

Abstract
We performed patch-clamp recordings on acutely isolated somata and dendritic segments of rat neocortical neurons, in order to compare the reversal potential (EGABA) and relative density of GABAA receptor-mediated Cl− currents in these two cellular compartments.
Currents were recorded with the Cl−-impermeable pore former gramicidin (25–75 μg ml−1) in HCO3−-free bath solution. Voltage ramps (−110 to −30 mV) from a holding potential (Vh) of −60 mV in the absence and presence of 2 μM GABA were used to construct instantaneous current-voltage relationships. Currents were abolished by co-application of GABA with the GABAA receptor antagonist bicuculline (40 μM).
GABA conductance, normalized to membrane surface area, was not different in somata and dendrites. In addition, EGABA was not different in the two compartments.
Replacement of intracellular K+ with Cs+ resulted in a significantly more depolarized EGABA in both somata and dendrites. These results suggest that the resting intracellular Cl− concentration ([Cl−]i) is similar in somata and dendrites and that an outward Cl− transporter system maintains low [Cl−]i.
In mature neocortex, GABAA receptor-mediated inhibitory postsynaptic potentials (IPSPs) have reversal potentials more negative than the resting membrane potential (Connors et al. 1988; van Brederode & Spain, 1995). The reversal potential for GABAA receptor-mediated neurotransmission (EGABA) depends on the transmembrane gradients for Cl− and HCO3− (Thompson et al. 1988; Kaila et al. 1993). Thus, the principal mechanism of synaptic inhibition is membrane hyperpolarization due to a net inward anion flux. Immature cortical neurons are strongly depolarized by GABAA receptor activation, however (Owens et al. 1996), and under these conditions GABAA receptor activation might paradoxically result in excitation. In addition, intense or prolonged GABA application in mature neocortex can also give rise to depolarizing responses (Staley et al. 1995; Cerne & Spain, 1997). Indirect evidence, obtained with intracellular recording electrodes, indicates that local application of GABA to dendrites elicits predominantly depolarizing responses (Barker & Ransom, 1978; Alger & Nicoll, 1982; Scharfmann & Sarvey, 1987).
Since the extracellular concentrations of Cl− and HCO3− are relatively constant, intracellular anion concentration and/or GABAA receptors with different permeability ratios for Cl− and HCO3− are most likely to underlie the observed variability of EGABA (Hara et al. 1992; Staley et al. 1995; Staley & Proctor, 1999; Perkins, 1999). Little direct evidence is available on the intracellular anion concentration in dendrites. In order to compare the mechanisms that control the direction and magnitude of dendritic and somatic GABAergic synaptic currents we examined the density of GABA currents and the regulation of intracellular Cl− homeostasis in these two compartments. For this purpose we used recordings from acutely dissociated dendritic segments and somata (Takigawa & Alzheimer, 1999). This allowed us to compare dendritic and somatic GABAA receptor-mediated Cl− currents under nominally HCO3−-free conditions, such that EGABA equals the Cl− reversal potential (ECl). In addition, by using the gramicidin-perforated patch technique (Ebihara et al. 1995), we were able to avoid internal dialysis of the cell and the resulting changes in [Cl−]i.
METHODS
Recordings were performed on acutely isolated somata and dendrites from 2- to 3-week-old Wistar rats. The animals were deeply anaesthetized with a ketamine-xylazine mixture (RBI; 1 ml kg−1i.p.) and decapitated. All experiments were carried out in accordance with established guidelines and with the approval of the Animal Care Committee at the University of Munich. The details of the surgery and dissociation procedures are given elsewhere (Takigawa & Alzheimer, 1999). Briefly, coronal slices, 400 μm thick, taken from a block of frontoparietal cortex were cut with a tissue slicer. The slices were then transferred to a holding chamber in Ringer solution for 30 min at 30 °C and another 30 min at room temperature (22–24 °C). Small pieces of cortical grey matter were dissected from each slice and put into papain solution (19 U ml−1; Sigma) in Hepes buffer solution for 90 min at 30 °C, bubbled with 100 % O2. After several rinses, cells were triturated using glass pipettes of progressively smaller bore. After trituration, cells were plated in Hepes buffer in a Petri dish mounted on the stage of an inverted microscope equipped with Hoffman optics. All recordings were performed at room temperature.
