

Abstract
The mechanisms of long-term potentiation (LTP) and long-term depression (LTD) induced by brief high frequency stimulation (HFS), paired with a particular pattern and amplitude of depolarisation has been investigated in the medial perforant pathway of the dentate gyrus of the 2- to 3-week-old rat hippocampus in vitro.
N-Methyl-D-aspartate (NMDA) receptor (NMDAR) activation was measured quantitatively during HFS-induced NMDAR-dependent LTP, LTD and at the LTD-LTP crossover point in order to test the hypothesis that the induction of the particular form of plasticity depends on the intensity of NMDAR activation.
The induction of LTD, the LTD-LTP crossover point and LTP was associated with an increasing NMDAR charge transfer.
In addition to the NMDAR-dependent LTD, a group I metabotropic glutamate receptor (mGluR)-dependent LTD could be induced by high intensity HFS paired with depolarisation under conditions of NMDAR inhibition.
The induction of mGluR-dependent LTD requires membrane depolarisation, Ca2+ influx via L-type Ca2+ channels and a rise in intracellular Ca2+.
Quantal analysis involving minimal stimulation demonstrated that the mGluR-dependent LTD induction was associated with a decrease in potency and an increase in failure rate.
LTP and LTD are long-lasting forms of synaptic plasticity involving an increase or decrease, respectively, in synaptic transmission. Both LTP and LTD have forms dependent upon the activation of NMDARs (Collingridge et al. 1983; Dudek & Bear, 1992a,b; Mulkey & Malenka, 1992). In addition, although initially controversial (Selig et al. 1995), there is now an abundance of evidence that certain forms of LTD induction are dependent upon the activation of mGluRs independently of NMDAR activation (Bolshakov & Siegelbaum, 1994, 1997; O'Mara et al. 1995; Oliet et al. 1997; Wang et al. 1997).
LTD is most commonly induced by prolonged (several minutes) low frequency stimulation (LFS) at 1–5 Hz, and LTP by brief high frequency stimulation (HFS) at 50–200 Hz (Bliss & Lomo, 1973; Bliss & Collingridge, 1993). The induction of LTD and LTP by such differing frequencies has been quantified formally in a frequency-response function (Bear, 1995), describing the smooth transition from LTD to LTP which occurs as the frequency of stimulation is increased from 1 to 200 Hz, with an LTD-LTP crossover point at 10–20 Hz. It has been hypothesised that the transition from LTD to LTP with increasing frequency of stimulation is associated with a higher level of activation of NMDARs and of associated Ca2+ influx (Bear & Malenka, 1994; Bear, 1995). Qualitative support for this theory was provided by the study of Cummings et al. (1996), in which LTP induced by brief HFS under control conditions was converted to LTD induction in the presence of partial NMDAR blockade by d-2-amino-phosphonopentanoate (AP5). However, quantitative measurements of NMDAR activation during the induction of plasticity have not so far been carried out to verify this theory. NMDAR activation is particularly difficult to measure during the induction of LTD by LFS because the NMDAR component of single excitatory postsynaptic currents (EPSCs) evoked at 1 Hz is very small. In the present study, we have devised a stimulation protocol for the induction of NMDAR-dependent LTD, LTP and the LTD-LTP crossover point in which a brief constant amplitude HFS is paired with postsynaptic depolarisation, the particular induction of LTP, LTD or the LTD-LTP crossover point being determined by changes in the pattern or amplitude of postsynaptic depolarisation. The presynaptic stimulation strength during the HFS, and therefore the number of synapses stimulated, was kept at a constant level. The summation of NMDAR-mediated currents which occurred during the HFS enabled quantitative measurements of NMDAR activation to be made and correlated with the amplitude and direction of synaptic plasticity. In addition, we have characterised an NMDAR-independent LTD which was evoked by HFS under conditions of blockade of NMDAR.
METHODS
All experiments were carried out on transverse slices of rat hippocampus (Wistar rats, aged 2–3 weeks, weight 40–80 g). Animal use was approved by the Bioresources Committee, Trinity College, Ireland. Rats were killed by decapitation and the brains rapidly removed and placed in cold oxygenated medium (95 % O2-5 % CO2). Slices were cut at a thickness of 350 μm using a Campden vibroslice and placed in a holding container containing oxygenated medium at room temperature (20–22 °C). The slices were then transferred as required into a submerged recording chamber and continuously superfused at a rate of 8 ml min−1 at 32 °C.
The control medium contained (mm): NaCl, 120; KCl, 2.5; NaH2PO4, 1.25; NaHCO3, 26; MgSO4, 2.0; CaCl2, 2.0, and d-glucose, 10. All solutions contained 100 μm picrotoxin (Sigma) to block GABAA receptor-mediated activity. AP5 was obtained from Tocris Cookson. The patch-clamp electrode, resistance 5–8 MΩ, contained (mm): potassium gluconate, 130; KCl, 10; EGTA, 10; CaCl2, 1; MgCl2, 3; Hepes, 20; Mg-ATP, 5; Na-GTP, 0.5; QX 314, 5; and the pH was adjusted to 7.2 (using KOH). The mGluR antagonists (R,S)-α-methyl-4-carboxyphenylglycine (MCPG), (R,S)-1-aminoindan-1,5-dicarboxylic acid (AIDA) and (2S)-α-ethylglutamic acid (EGLU) were obtained from Tocris Cookson.
Whole-cell recordings from dentate granule cells were made using an Axopatch-1D amplifier (3 kHz low pass Bessel filter), as described previously (O'Connor et al. 1995). Series resistance (Rs) varied from 15 to 25 MΩ, as measured directly from the amplifier or as measured directly in several cells from the peak amplitude of the resistive current Ic (without low pass filtering), as Rs = ER/Ic, where ER is the amplitude of the test pulse, usually 10 mV. The mean input resistance was 286 ± 36 MΩ, n = 53, and the mean resting potential −70 ± 4 mV, n = 57. The input resistance was monitored continuously, and the recording terminated if it varied by more than 10 %. Test EPSCs were recorded at a holding potential of −70 mV in response to stimulation of the medial perforant pathway at a control frequency of 0.05 Hz, with the stimulation intensity adjusted to evoke an EPSC which was about 30 % of the maximum amplitude, usually about 100 pA. Full experiments were carried out providing that certain criteria were met. These included a resting membrane potential of at least −65 mV, a high input resistance (at least 200 MΩ) and a low threshold and steep input-output curve for the EPSCs. Recordings were analysed using pCLAMP (Axon Instruments). Values are the means ±s.e.m., and Student's t test was used for statistical comparisons.
