

Abstract
Adenosine transport was measured in human cultured umbilical artery smooth muscle cells, isolated from non-diabetic or gestational diabetic pregnancies, under basal conditions and after pretreatment in vitro with insulin.
Adenosine transport in non-diabetic smooth muscle cells was significantly increased by insulin (half-maximal stimulation at 0.33 ± 0.02 nm, 8 h) and characterized by a higher maximal rate (Vmax) for nitrobenzylthioinosine (NBMPR)-sensitive (es) saturable nucleoside transport (17 ± 5 vs. 52 ± 12 pmol (μg protein)−1 min−1, control vs. insulin, respectively) and maximal binding sites (Bmax) for [3H]NBMPR (0.66 ± 0.07 vs. 1.1 ± 0.1 fmol (μg protein)−1, control vs. insulin, respectively), with no significant changes in Michaelis-Menten (Km) and dissociation (Kd) constants.
In contrast, in smooth muscle cells from diabetic pregnancies, where the values of Vmax for adenosine transport (59 ± 4 pmol (μg protein)−1 min−1) and Bmax for [3H]NBMPR binding (1.62 ± 0.16 fmol (μg protein)−1) were significantly elevated by comparison with non-diabetic cells, insulin treatment (1 nm, 8 h) reduced the Vmax for adenosine transport and Bmax for [3H]NBMPR binding to levels detected in non-diabetic cells.
In non-diabetic cells, the stimulatory effect of insulin on adenosine transport was mimicked by dibutyryl cGMP (100 nm) and reduced by inhibitors of phosphatidylinositol 3-kinase (10 nm wortmannin), nitric oxide synthase (100 μmNG-nitro-l-arginine methyl ester, l-NAME) or protein synthesis (1 μm cycloheximide), whereas inhibition of adenylyl cyclase (100 μm SQ-22536) had no effect.
Wortmannin or SQ-22536, but not l-NAME or cycloheximide, attenuated the inhibitory action of insulin on the diabetes-induced stimulation of adenosine transport.
Protein levels of inducible NO synthase (iNOS) were similar in non-diabetic and diabetic cells, but were increased by insulin (1 nm, 8 h) only in non-diabetic smooth muscle cells.
Our results suggest that adenosine transport via the es nucleoside transporter is modulated differentially by insulin in either cell type. Insulin increased adenosine transport in non-diabetic cells via NO and cGMP, but inhibited the diabetes-elevated adenosine transport via activation of adenylyl cyclase, suggesting that the biological actions of adenosine may be altered under conditions of sustained hyperglycaemia in uncontrolled diabetes.
The biological actions of the endogenous nucleoside adenosine depend on its extracellular concentration and are mediated mainly by cAMP in vascular smooth muscle and endothelium (for reviews, see Olsson & Pearson, 1990; Ralevic & Burnstock, 1998). Extracellular levels of adenosine are regulated by an efficient uptake and metabolism in erythrocytes and endothelial cells, the latter having a particularly high density of es nucleoside transporters (1.2 × 106 transporters cell−1; see Sobrevia et al. 1994). Adenosine transport is mediated by a similar high affinity transport system in smooth muscle cells (Pearson et al. 1978; Beck et al. 1983; Foga et al. 1996; Borgland & Parkinson, 1998). We have recently characterized adenosine transport in human smooth muscle cells cultured from explants obtained from umbilical artery from normal or gestational diabetic pregnancies (Aguayo & Sobrevia, 2000). The maximal transport rate (Vmax) for adenosine via system es in this cell type was found to be increased by ≈2-fold in cells from diabetic pregnancies. This finding contrasts with the reduced Vmax for adenosine transport via system es determined in human umbilical vein endothelium isolated from gestational diabetes (Sobrevia et al. 1994).
