sinauer Associates; 1992. [Google Scholar]
- Hirano Y, Hiraoka M. Barium-induced automatic activity in isolated ventricular myocytes from guinea-pig hearts. Journal of Physiology. 1988;395:455–472. doi: 10.1113/jphysiol.1988.sp016929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horio Y, Morishige K, Takahashi N, Kurachi Y. Differential distribution of classical inwardly rectifying potassium channel mRNAs in the brain: comparison of IRK2 with IRK1 and IRK3. FEBS Letters. 1996;379:239–243. doi: 10.1016/0014-5793(95)01519-1. [DOI] [PubMed] [Google Scholar]
- Huynh TV, Chen F, Wetzel GT, Friedman WF, Klitzner TS. Developmental changes in membrane Ca2+ and K+ currents in fetal, neonatal, and adult rabbit ventricular myocytes. Circulation Research. 1992;70:508–515. doi: 10.1161/01.res.70.3.508. [DOI] [PubMed] [Google Scholar]
- Ibarra J, Morley GE, Delmar M. Dynamics of the inward rectifier K+ current during the action potential of guinea pig ventricular myocytes. Biophysical Journal. 1991;60:1534–1539. doi: 10.1016/S0006-3495(91)82187-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara K. Time-dependent outward currents through the inward rectifier potassium channel IRK1. The role of weak blocking molecules. Journal of General Physiology. 1997;109:229–243. doi: 10.1085/jgp.109.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara K, Ehara T. A repolarization-induced transient increase in the outward current of the inward rectifier K+ channel in guinea-pig cardiac myocytes. Journal of Physiology. 1998;510:755–771. doi: 10.1111/j.1469-7793.1998.755bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomoto S, Kondo C, Kurachi Y. Inwardly rectifying potassium channels: their molecular heterogeneity and function. Japanese Journal of Physiology. 1997;47:11–39. doi: 10.2170/jjphysiol.47.11. [DOI] [PubMed] [Google Scholar]
- Karschin C, Dissmann E, Stuhmer W, Karschin A. IRK(1–3) and GIRK(1–4) inwardly rectifying K+ channel mRNAs are differentially expressed in the adult rat brain. Journal of Neuroscience. 1996;16:3559–3570. doi: 10.1523/JNEUROSCI.16-11-03559.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G, Gordon EA, Wickman K, Velimirovic B, Krapivinsky L, Clapham DE. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature. 1995;374:135–141. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Baldwin TJ, Jan YN, Jan LY. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 1993;362:127–133. doi: 10.1038/362127a0. [DOI] [PubMed] [Google Scholar]
- Kurachi Y. Voltage-dependent activation of the inward-rectifier potassium channel in the ventricular cell membrane of guinea-pig heart. Journal of Physiology. 1985;366:365–385. doi: 10.1113/jphysiol.1985.sp015803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London B, Jeron A, Zhou J, Buckett P, Han X, Mitchell GF, Koren G. Long QT and ventricular arrhythmias in transgenic mice expressing the N terminus and first transmembrane segment of a voltage-gated potassium channel. Proceedings of the National Academy of Sciences of the USA. 1998;95:2926–2931. doi: 10.1073/pnas.95.6.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopatin AN, Makhina EN, Nichols CG. The mechanism of inward rectification of potassium channels: “long-pore plugging” by cytoplasmic polyamines. Journal of General Physiology. 1995;106:923–955. doi: 10.1085/jgp.106.5.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda H. Open-state substructure of inwardly rectifying potassium channels revealed by magnesium block in guinea-pig heart cells. Journal of Physiology. 1988;397:237–258. doi: 10.1113/jphysiol.1988.sp016998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda H. Effects of external and internal K+ ions on magnesium block of inwardly rectifying K+ channels in guinea-pig heart cells. Journal of Physiology. 1991;435:83–99. doi: 10.1113/jphysiol.1991.sp018499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda H, Saigusa A, Irisawa H. Ohmic conductance through the inwardly rectifying K channel and blocking by internal Mg2+ Nature. 1987;325:156–159. doi: 10.1038/325156a0. [DOI] [PubMed] [Google Scholar]
- Masuda H, Sperelakis N. Inwardly rectifying potassium current in rat fetal and neonatal ventricular cardiomyocytes. American Journal of Physiology. 1993;265:H1107–H1111. doi: 10.1152/ajpheart.1993.265.4.H1107. [DOI] [PubMed] [Google Scholar]
- Morishige K, Takahashi N, Jahangir A, Yamada M, Koyama H, Zanelli JS, Kurachi Y. Molecular cloning and functional expression of a novel brain-specific inward rectifier potassium channel. FEBS Letters. 1994;346:251–256. doi: 10.1016/0014-5793(94)00483-8. [DOI] [PubMed] [Google Scholar]