The presynaptic level of stimulation during baseline recordings was set to evoke test EPSCs of ˜100 pA. Synaptic plasticity was evoked by HFS stimulation protocols, consisting of the pairing of a series of five trains of HFS (each of eight stimuli at 200 Hz, inter-train interval 200 ms) with step depolarisations from a holding potential of −70 mV. The presynaptic level of stimulation during HFS was increased threefold from the test level, sufficient to evoke EPSCs of ˜300 pA, and was set at the same intensity for the induction of LTP, LTD and the LTD-LTP crossover point. The holding potential was always −70 mV, and the imposed postsynaptic depolarisation was one of three types. Firstly, for the induction of LTP, a single step of 1.1 s to −30 mV was applied during the HFS trains (with no repolarisation after each train). Secondly, for the induction of the LTD-LTP crossover point, the stimulus was a series of five steps to −30 mV, with each step depolarisation of 40 ms duration applied at 5 Hz and the depolarisation phase coinciding with each train of HFS. Thirdly, for the induction of LTD, a single step of 1.1 s to −50 mV was applied during the HFS trains. All values of plasticity were measured at 25 min post-HFS. In order to avoid any long-term changes of NMDAR activity by HFS, the summated NMDAR component was measured in one of two ways. Either HFS was initially given in the presence of AP5 to block long-term changes, the AMPA receptor (AMPAR) component measured, and then subtracted from the total AMPAR and NMDAR component obtained from the second HFS given after washout of AP5, or the NMDAR component was measured in cells from different slices of the same animals, using identical conditions to experiments in which LTP was induced. The two methods gave very similar values of the NMDAR component.
In minimal stimulation experiments, the frequency of test stimulation was set at a higher level of 0.1–0.2 Hz in order to ensure an adequate number of EPSCs for analysis. The initial stimulus intensity was set at a level below which EPSCs were evoked. The intensity was then increased very slowly until the lowest level that evoked EPSCs and failures was detected. Minimal stimulation was only accepted if at least 10 % of trials resulted in failures.
RESULTS
Induction of LTP by a HFS-depolarisation pairing protocol evoking large NMDAR activation
A pairing of HFS and postsynaptic depolarisation was an effective stimulus for the induction of NMDAR-dependent LTP providing that large NMDAR activation was evoked by the pairing procedure. This was achieved by pairing a series of HFS trains with a single strong step depolarisation from −70 to −30 mV and duration 1.1 s (see Methods for details). LTP was induced by this protocol with a mean amplitude of 165 ± 19.1 % (n = 7, P < 0.05; Fig. 1A). The LTP was NMDAR dependent, being blocked by AP5. Thus, the pairing of HFS and depolarisation in the presence of the NMDAR antagonist AP5 (100 μm) did not induce LTP, but rather an NMDAR-independent LTD measuring 37.9 ± 15.6 %, P < 0.05 (n = 6; Fig. 1B). The properties of this LTD are investigated below.
Figure 1. Induction of NMDAR-dependent LTP by a pairing protocol consisting of HFS and a single step postsynaptic depolarisation.
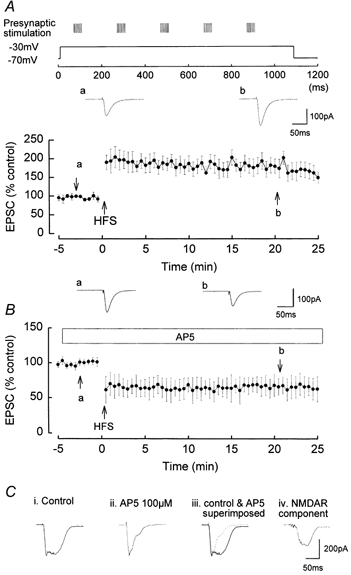
Presynaptic stimulation was set to evoke EPSCs of ˜100 pA amplitude during test stimulation. Five trains of HFS were given, each train consisting of eight stimuli at 200 Hz and the inter-train interval of 200 ms. The presynaptic stimulation was increased to evoke ˜300 pA EPSCs during the HFS. The holding potential of the postsynaptic cell was −70 mV, and a single depolarisation to −30 mV and of 1.1 s duration was applied during the HFS. A, in control media, LTP was induced with a mean amplitude of 165 ± 19.1 %, n = 7. a and b are original traces of EPSCs prior to and following LTP induction. B, in the presence of the NMDAR antagonist AP5 (100 μm), LTP was completely blocked, and an NMDAR-independent LTD was evoked with a mean amplitude of 37.9 ± 15.6 %, n = 6. a and b are original traces of EPSCs prior to and following LTD induction. C, examples of summated EPSCs during a single train of HFS. C i shows the summated EPSCs during the initial HFS in control media, the waveform being composed of both AMPAR- and NMDAR-mediated components. C ii shows the summated AMPAR component of the EPSCs in the presence of AP5. Note the much shorter time course of the summated EPSCs. C iii shows the superimposition of the summated EPSPs in control and in AP5. C iv shows the NMDAR-mediated component of the summated EPSCs of the train, obtained by subtracting the summated EPSCs in AP5 from the control summated EPSCs.
During each HFS in control media, successive EPSCs summated temporally to form a prolonged current waveform, composed of both AMPAR- and NMDAR-mediated components (Fig. 1C i) (the amplitude of individual EPSCs declined rapidly during each train due to strong presynaptic short-term depression at this synapse; McNaughton, 1980). Similar summation of AMPAR- and NMDAR-mediated EPSPs during HFS has been observed previously, for example, in the CA1 region of the hippocampus (Herron et al. 1986) and cerebellar granule cells (D'Angelo et al. 1995). The summated multiple EPSC waveforms in the present studies have an initial amplitude of ˜300 pA and a half-time of decay of 53.9 ± 3.5 ms, n = 5. In the presence of AP5 and the resulting block of NMDARs, the remaining summated AMPAR-mediated EPSCs during each HFS had a much shorter duration than in the absence of AP5 (Fig. 1C ii), with the half-time of decay of the summated AMPAR-mediated EPSC waveform during the HFS being 30.5 ± 2.1 ms (n = 5), a value significantly smaller than in the absence of AP5 (P < 0.05). The much shorter duration of the summated EPSC during HFS in AP5 compared with control is clearly shown in Fig. 1C iii, in which the summated waveform in control and in AP5 are superimposed. Subtraction of the summated EPSC waveform in the presence of AP5 from the control summated EPSC waveform revealed the NMDAR component of the waveform, i.e. the summated NMDAR-mediated EPSC (Fig. 1C iv), which had a peak amplitude of ˜200 pA and a decay half-time of 46.2 ± 6.3 ms (n = 5), a significantly longer duration than the isolated summated AMPAR-mediated EPSC (P < 0.05).