Insulin increases transport activity of amino acid system A in rat aorta smooth muscle cells (Obata et al. 1996), system y+/CAT-1 in human umbilical vein endothelium (Sobrevia et al. 1998), perfused rat pancreas (Muñoz et al. 1995) and rabbit isolated gastric glands (Contreras et al. 1997), and glucose transporter GLUT-4 in skeletal muscle (Czech & Corvera, 1999). The effects of insulin on endothelial cells include increased synthesis of nitric oxide (NO) and higher intracellular levels of cGMP (for review, see Baron, 1999). Insulin also induces relaxation of vascular smooth muscle by increasing cGMP levels and expression of the inducible/Ca2+-insensitive NO synthase iNOS (Kahn et al. 1997; Begum et al. 1998; Trovati et al. 1999). In diabetes mellitus, a disease characterized by high plasma levels of d-glucose, modulation of vascular tone is altered and the sensitivity of vascular smooth muscle to insulin or endothelium-derived NO appears to be reduced (for reviews, see Poston & Taylor, 1995; Sobrevia & Mann, 1997). However, there are no reports on the role that insulin may play in modulating the activity of the es nucleoside transporter for adenosine in vascular smooth muscle cells.
In the present study we have characterized the effect of insulin on adenosine transport in human umbilical artery smooth muscle cells (HUASMCs) isolated from normal or uncontrolled gestational diabetic pregnancies. Our results suggest that adenosine transport is modulated differentially by insulin in these two cell types. Insulin increased adenosine transport in non-diabetic cells via NO and cGMP, but inhibited the diabetes-elevated adenosine transport via activation of adenylyl cyclase. Preliminary accounts of this study have been reported in abstract form (Aguayo et al. 2000a,b).
METHODS
Diabetic patients and newborns
Paired experiments were conducted using umbilical cords from eight full-term normal and eight gestational diabetic pregnancies with normal spontaneous vaginal delivery (Clínica Francesa and Hospital Regional-Concepción, Chile). Written authorization from the hospitals and the patients for use of the umbilical cords was obtained. All diabetic patients were normotensive, exhibited no albuminuria or glucosuria and had a mean glycosylated haemoglobin (HbA1) plasma level of 8.9 ± 0.4 % (HbA1 for normal patients, 3.9 ± 0.4 %). Patients selected for this study were diagnosed as gestational diabetics when they were presented to the labour ward and the mean gestational period was 38 ± 0.5 weeks (normal patients, 38 ± 0.4 weeks). None of the patients received treatment for the disease before labour. Newborns did not present with symptoms of asphyxia, and the mean newborn weight was 2490 g (range 1910-3117 g) or 4201 g (range 3012-5843 g) for normal or gestational diabetic pregnancies, respectively. The mean blood glucose concentration in the umbilical vein was higher for gestational diabetes compared with normal pregnancies (3.7 ± 0.4 vs. 2.2 ± 0.3 mm, respectively), but was similar in the umbilical artery (2.1 ± 0.4 vs. 2.5 ± 0.3 mm, respectively).
Smooth muscle cell culture
Explants from human umbilical artery were obtained from normal or gestational diabetes full-term pregnancies and cultured in medium 199 (M199) containing 5 mmd-glucose, 3.2 mml-glutamine, 10 % fetal calf serum, 100 i.u. ml−1 penicillin-streptomycin, at 37 °C in a 5 % CO2 atmosphere (Aguayo & Sobrevia, 2000). Confluent second passage cells were resuspended (104 cells ml−1) and plated into 96- or 24-well plates, and 24 h prior to an experiment the incubation medium was changed to one free of fetal calf serum. Cells were confirmed as smooth muscle by their typical multilayered ‘hill and valley’ morphology and positive immunofluorescence staining with a monoclonal antibody against human α-smooth muscle actin or desmin. Cell viability was determined by trypan blue dye exclusion. Less than 1 % of smooth muscle cells took up the dye under control or experimental conditions, and the total protein content of cell wells was not altered by any of the treatments used (not shown).