Recording pipettes were pulled from borosilicate glass. Pipette resistance (measured in the bath) was 4–8 MΩ for somatic and 10–15 MΩ for dendritic recordings. The standard pipette solution contained (mm): KCl 130, Hepes 10, CaCl2 2, EGTA 11, pH 7.3, 280 mosmol l−1. In some recordings KCl in the pipette solution was replaced by an equimolar amount of CsCl. Gramicidin (Sigma) was dissolved in DMSO at a concentration of 25–100 mg ml−1. This stock solution was diluted in the pipette solution for a final concentration of 25–75 μg ml−1. Currents were recorded using an Axopatch 200 amplifier (Axon Instruments) in whole-cell voltage-clamp mode. In most experiments 70–75 % series resistance compensation was used. Currents were low-pass filtered at 1 or 2 kHz with a 4-pole Bessel filter. Data were A–D converted, stored and analysed using pCLAMP software (Axon Instruments). After establishing a gigaohm seal we injected 5 mV hyperpolarizing voltage steps and monitored the gradual decrease in series resistance due to membrane perforation by gramicidin. We waited until series resistance dropped below 60 MΩ, usually after > 20 min.
Local drug application was achieved by means of a remotely controlled, solenoid-operated Y-tube system. Local superfusion was accomplished by positioning the perfusion tube within 50–70 μm of the soma or dendrite with the aid of a micromanipulator. The composition of the bath and local perfusion solutions was (mm): NaCl 150, KCl 3, CaCl2 2, MgCl2 2, Hepes 10 and d-glucose 10 (pH 7.4; 300 mosmol l−1). To this solution we added tetrodotoxin (TTX; 1 μm; Sigma) and 30 μm LaCl3 in order to block voltage-activated Na+ and Ca2+ currents, respectively, and 2 μm CGP 54845A (Tocris Cookson) to block GABAB receptor-mediated currents. The GABAB antagonist was omitted when the Cs+-based patch solution was used. GABA (2 μm; Sigma) was added to the perfusion solution from a 10 mm stock solution in Hepes buffer. Bicuculline methiodide (BIC) was co-applied with GABA in some experiments (40 μm; Sigma).
Gramicidin perforated-patch recordings were performed on visually identified dendritic segments or cell somata. Liquid junction potentials were recorded and corrected in the bath solution just before seal formation. For calculation of GABA current density, cell surface area was estimated by analysing the exponential decay of the capacitative charging current. A −10 mV pulse was applied from a membrane potential of −60 mV and four capacitative transients were recorded and averaged to give the total membrane capacitance (assuming a specific membrane capacitance of 1 μF cm−2). This approach to estimating cell surface area has been validated by Takigawa & Alzheimer (1999).
Measured values are reported as the mean ± standard error of the mean unless noted otherwise. Statistical comparisons were performed using Student's two-tailed t test with significance set at P < 0.05.
RESULTS
A representative example of a dendritic recording using the acutely isolated cell preparation is illustrated in Fig. 1A. The mean surface area (determined from capacitance transients, see Methods) for dendritic segments (456.0 ± 60.8 μm2; n = 12) was significantly smaller than that for isolated somata (1090.4 ± 56.2 μm2; n = 18). Typically, the diameter of the dendritic segments was between 2 and 3 μm. A more detailed description of the morphological and electrophysiological properties of isolated dendritic segments is given by Takigawa & Alzheimer (1999).
Figure 1. Properties of GABA currents recorded in somata and dendrites.