Induction of LTD by a HFS-depolarisation pairing protocol evoking a low level of NMDAR activation
A pairing of HFS and postsynaptic depolarisation was an effective stimulus for the induction of NMDAR-dependent LTD providing that a low level of NMDAR activation was evoked by the pairing procedure. This was achieved by pairing the same series of HFS used to evoke LTP with a single mild step depolarisation of 1.1 s from −70 to −50 mV (see Methods). LTD was induced by this protocol with a mean amplitude of 22.1 ± 9.6 % (n = 5, P < 0.05; Fig. 2A). Such LTD was NMDAR dependent as it was blocked in the presence of AP5, the EPSC measuring 104.1 ± 4.1 %, P > 0.05, at 20 min post-pairing (Fig. 2B).
Figure 2. Induction of NMDAR-dependent LTD by the pairing of HFS and a single step depolarisation of 1.1 s duration and mild amplitude (from −70 to −50 mV).
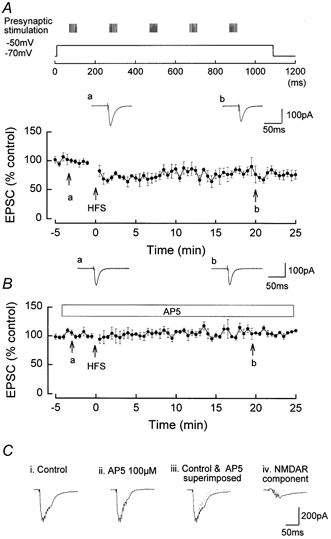
The presynaptic stimulation was increased to evoke ˜300 pA EPSCs during the HFS. A, LTD of mean amplitude 22.1 ± 9.6 %, n = 5, induced by HFS applied on the depolarising phase of a single step depolarisation of 1.1 s duration. a and b are original traces of EPSCs prior to and following LTD induction. B, inhibition of the LTD by AP5 (100 μm), mean amplitude 104.1 ± 4.1 %, n = 6. a and b are original traces of EPSCs prior to and following HFS. C i shows the summated EPSCs during the initial HFS in control media, composed of both AMPAR- and NMDAR-mediated components. C ii shows the summated AMPA receptor component of the EPSCs in the presence of AP5. C iii shows the superimposition of the summated EPSPs in control and in AP5. C iv shows the NMDAR component of the summated EPSCs of the train, obtained by subtracting the summated EPSCs in AP5 from the control summated EPSCs.
The summated multiple EPSC waveform during each HFS (Fig. 2C i) in control medium had a smaller amplitude (−60 pA) and a more rapid half-time of decay (˜35 ms) than in the previous experiments involving LTP induction. In the presence of AP5, the summated multiple AMPAR-mediated EPSC waveform during each HFS had a much shorter duration than in the absence of AP5 (Fig. 2C ii), with a half-time of 27.5 ± 5.1 ms, n = 5, P < 0.05. The much shorter duration of the summated EPSC during HFS in AP5 compared with control is clearly shown in Fig. 2C iii, in which the summated waveform in control and in AP5 are superimposed. Subtraction of the summated EPSC waveform in the presence of AP5 from the control summated EPSC waveform revealed the NMDAR-component of the waveform, i.e. the summated NMDAR-mediated EPSCs (Fig. 2C iv), which had a very small peak amplitude (˜30 pA) and a decay half-time of 39.8 ± 3.6 ms (n = 5). The peak amplitude and duration of this summated NMDAR-mediated EPSC was much smaller than that occurring during LTP induction.
Induction of the LTD-LTP crossover point by a HFS-depolarisation pairing protocol evoking moderate NMDAR activation
A pairing of HFS and postsynaptic depolarisation was an effective stimulus for the induction of the NMDAR-dependent LTD-LTP crossover point providing that moderate NMDAR activation was evoked by the pairing procedure. This was achieved by pairing an identical series of HFS trains used to induce LTP and LTD with a series of short step depolarisations (see Methods), designed to abbreviate activation of the NMDARs. Five strong intensity HFS trains were paired with the depolarising phase of five short (40 ms) depolarising steps each from −70 to −30 mV. This protocol resulted in an unchanged test EPSC, with no significant induction of LTP or LTD, the post-stimulus EPSC measuring 107.1 ± 7.8 % (n = 11, P > 0.05; Fig. 3A). However, although there was a lack of induction of synaptic plasticity, NMDARs were moderately activated during this stimulation protocol, as shown by isolation of the NMDAR current during the HFS. The summated multiple EPSC waveform, composed of both AMPAR and NMDAR components, had an initial large amplitude (˜300 pA) but a relatively short duration (˜40 ms) as the imposed short step depolarisations resulted in an abrupt termination of the NMDAR component of the waveform (Fig. 3B i). In the presence of AP5, the summated multiple AMPAR-mediated EPSCs during each HFS had a much reduced duration (Fig. 3B ii and iii). Subtraction of the summated EPSC waveform in the presence of AP5 from the control summated EPSC waveform revealed the NMDAR-component of the waveform, i.e. the summated NMDAR-mediated EPSCs (Fig. 3B iv), which had a maximum amplitude of ˜200 pA and a decay half-time of 40.3 ± 9.5 ms, n = 5.
Figure 3. Induction of the LTD-LTP crossover point by the pairing of HFS and a series of depolarising trains.
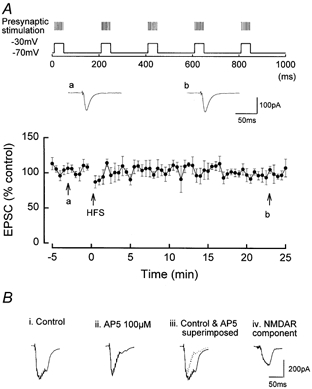
A, induction of the LTD-LTP crossover point by a protocol in which the presynaptic stimulation was increased to evoke ˜300 pA EPSCs during the trains of HFS which were applied on the depolarising phase of a series of 40 ms duration depolarising steps from −70 to −30 mV. Mean amplitude of the EPSC at 25 min HFS was not significantly altered from the test level (107.1 ± 7.8 %, n = 11). a and b are original traces of EPSCs prior to and following HFS. B i shows the summated EPSCs during the initial HFS in control media, composed of both AMPAR- and NMDAR-mediated components. B ii shows the summated AMPAR component of the EPSCs in the presence of AP5. B iii shows the superimposition of the summated EPSPs in control and in AP5. B iv shows the NMDAR component of the summated EPSCs of the train, obtained by subtracting the AMPAR summated EPSCs from the combined AMPAR and NMDAR summated EPSCs.