Adenosine transport experiments
Transport assays were performed in confluent second passage cells rinsed with warmed (37 °C) Krebs solution (mm): NaCl 131, KCl 5.6, NaHCO3 25, NaH2PO4 1, CaCl2 2.5, MgCl2 1, d-glucose 5, Hepes 20 (pH 7.4), containing 100 μml-arginine as described previously (Montecinos et al. 2000; Aguayo & Sobrevia, 2000). Cells were then preincubated for 30 min at 22 °C in the same medium or medium containing the es transport inhibitor nitrobenzylthioinosine (NBMPR, 0.01-10 μm). After removal of the preincubation medium, inward fluxes of [3H]adenosine (22 °C) were determined by the addition of 100 μl of medium containing [3H]adenosine (4 μCi ml−1) and d-[14C]mannitol (0.05-0.4 μCi, an extracellular marker). Kinetics of adenosine transport were measured under similar conditions in cells incubated with increasing concentrations of adenosine (0.19-500 μm) for periods of 20 s at 22 °C in Krebs solution. Uptake of adenosine was terminated by removal of the uptake medium 1 s before the addition of 200 μl of ice-cold Krebs containing 10 μm NBMPR. The cell monolayers were rinsed with a further three washes of ice-cold stop solution. Radioactivity associated with monolayers at time zero was determined by exposing the cell layer simultaneously to radiolabelled medium and ice-cold stop solution.
In order to determine the effect of insulin, cells were incubated in M199 with increasing concentrations of human insulin (0.001-10 nm) added during the last 8 h of a 24 h incubation period (see Sobrevia et al. 1998). In some experiments, smooth muscle cells were incubated with the phosphatidylinositol 3-kinase (PI3-kinase) inhibitor wortmannin (10 nm, 8 h), to establish whether the effect of insulin was mediated by the PI3-kinase signalling pathway. Cycloheximide (1 μm, 8 h) was also added to the incubation medium to establish whether the effect of insulin was dependent on protein synthesis.
The effect of NO on adenosine transport was assayed by incubating the smooth muscle cells with the inhibitor of NO synthase NG-nitro-l-arginine methyl ester (l-NAME, 100 μm, 30 min), or the NO donor S-nitroso-N-acetyl-l,d-penicillamine (SNAP, 100 μm, 30 min). Adenosine transport was also measured in cells exposed to the membrane-permeable forms of cGMP (dibutyryl cGMP, 100 nm, 30 min) or cAMP (dibutyryl cAMP, 1 μm, 30 min). Concentrations of drugs were selected from dose-dependence curves (not shown) for inhibition or stimulation of 10 μm adenosine transport (Aguayo & Sobrevia, 2000).
For determination of total cell protein content, smooth muscle cells were exposed to 1 n KOH (100 μl well−1) for 60 min at room temperature. Aliquots of KOH cell extracts (1-10 μl) or bovine serum albumin (BSA) standards were mixed with 100 μl Coomassie blue protein reagent diluted 1:10, and absorbances were measured at 620 nm in a Multiskan plate reader (Flow Laboratories, Irvine, UK) as previously described (Sobrevia et al. 1995; Aguayo & Sobrevia, 2000). Remaining KOH cell extracts were exposed to 100 μl formic acid and radioactivity was determined by liquid scintillation counting. Uptake values were corrected for 3H in the extracellular space and expressed as pmoles per microgram of protein. Kinetic data were analysed using the computer programs Enzfitter and Ultra Fit (Elsevier, Biosoft) and fitted best by a Michaelis-Menten equation.