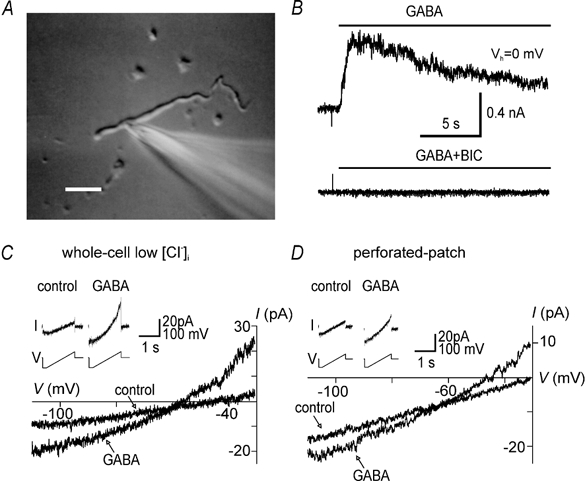
A, photograph of a dendritic recording. The recording electrode is shown on the right. Scale bar, 20 μm. B, currents recorded in a dendrite in perforated-patch mode in response to local superfusion with 2 μm GABA (top) and GABA in combination with 40 μm bicuculline (BIC; bottom). The holding potential (Vh) was 0 mV. The horizontal bars indicate the duration of the drug applications. C, ramp currents and corresponding I-V relationships recorded in conventional whole-cell mode in a dendrite in control solution or 2 μm GABA. Currents were recorded in response to a voltage ramp from −110 to −30 mV from a holding potential of −60 mV (inset). This record was obtained with a low Cl− (9 mm), Cs+-based patch solution. D, ramp currents and corresponding I–V relationships recorded in a different dendrite in gramicidin perforated-patch mode. Note the similar shape and outward rectification of GABA currents in C and D.
In the perforated-patch configuration, we found that local superfusion of isolated neurons with GABA (2 μm) resulted in an outward current at a holding potential (Vh) of 0 mV (Fig. 1B). This current was blocked by co-application of GABA with bicuculline (n = 3), suggesting that it was mediated by activation of the GABAA receptors (Fig. 1B). In these and the following experiments we used a relatively low concentration of GABA in order to minimize receptor desensitization. In several recordings we were able to establish the conventional whole-cell configuration by rupturing the patch after measuring GABA currents in the gramicidin-perforated patch configuration. After patch rupture GABA application at a Vh of −60 mV resulted in a large inward current, suggesting dialysis of the cell interior by the high Cl− concentration of the recording electrode (data not shown). This difference in EGABA between conventional whole-cell and gramicidin perforated-patch modes served as a control for accidental rupture of the patch and experiments that showed this effect were discarded.
Current-voltage (I–V) relationships of GABA-evoked currents were studied using voltage ramps. Membrane voltage (Vh−60 mV) was first stepped to −110 mV for 200 ms and then ramped from −110 mV to −30 mV in 1400 ms (Fig. 1 and Fig. 2). Instantaneous I–V curves, obtained by plotting ramp current in GABA solution against voltage, showed outward rectification (Fig. 1C), but the amount of rectification varied from cell to cell. I-V relationships recorded in perforated-patch mode were similar in shape to those recorded in conventional whole-cell mode with a low Cl− patch solution (compare Fig. 1C and D). Outward rectification of whole-cell GABA currents is most likely to be the result of asymmetrical Cl− concentrations - as predicted by the constant field equation - since I–V relationships of GABAA receptors themselves are linear (Bormann et al. 1987).
Figure 2. A comparison of GABA currents recorded in perforated-patch mode in somata and dendrites.
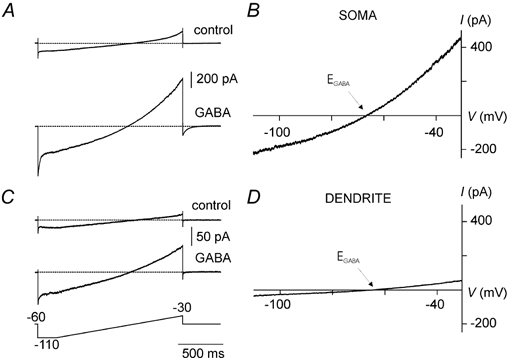
Representative examples of ramp currents recorded in control solution and in 2 μm GABA in a soma (A) and a dendrite (C) using voltage ramps (−110 to −30 mV; Vh =−60 mV, shown in bottom trace). B and D, corresponding instantaneous I-V relationships calculated by subtracting ramp currents recorded in control solution from currents in GABA. A straight line was fitted through a short segment of data points (within 2–3 mV at either side of the zero-current level). The zero-current intercept and slope of this line were used to calculate EGABA and slope conductance, respectively.