Lack of LTP or LTD induction by a HFS-hyperpolarisation pairing protocol which did not evoke NMDAR activation
Pairing high intensity HFS with a single hyperpolarising step of 1.1 s from −70 to −90 mV did not induce significant LTP or LTD, the post-stimulus EPSCs measuring 98.4 ± 8.7 % (P > 0.05, n = 5), at 25 min post- HFS (Fig. 4A). The absence of induction of LTP or LTD was associated with a lack of activation of NMDARs during the HFS. The summated multiple EPSC waveform had an initial large amplitude (˜300 pA) but a relatively short duration (˜40 ms; Fig. 4B i). In the presence of AP5, in which there was also no induction of LTP or LTD (99.1 ± 4.7 %), the summated multiple AMPA receptor-mediated EPSCs during each HFS had a very similar amplitude and duration to control (Fig. 4B ii and iii). Subtraction of the summated EPSC waveform in the presence of AP5 from the control summated EPSC waveform resulted in an absence of NMDAR-mediated current (Fig. 4B iv).
Figure 4. The absence of induction of LTP or LTD by a pairing protocol consisting of high intensity presynaptic HFS and a single step postsynaptic hyperpolarisation.
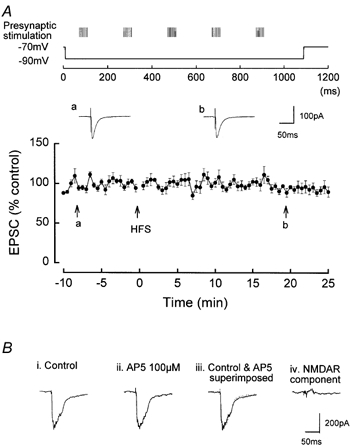
The presynaptic stimulation was increased to evoke ˜300 pA EPSCs during the HFS. The holding potential of the postsynaptic cell was −70 mV, and a single step hyperpolarisation to −90 mV and of 1.1 s duration was applied during the HFS. A, no LTP or LTD was induced by the HFS, the post-stimulus EPSCs measuring 98.4 ± 8.7 % (P > 0.05, n = 5) at 25 min post-HFS. a and b are original traces of EPSCs prior to and following HFS. B, examples of summated EPSCs during a single train of HFS. Bi shows the summated EPSCs during the initial HFS in control media. Bii shows the summated AMPAR component of the EPSCs in the presence of AP5. Biii shows the superimposition of the summated EPSPs in control and in AP5. Note that the two traces are virtually identical. Biv shows that subtracting the waveform of the summated EPSCs in AP5 from the control summated EPSCs results in no NMDAR-mediated current.
Correlation of NMDAR activation with induction of plasticity
In order to correlate quantitatively the degree of activation of the NMDAR with the induction of synaptic plasticity, the total NMDAR charge transfer was measured from the isolated NMDAR currents evoked under the different stimulation protocols which induced LTP, LTD or the LTD-LTP crossover point. This measure of NMDAR charge transfer was plotted against the amplitude and direction of plasticity, as shown in Fig. 5, in a plasticity versus NMDAR activation curve. It can be seen that a transition from induction of LTD to LTP via a crossover point occurs as the charge transfer via the NMDAR is increased. Particularly notable is that NMDAR-dependent LTD is associated with a very small NMDAR charge transfer, and that the LTD-LTP crossover point is associated with relatively large NMDAR activation, although less than that occurring during LTP induction.
Figure 5. Synaptic plasticity-NMDAR activation curve.
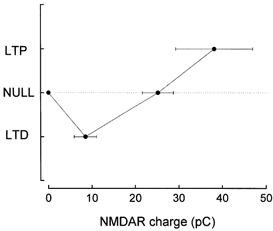
The amplitude of LTP, LTD or zero plasticity induced by the different stimulation protocols was plotted against the NMDAR charge transfer. Each point is the mean of n = 5. Note the increasing NMDAR charge transfer as plasticity alters from induction of LTD to the LTD-LTP crossover point and to LTP.
High intensity HFS paired with depolarisation induces group I mGluR-dependent LTD under conditions of NMDAR inhibition
As shown in Fig. 1B, the pairing protocol consisting of HFS and a single step depolarisation from −70 to −30 mV induced an NMDAR-independent LTD in the presence of AP5. Such LTD was found to be dependent upon activation of mGluR, being inhibited by the mGluR antagonist MCPG (500 μm). Thus in MCPG, perfused for at least 30 min prior to HFS in the presence of AP5, the EPSC measured 102.5 ± 4.7 % at 25 min post-HFS, a value not significantly different from the pre-pairing baseline value (Fig. 6A).
Figure 6. NMDAR-independent LTD induced by high intensity HFS paired with a single depolarisation from −70 to −30 mV is dependent upon activation of group I mGluR, but not group II.
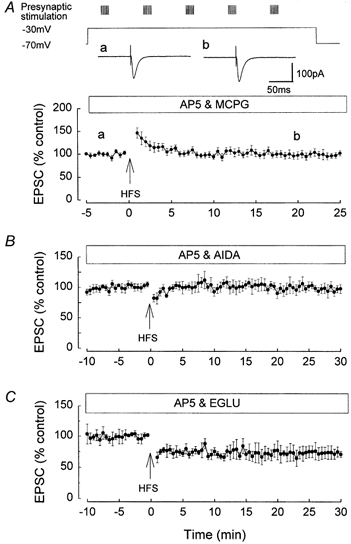
A, the induction of NMDAR-independent LTD by HFS was inhibited by the mGluR antagonist MCPG. The traces a and b show the original EPSCs prior to and following HFS, and are not significantly different. B, the induction of NMDAR-independent LTD was inhibited by the group I mGluR antagonist AIDA. C, the induction of NMDAR-independent LTD was not inhibited by the group II mGluR antagonist EGLU.