Binding of nitrobenzylthioinosine (NBMPR)
The nucleoside analogue NBMPR binds specifically to and inhibits adenosine transport by es transporters, but is not transported itself, and therefore can be used to estimate the surface density of es transporters in intact cells (Paterson et al. 1981). Smooth muscle cells in triplicate wells were cultured in M199 containing 5 mmd-glucose for 24 h in the absence or in the presence of insulin (1 nm), added to the culture medium for the last 8 h of this period. After this period, cells were prepared for [3H]NBMPR equilibrium binding studies following two rinses with Krebs solution followed by a 15 min incubation at 22 °C in Krebs or insulin-containing Krebs solution, in the presence or absence of 10 μm NBMPR (Sobrevia et al. 1994; Montecinos et al. 2000; Aguayo & Sobrevia, 2000). Smooth muscle cells were then incubated with 800-1500 μl of [3H]NBMPR or [3H]NBMPR plus 10 μm NBMPR for 30 min at 22 °C in the absence or in the continuous presence of insulin. At the end of the incubation period, 200 μl of the supernatant were retained for radioactivity determinations and the cells were rapidly rinsed three times with ice-cold phosphate-buffered saline (PBS). Radioactivity associated with the cells was determined as described above for the transport assays. Specific binding was defined as the difference in the binding in the presence and absence of 10 μm NBMPR.
Intracellular levels of cGMP and cAMP
Confluent cells in 24-well plates were preincubated for 25 min with Krebs solution (37 °C) containing 100 μml-arginine and the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX, 0.05-0.5 mm), in the absence or in the presence of insulin (1 nm). The preincubation medium was removed and 500 μl Krebs solution containing IBMX or IBMX and 1 nm insulin were added to the wells for a further 5 min (37 °C). Cells were then placed on ice and incubated with 0.1 n HCl (1 ml well−1, 60 min) and 800 μl of the HCl cell extract were stored at −20 °C for radioimmunoassay of cGMP or cAMP following acetylation as previously described (Sobrevia et al. 1995; Aguayo & Sobrevia, 2000). cAMP levels were also determined in cells incubated with the activator forskolin (1 μm, 30 min) or the inhibitor SQ-22536 (100 μm, 30 min).
Conversion of l-[3H]arginine into l-[3H]citrulline
For determinations of conversion of l-[3H]arginine into l-[3H]citrulline, smooth muscle cells cultured in 24-well plates in M199 containing 5 mmd-glucose (24 h) were incubated with 100 μml-[3H]arginine (4 μCi ml−1, 30 min, 37 °C) in the absence or in the presence of 100 μml-NAME and/or insulin (1 nm). Aliquots of 100 g of the cation ion-exchange resin Dowex50W (50X8-200) in its protonated form were converted into the sodium ion form by incubation with 200 ml 1 n NaOH. After calibration of the Dowex column, 200 μl of smooth muscle cells digested in 95 % formic acid (≈40 μg protein) were passed through the column (0.5 g Dowex50W) and eluates of H2O and NaOH were collected (Contreras et al. 1997; Sobrevia et al. 1998; Montecinos et al. 2000; Aguayo & Sobrevia, 2000). The amount of l-[3H]citrulline produced after 30 min incubation with l-[3H]arginine was determined in the H2O eluate and expressed as disintegrations per minute per 106 cells per 30 min (d.p.m. (μg protein)−1 (30 min)−1).
Immunoblot for iNOS
Confluent (passage 2) smooth muscle cells in 24-well plates were deprived of serum for 24 h in the absence or presence of 1 nm insulin (added for the last 8 h of the 24 h incubation period). Smooth muscle cells were washed twice with Krebs buffer (37 °C) and lysed in buffer containing 63.5 mm Tris-HCl (pH 6.8), 10 % glycerol, 2 % sodium dodecyl sulphate (SDS), 1 mm sodium orthovanadate, 1 mm 4-(2-aminoethyl)benzenesulphonyl fluoride (AEBSF), 50 μg ml−1 leupeptin and 5 % 2-mercaptoethanol. Protein cell lysates were boiled for 3 min and equal amounts (20-30 μg) were separated by 13 % polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to Immobilon-P, 0.45 μm pore size polyvinylidene difluoride (PVDF) membranes which were blocked for 3 h in bovine serum albumin (BSA, 3 % in Tris buffered saline Tween (TBST) containing 50 mm Tris-HCl, 150 mm NaCl, 0.02 % (v/v) Tween 20, pH 7.4) and probed with a primary polyclonal rabbit anti-iNOS (1:100). Membranes were then washed (× 6) in TBST and incubated for 1 h in TBST-0.2 % BSA containing primary horseradish peroxidase-conjugated goat anti-rabbit antibody (1: 1000). Protein bands were detected using enhanced chemiluminescence (ECL) detection reagents (Montecinos et al. 2000; Aguayo & Sobrevia, 2000).