In order to compare the density of GABAA receptor-mediated currents between somata and dendrites we subtracted ramp currents in control solution from ramp currents obtained during superfusion with GABA. We then calculated the slope conductance at EGABA from these instantaneous I–V curves (Fig. 2). Although absolute slope conductance was larger in somata than in dendrites, slope conductances were not significantly different after normalization for membrane surface area (somata: 2.99 ± 0.95 pS μm−2, n = 11 vs. dendrites: 2.51 ± 0.50 pS μm−2, n = 7).
Mean EGABA in somata and dendrites was not significantly different using a K+-based patch solution (Fig. 3). Mean values were −65.8 ± 0.6 mV in somata (n = 14) and −68.4 ± 1.8 mV in dendrites (n = 12). These values for EGABA were slightly more negative than the holding potential (−60 mV), suggesting that [Cl−]i is maintained at levels lower than predicted if Cl− were passively distributed across the cell membrane.
Figure 3. The effects of intracellular [K+] on EGABA.
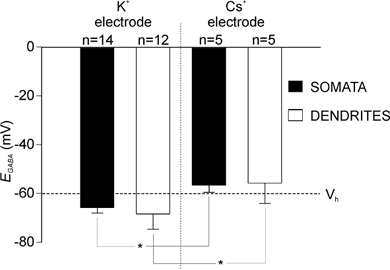
Height of the bar indicates mean values of EGABA in somata and dendrites recorded with a K+-based and Cs+-based patch solution in perforated-patch mode. Error bars represent s.d. EGABA was not significantly different between somata and dendrites, but EGABA was significantly more depolarized when determined with Cs+ as the major cation in the electrode compared to K+ in both dendrites and somata (*P < 0.05, t test).
It has been shown that [Cl−]i and EGABA are sensitive to the intracellular K+ concentration, presumably due to the dependence of the electroneutral K+-Cl− co-transporter system KCC2 on the intracellular K+ concentration (Payne, 1997; DeFazio et al. 2000; Kakazu et al. 2000). To compare the functional role that this transporter system plays in maintaining low resting [Cl−]i in dendrites and somata, we compared the values of EGABA recorded with a K+-based and Cs+-based patch solution. Mean EGABA with the Cs+-based patch solution was −56.5 ± 1.3 mV (somata; n = 5) and −55.8 ± 3.7 mV (dendrites; n = 5). These values did not differ from each other, but were significantly more positive than those recorded in each cellular compartment with a K+-based patch solution and more positive than Vh (Fig. 3). These results suggest that there are K+-sensitive transport mechanisms present in both somata and dendrites that function to extrude Cl− and function to keep EGABA below resting membrane potential.
DISCUSSION
This study provides direct evidence that [Cl−]i is maintained at similar levels in somata and dendrites of neocortical pyramidal cells, based on measurements of EGABA. The Nernst equation predicts a resting [Cl−]i of 13 and 11 mm in somata and dendrites, respectively (22.5 °C, [Cl−]o = 161 mm), which is slightly less than would be expected if Cl− were passively distributed across the cell membrane (i.e. [Cl−]i = 15 mm). Similar values for somatic EGABA have been obtained with gramicidin perforated-patch recordings in acutely dissociated rat substantia nigra neurons (Ebihara et al. 1995).
It is conceivable that mechanisms that maintain low [Cl−]i were disrupted by the dissociation procedure. We believe that this is unlikely, however. First, our values for ECl are similar to what has been described in a more intact preparation, such as neocortical brain slices (Owens et al. 1996). In addition, previous studies have shown that functional voltage-dependent Na+, K+ and Ca2+ channels are present in dendrosomes, and that receptor agonists are capable of inducing GIRK current, indicating that transduction mechanisms in the membrane are preserved (Kavalali et al. 1997; Takigawa & Alzheimer, 1999). We are not aware of any reports showing that Cl− transport mechanisms are selectively destroyed by the dissociation procedure.