The effects of subgroup-selective mGluR antagonists were examined on the induction of the NMDAR-independent LTD. The NMDAR-independent LTD was blocked by the selective group I mGluR antagonist AIDA (Pelliacciari et al. 1996). Thus in the presence of AIDA (450 μm), perfused for at least 30 min prior to pairing, the EPSC measured 101.8 ± 7.4 %, n = 5, at 25 min post-pairing, not significantly different from the pre-pairing baseline value (Fig. 6B).
The mGluR-dependent LTD was not blocked by the selective group II mGluR antagonist EGLU (Jane et al. 1996). In the presence of EGLU (250 μm), which was perfused for at least 30 min pre-HFS, the pairing of high intensity HFS and depolarisation induced LTD, the ESPC measuring 75.4 ± 9.5 % (n = 5), at 25 min post-HFS, a value not significantly different from control (Fig. 6C).
Induction of mGluR-dependent LTD Ca2+ influx via L-type Ca2+ channels and a rise in intracellular Ca2+
The experiments shown in Fig. 4A demonstrated that the mGluR-dependent LTD induction required depolarisation, suggesting that Ca2+ influx is necessary for the LTD induction. Accordingly, the effects of the L-type voltage-gated Ca2+ channel inhibitor nifedipine were investigated on the induction of the LTD. The mGluR-dependent LTD was blocked by nifedipine. In the presence of nifedipine (10 μm), which was perfused for at least 30 min pre-pairing, the pairing of high intensity HFS and depolarisation failed to induce LTD, the EPSC measuring 97.8 ± 6.8 % (n = 6) at 25 min post-HFS (Fig. 7A).
Figure 7. NMDAR-independent LTD induction is dependent upon an influx of Ca2+ via L-type Ca2+ channels and an increase in intracellular Ca2+.
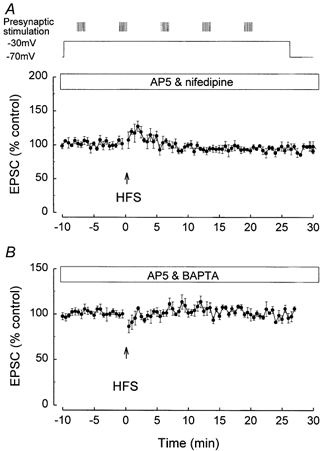
A, the induction of NMDAR-independent LTD was inhibited by the L-type Ca2+ channel blocker nifedipine (10 μm). B, the induction of NMDAR-independent LTD was inhibited by the Ca2+ chelator BAPTA.
The mGluR-dependent LTD was also blocked if a rise in postsynaptic intracellular Ca2+ was prevented. Following postsynaptic injection of the Ca2+ chelator BAPTA (20 mm), the HFS pairing failed to induce LTD, the EPSC measuring 98.7 ± 5.6 % (n = 6), at 25 min post-HFS (Fig. 7B).
Quantal analysis involving minimal stimulation
In order to obtain information about the site of expression of mGluR-dependent LTD, quantal analysis involving minimal stimulation techniques was carried out. Both the failure rate and potency (defined as the EPSC amplitude excluding failures (Stevens & Wang, 1994) were measured before and following mGluR-dependent LTD induction.
In all cells LTD was associated with both a decrease in potency and an increase in failure rate. Figure 8A shows the complete time course of one typical experiment. The mean EPSC amplitude decreased by 64 % in this cell following the induction of LTD. LTD was accompanied by an increase in the failure rate, from 14 % during the baseline period to 64 % after the pairing, and by a decrease in the potency, from 11.5 pA at baseline to 4.1 pA after the pairing. The plot of the pre-pairing period and post-pairing period data in the form of amplitude histograms confirmed the increase in failure rate and the decrease in potency occurring following the induction of LTD (Fig. 8B). For all the five cells in which LTD was successfully induced using minimal stimulation, failure rate increased from 20.0 ± 5.4 to 52.6 ± 8.2 %, and the potency decreased from 24.5 ± 3.2 to 12.7 ± 2.4 pA (P < 0.005).
Figure 8. mGluR-dependent LTD is associated with a decrease in potency and an increase in failure rate.
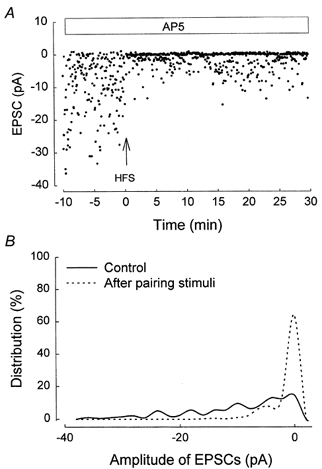
An example of an experiment using minimal stimulation. A, individual EPSC amplitude plotted against time during the whole experiment. B, amplitude histogram for this experiment (bin width =1 pA).
In order to ensure that the pairing procedure itself did not result in a change in potency or failure rate in the absence of LTD induction, minimal stimulation experiments were also carried out under conditions in which LTD induction was blocked by MCPG. Figure 9A shows the complete time course of one experiment. No significant change of the mean EPSC, the failure rate or the potency was induced by pairing in the presence of MCPG. This lack of significant change was confirmed in the plot of the amplitude histogram for pre-pairing and post-pairing periods (Fig. 9B). Thus in all the five cells in which stimulation was paired in the presence of MCPG, the failure rate was 25.5 ± 5.7 % pre-pairing and 25.3 ± 6.6 % post-pairing. The potency was 18.7 ± 1.9 pA pre-pairing and 18.9 ± 0.9 pA post-pairing.
Figure 9. Inhibition of mGluR-dependent LTD by MCPG prevents the change in potency and failure rate following HFS.
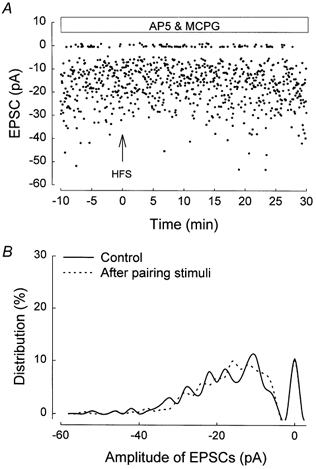
An example of an experiment using minimal stimulation. A, individual EPSC amplitude plotted against time during the whole experiment in the presence of MCPG. B, amplitude histogram for this experiment (bin width =1 pA).