- Nakamura TY, Artman M, Rudy B, Coetzee WA. Inhibition of rat ventricular IK1 with antisense oligonucleotides targeted to Kir2. 1 mRNA. American Journal of Physiology. 1998;274:H892–H900. doi: 10.1152/ajpheart.1998.274.3.H892. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Makhina EN, Pearson WL, Sha Q, Lopatin AN. Inward rectification and implications for cardiac excitability. Circulation Research. 1996;78:1–7. doi: 10.1161/01.res.78.1.1. [DOI] [PubMed] [Google Scholar]
- Noble D. Ionic mechanisms controlling the action potential duration and the timing of repolarization. Japanese Heart Journal. 1986;27(1):3–19. [PubMed] [Google Scholar]
- Noma A, Nakayama T, Kurachi Y, Irisawa H. Resting K conductances in pacemaker and nonpacemaker heart cells of the rabbit. Japanese Journal of Physiology. 1984;34:245–254. doi: 10.2170/jjphysiol.34.245. [DOI] [PubMed] [Google Scholar]
- Nuss HB, Marban E. Electrophysiological properties of neonatal mouse cardiac myocytes in primary culture. Journal of Physiology. 1994;479:265–279. doi: 10.1113/jphysiol.1994.sp020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakmann B, Trube G. Conductance properties of single inwardly rectifying potassium channels in ventricular cells from guinea-pig heart. Journal of Physiology. 1984;347:641–657. doi: 10.1113/jphysiol.1984.sp015088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng SL, Sha Q, Ferrigni T, Lopatin AN, Nichols CG. Depletion of intracellular polyamines relieves inward rectification of potassium channels. Proceedings of the National Academy of Sciences of the USA. 1996;93:12014–12019. doi: 10.1073/pnas.93.21.12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver MR, Decoursey TE. Intrinsic gating of inward rectifier in bovine pulmonary artery endothelial cells in the presence or absence of internal Mg2+ Journal of General Physiology. 1990;96:109–133. doi: 10.1085/jgp.96.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Morishige K, Jahangir A, Yamada M, Findlay I, Koyama H, Kurachi Y. Molecular cloning and functional expression of cDNA encoding a second class of inward rectifier potassium channels in the mouse brain. Journal of Biological Chemistry. 1994;269:23274–23279. [PubMed] [Google Scholar]
- Tinker A, Jan YN, Jan LY. Regions responsible for the assembly of inwardly rectifying potassium channels. Cell. 1996;87:857–868. doi: 10.1016/s0092-8674(00)81993-5. [DOI] [PubMed] [Google Scholar]
- Topert C, Doring F, Wischmeyer E, Karschin C, Brockhaus J, Ballanyi K, Derst C, Karschin A. Kir2.4: a novel K+ inward rectifier channel associated with motoneurons of cranial nerve nuclei. Journal of Neuroscience. 1998;18:4096–4105. doi: 10.1523/JNEUROSCI.18-11-04096.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela F, Vassalle M. Role of membrane potential in Ba2+ induced automaticity in guinea pig cardiac myocytes. Cardiovascular Research. 1991;25:421–430. doi: 10.1093/cvr/25.5.421. [DOI] [PubMed] [Google Scholar]
- Vandenberg CA. Inward rectification of a potassium channel in cardiac ventricular cells depends on internal magnesium ions. Proceedings of the National Academy of Sciences of the USA. 1987;84:2560–2564. doi: 10.1073/pnas.84.8.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varro A, Papp JG. The impact of single cell voltage clamp on the understanding of the cardiac ventricular action potential. Cardioscience. 1992;3:131–144. [PubMed] [Google Scholar]
- Wahler GM. Developmental increases in the inwardly rectifying potassium current of rat ventricular myocytes. American Journal of Physiology. 1992;262:C1266–C1272. doi: 10.1152/ajpcell.1992.262.5.C1266. [DOI] [PubMed] [Google Scholar]
- Walker MJA, Pugsley MK. Methods in Cardiac Electro-Physiology. Boca Raton, FL, USA: CRC Press; 1998. [Google Scholar]
- Xu H, Dixon JE, Barry DM, Trimmer JS, Merlie JP, Mckinnon D, Nerbonne JM. Developmental analysis reveals mismatches in the expression of K+ channel α subunits and voltage-gated K+ channel currents in rat ventricular myocytes. Journal of General Physiology. 1996;108:405–419. doi: 10.1085/jgp.108.5.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Jan YN, Jan LY. Determination of the subunit stoichiometry of an inwardly rectifying potassium channel. Neuron. 1995;15:1441–1447. doi: 10.1016/0896-6273(95)90021-7. [DOI] [PubMed] [Google Scholar]
- Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K+ current in K+-mediated vasodilation. Circulation Research. 2000;87:160–166. doi: 10.1161/01.res.87.2.160. [DOI] [PubMed] [Google Scholar]