Whereas it is widely accepted that somatic EGABA is typically maintained below passive ECl in many adult CNS neurons, studies investigating GABA responses and/or Cl− concentrations in somatic vs. dendritic compartments have yielded apparently conflicting results. For example, Hara et al. (1992) and Kuner & Augustine (2000), who used Cl−-sensitive fluorescent probes, reported higher resting [Cl−]i in dendrites than in somata of hippocampal neurons in culture. In contrast, Jarolimek et al. (1999) found a more negative ECl in the dendrite than in the soma of cultured midbrain neurons, based on somatic whole-cell recordings of spontaneous GABAergic synaptic currents. Somewhat in between these two opposing findings is the study by Morishita & Alger (2001), who noted that dendritically recorded IPSPs (in the presence of HCO3−) were slightly more hyperpolarized than somatic IPSPs. In our experiments, EGABA in dendrites was also slightly more negative than in somata, albeit not significantly so. Finally, variations in the direction of dendritic GABA responses have been observed depending on the position of the application pipette in the dendritic tree (Alger & Nicoll, 1982; Scharfman & Sarvey, 1987), suggesting regional variability in [Cl−]i along the dendritic tree. The reasons for these divergent results are not immediately obvious, but they might reflect the use of different technical approaches as well as differences inherent in the various preparations used. In the hippocampus, responses to uncaged GABA have been shown to be spatially non-uniform, indicating regional variations in receptor distribution between different types of neurons, between somata and dendrites and even between different types of dendrites (Pettit & Augustine, 2000). Since our technical approach did not allow us to determine the source (i.e. pyramidal or non-pyramidal, apical or basal) of the dendritic segments used for whole-cell recordings, possible regional variations in GABA conductance or ECl might have remained undetected.
Here we used isolated cellular compartments, which offer maximum control of membrane voltage and ion gradients. Using this preparation, it was shown that the GABAB receptor agonist baclofen evokes a larger conductance in the dendrites than in somata (Takigawa & Alzheimer, 1999). In the present study, we found that the GABAA conductance in somata and dendrites is similar. Our results suggest that activation of GABAA receptors in the two cellular compartments gives rise to Cl− fluxes of similar magnitude for a given concentration of GABA. How can this be reconciled with the aforementioned observation (see Introduction) that intense GABAergic stimulation gives rise to depolarizing responses, particularly in the dendrites? At resting membrane potential GABAA receptor activation leads to an inward flux of Cl− and an outward flux of HCO3− (Kaila et al. 1993; Staley et al. 1995). Thus, even when the membrane potential equals EGABA, anion currents continue to flow, since membrane potential cannot reach ECl or EHCO3. The transmembrane gradient for HCO3− is relatively stable, but sufficient Cl− accumulation can occur to alter [Cl−]i, due to the limited capacity of Cl− extrusion mechanisms (Thompson & Gahwiler, 1989a,Thompson & Gahwiler, 1989b; Dallwig et al. 1999). The resulting change in [Cl−]i is larger in structures with a low volume to receptor density, such as dendrites. This diminishes the driving force for inward flux of Cl− and makes the outward flux of HCO3− - and the associated membrane depolarization - more dominant in dendrites (Staley & Proctor, 1999).
Our results agree with previous studies that showed that EGABA is more positive when determined with Cs+- than with K+-filled electrodes (Thompson & Gahwiler, 1989b). This effect has been attributed to a displacement of K+ by Cs+, resulting in a reduced effectiveness of the K+ gradient-sensitive Cl− extrusion mechanism KCC2 (Kakazu et al. 2000). With Cs+-filled electrodes, EGABA is maintained at levels slightly more positive than the holding potential, suggesting the presence of an inwardly directed Cl− transporter system (Ballanyi & Grafe, 1985). Such a transporter system (NKCC1) has been characterized in neurons (Kakazu et al. 2000) and has recently been identified in the neocortex (Clayton et al. 1998).