DISCUSSION
The present studies have shown that a pairing stimulation procedure of brief HFS and depolarisation can induce NMDAR-dependent LTP, LTD, LTD-LTP crossover and also an mGluR-dependent LTD depending on the particular protocol of the induction stimulation.
NMDAR activation during the induction of plasticity
The quantitative measurement of NMDAR activity during induction of LTP, LTD and the LTP-LTD crossover point was made under conditions in which all three types of plasticity were induced by an identical level of presynaptic stimulation in order to ensure that the same number of synapses were stimulated during the induction of the three types of plasticity. Thus only the level and pattern of postsynaptic depolarisation were modified to induce either LTP, LTD or the LTD-LTP crossover point.
NMDAR charge transfer was found to increase as the NMDAR-dependent plasticity underwent a transition from no plasticity to LTD, and then from LTD to LTP via an LTD-LTP crossover point, as shown in Fig. 5. Particularly notable is the very low level of NMDAR activation associated with the induction of LTD compared with that during the induction of LTP, the NMDAR charge transfer ratio for LTD compared to LTP being 0.22. Also notable is that the LTD-LTP crossover point was associated with a relatively high level of activation of NMDARs compared to that for the induction of LTD. No LTD or LTP was induced if HFS was applied at a hyperpolarised membrane potential, in agreement with several previous studies (Malinow & Miller, 1986; Artola et al. 1990; Mulkey & Malenka, 1992; Ngezahayo et al. 2000), demonstrating that LTD/LTP induction requires a minimum level of membrane depolarisation. The inability of HFS given at hyperpolarised potentials to induce LTP/LTD was shown in the present studies to be associated with a complete absence of NMDAR activation.
The curve relating plasticity to NMDAR activation shown in Fig. 5 shows a strong resemblance to the modified Bienenstock-Cooper-Munro (BCM) function (Bear, 1995), in which a smooth transition from LTD to LTP induction occurs as the frequency of stimulation is increased. The present experiments are strong quantitative support for the theory of Bear (1995) that the transition from LTD to LTP with increased frequency is due to an increased NMDAR activation, and that the LTP-LTD crossover frequency is associated with a critical level of NMDAR activation (reviewed by Bear & Malenka, 1994; Bear, 1995).
It is well established that Ca2+ influx via NMDARs is required to trigger the induction of NMDAR-dependent LTP or LTD (Lynch et al. 1983; Malenka et al. 1983; Artola & Singer, 1993). According to the differential threshold hypothesis, LTP has a high intrinsic Ca2+ threshold and is induced by a large rise in Ca2+, whereas LTD has a low intrinsic Ca2+ threshold and is selectively induced by a lower rise in Ca2+ (Lisman, 1989; Artola & Singer, 1993). Measurements of Ca2+ increase induced by stimulation protocols which induce either LTP or LTD have supported this theory, although recent studies have shown that the rate of rise and/or the duration of Ca2+ increase is also important in determining the polarity of synaptic plasticity (Hansel et al. 1997; Otani & Connor, 1998; Yang et al. 1999). Peak dendritic Ca2+ levels have been shown to rise by 37 % and 62 % (ratio 0.6) following LTD and LTP induction, respectively, in the visual cortex (Hansel et al. 1997), and to 464 nm and 1.25 μm (ratio 0.37) during LTD and LTP, respectively, in the CA1 region of the hippocampus (Otani & Connor, 1998). The ratio of Ca2+ increase associated with LTD/LTP induction is somewhat larger in these studies than the ratio of NMDAR activation associated with LTD/LTP induction measured in the present experiments (0.22). This suggests that the increase in Ca2+ levels occurring during LTD induction may be due in part to Ca2+-induced Ca2+ release from intracellular Ca2+ stores, with Ca2+ influx the via NMDAR serving as the trigger for Ca2+ release. The necessity of Ca2+-induced Ca2+ release from intracellular ryanodine-sensitive Ca2+ stores for the induction of LTD has been previously demonstrated in the dentate gyrus using pharmacological inhibitors of the ryanodine receptor (O'Mara et al. 1995; Wang et al. 1997).
Induction of LTD by HFS
Most previous studies have shown a clear division of the stimulation parameters resulting in LTP and LTD in the CA1 or dentate gyrus of the hippocampus, with NMDAR-dependent LTP induced by brief HFS of 50–200 Hz or the pairing of brief LFS and large postsynaptic depolarisation (Bliss & Lomo, 1973; Wigstrom et al. 1985; Kelso et al. 1986; reviewed by Bliss & Collingridge, 1993), and NMDAR-dependent LTD induced by several minutes of LFS at 1–5 Hz (Dudek & Bear, 1992a,b; Mulkey & Malenka, 1992; O'Mara et al. 1995; Huang et al. 1997; reviewed by Linden & Connor, 1995; Bear & Abraham, 1996; Anwyl, 1999). In the present studies, either LTP or LTD could be induced by brief HFS protocols without media or pharmacological changes simply by modifying the duration or amplitude of postsynaptic depolarisation. This finding supports other recent studies in which LTD/depotentiation could be induced by HFS in vitro, although only under altered media or pharmacological conditions. Thus HFS applied in the presence of a low concentration of AP5 (Cummings et al. 1996), or HFS applied on the trough of a cholinergically evoked theta rhythm (Huerta & Lisman, 1995), resulted in the induction of LTD and depotentiation, respectively. In addition, HFS applied in vivo was found to induce depotentiation when applied on the trough of the tail pinch-evoked theta rhythm (Holscher et al. 1997). Such findings raise the possibility that HFS may be a strong physiological stimulus for the induction of LTD as well as LTP in vivo, with the level of presynaptic activation and the pattern and amplitude of postsynaptic depolarisation determining the direction of plasticity.
The present studies showing that an mGluR-dependent form of LTD induction can be induced in the dentate gyrus in addition to an NMDAR-dependent LTD induction adds strongly to the growing evidence for an mGluR-dependent form of LTD which is distinct from an NMDAR-dependent LTD (Bolshakov & Siegelbaum, 1994; O'Mara et al. 1995; Oliet & Nicoll, 1997; Malenka, 1998; Camodeca et al. 1999). In the present studies, the mGluR-dependent LTD induction was only induced by a pairing procedure involving high intensity presynaptic stimulation. Such stimulation may result in spillover of extracellular glutamate to the perisynaptic location of the group I mGluRs (Lujan et al. 1996) on adjacent axon terminals. The demonstration of an involvement of group I mGluRs in the LTD induced by HFS is in agreement with previous studies showing that LFS-induced LTD can be induced via the activation of group I mGluRs in the medial perforant path of the dentate gyrus (Camodeca et al. 1999) and CA1 (Oliet et al. 1997). The lack of involvement of group II mGluRs in HFS-induced LTD in the present studies is surprising in view of the high density of such receptors at the medial perforant path synapses (Shigemoto et al. 1997) and that prolonged LFS-induced LTD was found to involve group II mGluRs at this synapse (Huang et al. 1997, 1999). It is possible that the HFS induction protocol for LTD used in the present studies is too brief for the sufficient activation of the group II mGluRs.