Reagents and radioactive molecules
Fetal calf serum was purchased from Gibco and all other reagents, except the following, were from Sigma. Bradford protein reagent was from BioRad Laboratories (Herts, UK). [2,8,5′-3H]Adenosine (60 Ci mmol−1) and d-[1-14C]mannitol (49.3 mCi mmol−1) were from NEN (Dreieich, FRG). [3H]NBMPR was a gift from Dr S. M. Jarvis (University of Kent, UK). Agonists and antagonists were obtained from Research Biochemicals International (UK). iNOS antibody was obtained from Transduction Laboratories (USA).
Statistics
Values are expressed as means ±s.e.m., where n indicates the number of different umbilical artery smooth muscle cell cultures with 3-6 replicate measurements per experiment. Statistical analyses were carried out on raw data using the Peritz F multiple means comparison test (Harper, 1984). Student's t test was applied for unpaired data and P < 0.05 was considered statistically significant.
RESULTS
Effect of insulin on adenosine transport
When smooth muscle cells, derived from non-diabetic pregnancies, were incubated in the presence of insulin (8 h), NBMPR-sensitive adenosine (10 μm) transport was increased in a concentration-dependent manner, with a half-maximal effect (K1/2) reached at 0.33 ± 0.02 nm insulin. By contrast, in smooth muscle cells derived from gestational diabetic pregnancies, where transport was significantly higher than in non-diabetic cells, insulin significantly attenuated (K1/2= 0.01 ± 0.002 nm) the diabetes-induced stimulation of adenosine transport (Fig. 1A). When smooth muscle cells were incubated with wortmannin (10 nm), an inhibitor of PI3-kinase, the stimulatory and inhibitory effects of insulin in non-diabetic and diabetic cells, respectively, were abolished (Fig. 1B). Kinetic experiments revealed that in non-diabetic smooth muscle cells stimulation of adenosine transport by insulin was associated with an increase of the maximal rate of transport (Vmax), with negligible changes in the apparent Km, whereas insulin decreased the diabetes-mediated increase in Vmax for adenosine transport to values in non-diabetic cells (Table 1).
Figure 1. Effect of insulin on adenosine transport in human umbilical artery smooth muscle cells from non-diabetic or gestational diabetic pregnancies.
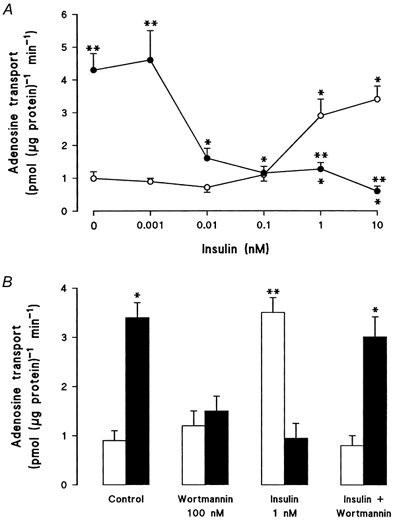
Overall transport of 10 μm adenosine (22 °C, 20 s) was determined in human umbilical artery smooth muscle cells (passage 2) isolated from non-diabetic (^, □) or diabetic (•, ▪) pregnancies. A, adenosine transport was determined after preincubation of cells for 8 h with varying concentrations of insulin (*P < 0.05 vs. corresponding control values; **P < 0.05 vs. corresponding values in non-diabetes). B, adenosine transport was determined after preincubation of cells for 8 h in the absence (Control) or in the presence of insulin, wortmannin, or insulin + wortmannin (*P < 0.05 vs. values in the presence of insulin or wortmannin in diabetic cells; **P < 0.05 vs. corresponding values in non-diabetes). Values are means ±s.e.m. of experiments in 3-9 different cell cultures.