In summary, this preparation presents us with an opportunity to study quantitatively the mechanisms that determine the magnitude and direction of GABAA receptor-mediated currents in dendrites. Our initial results suggest the presence of both inward and outward Cl− transport in dendrites. Further studies are needed to determine under what conditions these regulatory mechanisms break down and what the functional consequences might be in health and disease.
Acknowledgments
This work was supported by grants from the NIH (NS34769) to J.F.M.vB., and the DFG (SFB 391 A9) to C.A., and a VA Merit review (Dr W. J. Spain). C.A. is a Heisenberg-Fellow of the DFG.
References
- Alger BE, Nicoll RA. Pharmacological evidence for two kinds of GABA receptors on rat hippocampal pyramidal cells studied in vitro. Journal of Physiology. 1982;328:125–141. doi: 10.1113/jphysiol.1982.sp014256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballanyi K, Grafe P. An intracellular analysis of γ-aminobutyric-acid-associated ion movements in rat sympathetic neurones. Journal of Physiology. 1985;365:41–58. doi: 10.1113/jphysiol.1985.sp015758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker JL, Ransom BR. Amino acid pharmacology of mammalian central neurones grown in tissue culture. Journal of Physiology. 1978;280:331–354. doi: 10.1113/jphysiol.1978.sp012387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bormann J, Hamill OP, Sakmann B. Mechanism of anion permeation through channels gated by glycine and γ-aminobutyric acid in mouse cultured spinal neurones. Journal of Physiology. 1987;385:243–286. doi: 10.1113/jphysiol.1987.sp016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerne R, Spain WJ. GABAA mediated afterdepolarization in pyramidal neurons from rat neocortex. Journal of Neurophysiology. 1997;77:1039–1045. doi: 10.1152/jn.1997.77.2.1039. [DOI] [PubMed] [Google Scholar]
- Clayton GH, Owens GC, Wolff JS, Smith RL. Ontogeny of cation Cl− cotransporter expression in rat neocortex. Brain Research. Developmental Brain Research. 1998;109:281–292. doi: 10.1016/s0165-3806(98)00078-9. [DOI] [PubMed] [Google Scholar]
- Connors BW, Malenka RC, Silva LR. Two inhibitory postsynaptic potentials, and GABAA and GABAB receptor-mediated responses in neocortex of rat and cat. Journal of Physiology. 1988;406:443–468. doi: 10.1113/jphysiol.1988.sp017390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallwig R, Deitmer JW, Backus KH. On the mechanism of GABA-induced currents in cultured rat cortical neurons. Pflügers Archiv. 1999;437:289–297. doi: 10.1007/s004240050782. [DOI] [PubMed] [Google Scholar]
- Defazio RA, Keros S, Quick MW, Hablitz JJ. Potassium-coupled chloride cotransport controls intracellular chloride in rat neocortical pyramidal neurons. Journal of Neuroscience. 2000;20:8069–8076. doi: 10.1523/JNEUROSCI.20-21-08069.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara S, Shirato K, Harata N, Akaike N. Gramicidin-perforated patch recording: GABA response in mammalian neurones with intact intracellular chloride. Journal of Physiology. 1995;484:77–86. doi: 10.1113/jphysiol.1995.sp020649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara M, Inoue M, Yasukura T, Ohnishi S, Mikami Y, Inagaki C. Uneven distribution of intracellular Cl− in rat hippocampal neurons. Neuroscience Letters. 1992;143:135–138. doi: 10.1016/0304-3940(92)90250-b. [DOI] [PubMed] [Google Scholar]