The influx of Ca2+ required for the mGluR-dependent LTD was shown to be via L-type Ca2+ channels in the present study, in agreement with one previous study involving LFS-induced mGluR-dependent LTD in CA1 (Bolshakov & Siegelbaum, 1994). However, under certain conditions of stimulation, Ca2+ entry via T-type Ca2+ channels can support mGluR-dependent LTD. Thus in previous studies in the dentate gyrus (Wang et al. 1997, 1998) and CA1 (Oliet et al. 1997), mGluR-dependent LFS-induced LTD evoked under conditions of mild postsynaptic depolarisation was blocked by the T-type Ca2+ channel inhibitor Ni2+. It therefore appears that Ca2+ influx via L- or T-type Ca2+ channels can participate in mGluR-dependent LTD induction, with T-type Ca2+ channels involved when postsynaptic depolarisation is mild and L-type Ca2+ channels involved when depolarisation is strong.
The expression of the mGluR-dependent LTD was associated with both a decrease in potency and an increase in failure rate. The decrease in potency could be caused by a decrease in q (quantal size) or n (number of functional synapses) providing that a single synapse was being activated during the minimal stimulation, although if more than one synapse was being activated, then the decrease in potency could also be caused by a decrease in the probability of release. Although the increase in failure rate could classically be due to a decrease in the probability of release, under the silent synapse theory of Liao et al. (1995), it could also be caused by a postsynaptic change associated with a failure of detection of released transmitter as active synapses are converted into silent synapses. In previous studies in the hippocampus, a similar decrease in potency and increase in failure rate accompanying NMDAR-dependent LTD induction was detected (Man et al. 2000) and attributed to a postsynaptic decrease in the number of active synapses. Moreover, immunocytochemical studies have provided strong evidence that LTD is associated with a complete removal of AMPAR clusters (Carroll et al. 1999).
Acknowledgments
This research was supported by the Wellcome Trust and the European Union (BI04-CT96-0049).
References
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Research Reviews. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Artola, Abrocher S, Singer W. Different voltage-dependent thresholds for inducing long-term depression and long-term potentiation in slices of rat visual cortex. Nature. 1990;347:69–72. doi: 10.1038/347069a0. [DOI] [PubMed] [Google Scholar]
- Artola A, Singer W. Long-term depression of excitatory synaptic depression and its relationship to long-term potentiation. Trends in Neurosciences. 1993;16:480–487. doi: 10.1016/0166-2236(93)90081-v. [DOI] [PubMed] [Google Scholar]
- Bear MF. Mechanism for a sliding synaptic modification threshold. Neuron. 1995;15:1–4. doi: 10.1016/0896-6273(95)90056-x. [DOI] [PubMed] [Google Scholar]
- Bear MF, Abraham WC. Long-term depression in the hippocampus. Annual Review of Neuroscience. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Current Opinion in Neurobiology. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Bliss TV P, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bliss TV P, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetised rabbit following stimulation of the perforant path. Journal of Physiology. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science. 1994;264:1148–1152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- Camodeca N, Breakwell NA, Rowan MJ, Anwyl R. Induction of LTD by activation of group I mGluR in the dentate gyrus in vitro. Neuropharmacology. 1999;38:1597–1606. doi: 10.1016/s0028-3908(99)00093-3. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Lissin DV, Von zastrow M, Nicoll RA, Malenka RC. Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nature Neuroscience. 1999;2:454–460. doi: 10.1038/8123. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, Mclennan H. The antagonism of amino acid induced excitations of rat hippocampal CA1 neurons in vitro. Journal of Physiology. 1983;334:19–31. doi: 10.1113/jphysiol.1983.sp014477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+ signalling requirements for long-term depression in the hippocampus. Neuron. 1996;16:825–833. doi: 10.1016/s0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- D'angelo E, De filippi G, Rossi P, Taglietti V. Synaptic excitation of individual rat cerebellar granule cells in situ: evidence for the role of NMDA receptors. Journal of Physiology. 1995;484:397–413. doi: 10.1113/jphysiol.1995.sp020673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of the hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proceedings of the National Academy of Sciences of the USA. 1992a;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Bi-directional long-term modification of synaptic effectiveness in the adult and immature hippocampus. Journal of Neuroscience. 1992b;13:2910–2918. doi: 10.1523/JNEUROSCI.13-07-02910.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzjohn SM, Kingston AE, Lodge D, Collingridge GL. DHPG-induced LTD in area CA1 of juvenile rat hippocampus: characterization and sensitivity to novel mGluR antagonists. Neuropharmacology. 1999;38:1577–1583. doi: 10.1016/s0028-3908(99)00123-9. [DOI] [PubMed] [Google Scholar]
- Hansel C, Artola A, Singer W. Relation between dendritic Ca2+ levels and the polarity of synaptic long-term modifications in rat visual cortex neurons. European Journal of Neuroscience. 1997;9:2309–2322. doi: 10.1111/j.1460-9568.1997.tb01648.x. [DOI] [PubMed] [Google Scholar]
- Herron CE, Lester RAJ, Coan EJ, Collingridge GL. Frequency-dependent involvement of NMDA receptors in the hippocampus: a novel synaptic mechanism. Nature. 1986;322:265–268. doi: 10.1038/322265a0. [DOI] [PubMed] [Google Scholar]
- Holscher C, Anwyl R, Rowan MJ. Stimulation on the positive phase of hippocampal theta rhythm induces long-term potentiation that can be depotentiated by stimulation on the negative phase in area CA1 in vivo. Journal of Neuroscience. 1997;17:6470–6477. doi: 10.1523/JNEUROSCI.17-16-06470.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L-Q, Rowan MR, Anwyl R. mGluRII agonist inhibition of LTP induction, and mGluRII antagonist inhibition of LTD induction, in the dentate gyrus in vitro. NeuroReport. 1997;8:687–693. doi: 10.1097/00001756-199702100-00022. [DOI] [PubMed] [Google Scholar]