Table 1.
Kinetic parameters for adenosine transport in human umbilical artery smooth muscle cells
| Km | Vmax | |
|---|---|---|
| Non-diabetic | ||
| Control | 163 ± 26 | 17 ± 5 |
| l-NAME | 154 ± 22 | 19 ± 8 |
| Cycloheximide | 156 ± 35 | 15 ± 7 |
| SQ-22536 | 178 ± 17 | 74 ± 12* |
| Insulin | 185 ± 18 | 52 ± 12* |
| Insulin +l-NAME | 207 ± 24 | 19 ± 3† |
| Insulin + cycloheximide | 217 ± 32 | 33 ± 7* |
| Insulin + SQ-22563 | 178 ± 33 | 48 ± 6* |
| Diabetic | ||
| Control | 196 ± 31 | 59 ± 4* |
| l-NAME | 177 ± 31 | 22 ± 7‡ |
| Cycloheximide | 196 ± 65 | 85 ± 17* |
| SQ-22536 | 182 ± 22 | 63 ± 7* |
| Insulin | 177 ± 15 | 17 ± 3‡ |
| Insulin +l-NAME | 161 ± 12 | 16 ± 6‡ |
| Insulin + cycloheximide | 187 ± 23 | 17 ± 8‡ |
| Insulin + SQ-22536 | 196 ± 33 | 59 ± 12*§ |
Adenosine transport kinetics in non-diabetic or gestational diabetic smooth muscle cells was determined for NBMPR-sensitive adenosine transport. Smooth muscle cells were cultured in medium 199 in the absence (Control) or in the presence of insulin (1nM, 8 h), NG-nitro-L-arginine methylester (L-NAME, 100 μm, 30 min), cycloheximide (1μm, 8 h) or SQ-22536 (100 μm, 30 min), and transport was assayed in Krebs solution. Values for Km and Vmax are in μm and pmol (μg protein)−1 min−1, respectively, and are the means ±s.e.m. of experiments in 9 different cell cultures;
P < 0.05 vs. control values in non-diabetic cells,
P <0.05 vs. values in non-diabetic cells treated with insulin,
P < P < 0.05 vs. control values in non-diabetic cells,
P < P < 0.05 vs. values in diabetic cells treated with insulin.
In order to determine whether the effect of insulin on the Vmax for adenosine transport was due to changes in the number of available adenosine transport sites, equilibrium binding of [3H]NBMPR was determined. Table 2 shows that preincubation of non-diabetic smooth muscle cells with 1 nm insulin for 8 h increased by 1.7-fold the maximal binding (Bmax) of [3H]NBMPR, with no significant changes in the Kd. By contrast, insulin treatment of diabetic smooth muscle cells caused a 64 % reduction in the Bmax for [3H]NBMPR. Scatchard plots of the specific binding data were linear (not shown), indicating a single population of high-affinity NBMPR binding sites in non-diabetic and diabetic smooth muscle cells in the absence or in the presence of insulin.
Table 2.