- Jarolimek W, Lewen A, Misgeld U. A furosemide-sensitive K+-Cl− cotransporter counteracts intracellular Cl− accumulation and depletion in cultured rat midbrain neurons. Journal of Neuroscience. 1999;19:4695–4704. doi: 10.1523/JNEUROSCI.19-12-04695.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K, Voipio J, Paalasmaa P, Pasternack M, Deisz RA. The role of bicarbonate in GABAA receptor-mediated IPSPs of rat neocortical neurones. Journal of Physiology. 1993;464:273–289. doi: 10.1113/jphysiol.1993.sp019634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakazu Y, Uchida S, Nakagawa T, Akaike N, Nabekura J. Reversibility and cation selectivity of the K+-Cl− cotransport in rat central neurons. Journal of Neurophysiology. 2000;84:281–288. doi: 10.1152/jn.2000.84.1.281. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Zhuo M, Bito H, Tsien RW. Dendritic Ca2+ channels characterized by recordings from isolated hippocampal dendritic segments. Neuron. 1997;18:651–663. doi: 10.1016/s0896-6273(00)80305-0. [DOI] [PubMed] [Google Scholar]
- Kuner T, Augustine GJ. A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron. 2000;27:447–459. doi: 10.1016/s0896-6273(00)00056-8. [DOI] [PubMed] [Google Scholar]
- Morishita W, Alger BE. Direct depolarization and antidromic action potentials transiently suppress dendritic IPSPs in hippocampal CA1 pyramidal cells. Journal of Neurophysiology. 2001;85:480–484. doi: 10.1152/jn.2001.85.1.480. [DOI] [PubMed] [Google Scholar]
- Owens DF, Boyce LH, Davis MB E, Kriegstein A. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. Journal of Neuroscience. 1996;16:6416–6423. doi: 10.1523/JNEUROSCI.16-20-06414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JA. Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. American Journal of Physiology. 1997;273:C1516–C1525. doi: 10.1152/ajpcell.1997.273.5.C1516. [DOI] [PubMed] [Google Scholar]
- Perkins KL. Cl− accumulation does not account for the depolarizing phase of the synaptic GABA response in hippocampal pyramidal cells. Journal of Neurophysiology. 1999;82:768–777. doi: 10.1152/jn.1999.82.2.768. [DOI] [PubMed] [Google Scholar]
- Pettit DL, Augustine GJ. Distribution of functional glutamate and GABA receptors on hippocampal pyramidal cells and interneurons. Journal of Neurophysiology. 2000;84:28–38. doi: 10.1152/jn.2000.84.1.28. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Sarvey JM. Responses to GABA recorded from identified rat visual cortical neurons. Neuroscience. 1987;23:407–422. doi: 10.1016/0306-4522(87)90065-0. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Proctor WR. Modulation of mammalian dendritic GABAA receptor function by the kinetics of Cl− and HCO3− transport. Journal of Physiology. 1999;519:693–712. doi: 10.1111/j.1469-7793.1999.0693n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269:977–981. doi: 10.1126/science.7638623. [DOI] [PubMed] [Google Scholar]
- Takigawa T, Alzheimer C. G protein-activated inwardly rectifying K+ (GIRK) currents in dendrites of rat neocortical pyramidal cells. Journal of Physiology. 1999;517:385–390. doi: 10.1111/j.1469-7793.1999.0385t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SM, Deisz RA, Prince DA. Relative contributions of passive equilibrium and active transport to the distribution of chloride in mammalian cortical neurons. Journal of Neurophysiology. 1988;60:105–124. doi: 10.1152/jn.1988.60.1.105. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Gahwiler BH. Activity-dependent disinhibition. I. Repetitive stimulation reduces IPSP driving force and conductance in the hippocampus in vitro. Journal of Neurophysiology. 1989a;61:501–511. doi: 10.1152/jn.1989.61.3.501. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Gahwiler BH. Activity-dependent disinhibition. II. Effects of extracellular potassium, furosemide, and membrane potential on ECl- in hippocampal CA3 neurons. Journal of Neurophysiology. 1989b;61:512–523. doi: 10.1152/jn.1989.61.3.512. [DOI] [PubMed] [Google Scholar]
- Van brederode JFM, Spain WJ. Differences in inhibitory synaptic input between layer II-III and layer V neurons of the cat neocortex. Journal of Neurophysiology. 1995;74:1149–1166. doi: 10.1152/jn.1995.74.3.1149. [DOI] [PubMed] [Google Scholar]