- Huang L-Q, Rowan MR, Anwyl R. Activation of group II mGluR induces LTD via activation of protein kinase A and C in the rat dentate gyrus in vitro. Neuropharmacology. 1999;38:73–83. doi: 10.1016/s0028-3908(98)00168-3. [DOI] [PubMed] [Google Scholar]
- Huerta PT, Lisman JE. Bidirectional synaptic plasticity induced by a single burst during cholinergic theta oscillation in CA1 in vitro. Neuron. 1995;15:1053–1063. doi: 10.1016/0896-6273(95)90094-2. [DOI] [PubMed] [Google Scholar]
- Kelso SR, Ganong AH, Brown TH. Hebbian synapses in hipocampus. Proceedings of the National Academy of Sciences of the USA. 1986;83:5326–5330. doi: 10.1073/pnas.83.14.5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J. A mechanism for the Hebb and anti-Hebb processes underlying learning and memory. Proceedings of the National Academy of Sciences of the USA. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan R, Nusser Z, Roberts DB, Shigemoto R, Somogyi P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. European Journal of Neuroscience. 1996;8:1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- Lynch G, Larson J, Kelso S, Barrionuevo G, Schottler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983;305:719–721. doi: 10.1038/305719a0. [DOI] [PubMed] [Google Scholar]
- Mcnaughton BL. Evidence for two physiologically distinct perforant pathways to the dentate fascia. Brain Research. 1980;199:1–19. doi: 10.1016/0006-8993(80)90226-7. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Zucker RS, Nicoll RA. Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science. 1988;242:81–84. doi: 10.1126/science.2845577. [DOI] [PubMed] [Google Scholar]
- Malinow R, Miller JP. Postsynaptic hyperpolarization during conditioning reversibly blocks induction of long-term potentiation. Nature. 1986;320:529–530. doi: 10.1038/320529a0. [DOI] [PubMed] [Google Scholar]
- Man H-Y, Lin JW, Ju WH, Ahmadian G, Liu L, Becker LE, Sheng M, Wang YT. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron. 2000;25:649–662. doi: 10.1016/s0896-6273(00)81067-3. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- Neveu D, Zucker RS. Postsynaptic levels of [Ca2 +] needed to trigger LTD and LTP. Neuron. 1996;16:619–629. doi: 10.1016/s0896-6273(00)80081-1. [DOI] [PubMed] [Google Scholar]
- Ngezhayo A, Schachner M, Artola A. Synaptic activity modulates the induction of bidirectional synaptic changes in adult mouse hippocampus. Journal of Neuroscience. 2000;20:2451–2458. doi: 10.1523/JNEUROSCI.20-07-02451.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'connor J, Rowan MJ, Anwyl R. Tetanically induced LTP involves a similar increase in the AMPA and NMDA receptor components of the excitatory postsynaptic current: Investigations of the role of mGluR receptors. Journal of Neuroscience. 1995;15:2013–2020. doi: 10.1523/JNEUROSCI.15-03-02013.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'mara SM, Rowan MJ, Anwyl R. Dantrolene inhibits long-term depression and depotentiation of synaptic transmission in the rat dentate gyrus. Neuroscience. 1995a;68:621–624. doi: 10.1016/0306-4522(95)00233-9. [DOI] [PubMed] [Google Scholar]
- O'mara SM, Rowan MJ, Anwyl R. Metabotropic glutamate receptor induced long-term depression and potentiation in the dentate gyrus of the rat hippocampus in vitro. Neuropharmacology. 1995b;34:983–989. doi: 10.1016/0028-3908(95)00062-b. [DOI] [PubMed] [Google Scholar]
- Oliet SH R, Malenka RC, Nicoll RA. Two distinct forms of long-term depression co-exist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Otani S, Connor JA. Requirement of rapid Ca2+ entry and synaptic activation of metabotropic glutamate receptors for the induction of long-term depression in adult rat hippocampus. Journal of Physiology. 1998;511:761–770. doi: 10.1111/j.1469-7793.1998.761bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MJ, Irving AJ, Seabrook GR, Jane DE, Collingridge GL. The group I mGluR agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology. 1997;36:1517–1532. doi: 10.1016/s0028-3908(97)00181-0. [DOI] [PubMed] [Google Scholar]
- Pelliacciari R, Raimondo M, Marinozzi M, Natalini B, Constantino G, Thomsen C. (S)-(+)-2-(3′-Carboxybicyclo[1. 1.1]pentyl)-glycine, a structurally new group I metabotropic glutamate receptor antagonist. Journal of Medicinal Chemistry. 1996;39:2874–2876. doi: 10.1021/jm960254o. [DOI] [PubMed] [Google Scholar]
- Selig DK, Lee HK, Bear MF, Malenka RC. Re-examination of the effects of MCPG on hippocampal LTP, LTD and depotentiation. Journal of Neurophysiology. 1995;74:1075–1082. doi: 10.1152/jn.1995.74.3.1075. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Wang Y. Changes in reliability of synaptic function as a mechanism for plasticity. Nature. 1994;371:704–707. doi: 10.1038/371704a0. [DOI] [PubMed] [Google Scholar]
- Wang Y, Rowan MJ, Anwyl R. Induction of LTD in the dentate gyrus in vitro is NMDA receptor-independent, but dependent on Ca2+ influx via low-voltage-activated Ca2+ channels and release of Ca2+ from intracellular stores. Journal of Neurophysiology. 1997;77:812–825. doi: 10.1152/jn.1997.77.2.812. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wu J, Rowan MJ. Role of protein kinase C in the induction of homosynaptic long-term depression by brief low frequency stimulation in the dentate gyrus of the rat hippocampus in vitro. Journal of Physiology. 1998;513:467–475. doi: 10.1111/j.1469-7793.1998.467bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigstrom H, Gustaffson B, Huang Y-Y, Abraham WC. Hippocampal long-term potentiation is induced by pairing single afferent volleys with intracellularly injected depolarizing current pulses. Acta Physiologica Scandinavica. 1986;126:317–319. doi: 10.1111/j.1748-1716.1986.tb07822.x. [DOI] [PubMed] [Google Scholar]
- Yang S-N, Tang Y-G, Zucker RS. Selective induction of LTP and LTD by postsynaptic [Ca2+]i elevation. Journal of Neurophysiology. 1999;81:781–787. doi: 10.1152/jn.1999.81.2.781. [DOI] [PubMed] [Google Scholar]