Kinetic parameters for specific [3H]NBMPR binding in human umbilical artery smooth muscle cells
| Kd | Bmax | |
|---|---|---|
| Non-diabetic | ||
| Control | 0.16 ± 0.03 | 0.66 ± 0.07 |
| l-NAME | 0.12 ± 0.02 | 0.55 ± 0.01 |
| Cycloheximide | 0.12 ± 0.02 | 0.7 ± 0.02 |
| SQ-22536 | 0.14 ± 0.03 | 1.3 ± 0.02* |
| Insulin | 0.15 ± 0.01 | 1.1 ± 0.1* |
| Insulin +l-NAME | 0.17 ± 0.03 | 0.71 ± 0.09† |
| Insulin + cycloheximide | 0.17 ± 0.03 | 0.64 ± 0.04† |
| Insulin + SQ-22563 | 0.16 ± 0.01 | 1.1 ± 0.01* |
| Diabetic | ||
| Control | 0.15 ± 0.03 | 1.62 ± 0.16* |
| l-NAME | 0.12 ± 0.02 | 0.75 ± 0.11‡ |
| Cycloheximide | 0.13 ± 0.01 | 1.2 ± 0.1* |
| SQ-22536 | 0.14 ± 0.01 | 1.5 ± 0.09* |
| Insulin | 0.18 ± 0.01 | 0.6 ± 0.13‡ |
| Insulin +l-NAME | 0.10 ± 0.03 | 0.75 ± 0.09*§ |
| Insulin + cycloheximide | 0.11 ± 0.03 | 0.59 ± 0.03‡ |
| Insulin + SQ-22536 | 0.13 ± 0.01 | 1.50 ± 0.2*§ |
Kinetic parameters for equilibrative [3H]NBMPR binding (30 min, 22°C) were determined in smooth muscle cells incubated in medium 199 in the absence (Control) or in the presence of insulin (1nm, 8 h), NG-nitro-L-arginine methylester (l-NAME, 100 μm, 30 min), cycloheximide (1μm, 8 h) or SQ-22536 (100 μm, 30 min), and binding was assayed in Krebs solution. Values for Kd and Bmax are in nm and fmol (μg protein)−1, respectively, and are the means ±s.e.m. of experiments in 3–4 different cell cultures;
P < 0.05 vs. corresponding control values in non-diabetic cells,
P< P < 0.05 vs. values in non-diabetic cells treated with insulin,
P< P < 0.05 vs. corresponding control values in non-diabetic cells,
P < 0.05 vs. values in diabetic cells treated with insulin.
Involvement of nitric oxide in the effect of insulin on adenosine transport
The effect of insulin on NO synthesis was assayed by measuring intracellular cGMP levels or the formation of l-[3H]citrulline from l-[3H]arginine in the absence or presence of l-NAME. In non-diabetic smooth muscle cells, insulin (1 nm, 8 h) increased cGMP levels (Fig. 2A) and the formation of l-[3H]citrulline (Fig. 2B) by 2.5- and 2.7-fold, respectively, an effect that was completely blocked by l-NAME. In diabetic cells, the basal levels of cGMP and l-[3H]citrulline formation were significantly elevated (to levels similar to those produced by insulin stimulation of non-diabetic cells), and these levels were not further increased by insulin.
Figure 2. Effect of insulin on intracellular cGMP and l-citrulline formation in human umbilical artery smooth muscle cells from non-diabetic (□) or gestational diabetic (▪) pregnancies.
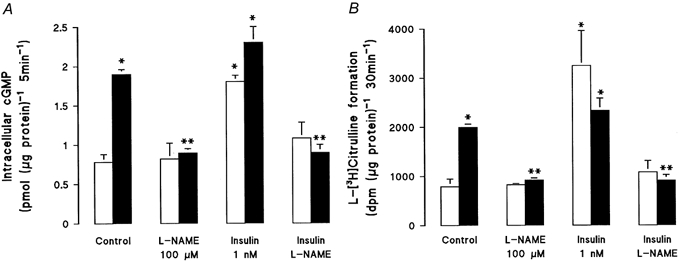
Cells were incubated (8 h) with medium 199 in the absence (Control) or in the presence of insulin and/or NG-nitro-l-arginine methyl ester (l-NAME), and then intra§cellular cGMP (in the presence of 0.5 mm isobutylmethylxanthine; A) or l-[3H]citrulline formation from l-[3H]arginine (30 min, 37 °C; B) was measured in cells incubated in Krebs solution containing 100 μml-arginine. Values are the means ±s.e.m. of experiments in 9 different cell cultures. *P < 0.05 vs. values in non-diabetic cells; **P < 0.05 vs. Control and insulin values in diabetic cells.
The stimulatory effect of 1 nm insulin on adenosine transport and NBMPR binding in non-diabetic smooth muscle cells was inhibited when cells were co-incubated with the NO synthase inhibitor l-NAME (Fig. 3, Tables 1 and 2). The pre-existing enhanced adenosine transport in diabetic cells was reduced to control levels by l-NAME, by insulin, or by their combination. The stimulatory effect of insulin on adenosine transport in non-diabetic cells was mimicked by 100 nm dibutyryl cGMP (dbcGMP, 3.1 ± 0.3 pmol (μg protein)−1 min−1, n = 4), as previously reported in this cell type (Aguayo & Sobrevia, 2000), and the inhibition of adenosine transport induced by insulin in diabetic smooth muscle cells was completely reversed by dbcGMP (Fig. 3). These results suggest that increased levels of NO, and hence cGMP, are necessary for the activation of adenosine transport by insulin in non-diabetic human umbilical artery smooth muscle cells. In diabetic cells, the inhibitory action of insulin on adenosine transport is presumably not cGMP mediated, and can be reversed by supplying cGMP exogenously. We have previously shown that the NO donor S-nitroso-N-acetyl-l,d-penicillamine (SNAP, 100 μm, 8 h) stimulates adenosine transport in non-diabetic smooth muscle cells, but does not alter the diabetes-induced increase in adenosine transport (see Aguayo & Sobrevia, 2000). Interestingly, SNAP-derived NO had no effect on insulin-stimulated adenosine transport in non-diabetic cells (2.7 ± 0.2 pmol (μg protein)−1 min−1, n = 6), but blocked the inhibition by insulin of adenosine transport in diabetic cells (3.7 ± 0.5 pmol (μg protein)−1 min−1, P < 0.05, n = 6).
Figure 3. Involvement of nitric oxide in the modulation of adenosine transport by insulin in human umbilical artery smooth muscle cells.
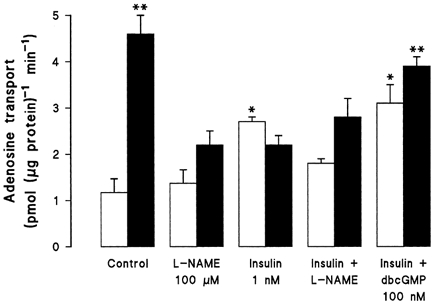
Adenosine transport (10 μm, 20 s, 22 °C) was measured in smooth muscle cells isolated from non-diabetic (□) or gestational diabetic (▪) pregnancies. Cells were pretreated for 8 h with insulin in the absence or presence of NG-nitro-l-arginine methyl ester (l-NAME). In parallel studies smooth muscle cells were incubated with dibutyryl cGMP (dbcGMP) for the last 30 min incubation period with insulin. Values are the means ±s.e.m. of experiments in 4-9 different cell cultures. *P < 0.05 vs. corresponding values in Control, l-NAME, or insulin +l-NAME; **P < 0.05 vs. corresponding values in l-NAME, insulin, or insulin +l-NAME.
Immunoblot studies demonstrated that insulin increased the expression of iNOS protein levels in non-diabetic cells, whereas iNOS levels were not altered in diabetic smooth muscle cells (Fig. 4). It is worth noting that basal protein levels for iNOS were similar in both cell types, despite the basally elevated NOS activity in diabetic cells. Stimulation of the Vmax for adenosine transport by insulin in non-diabetic smooth muscle cells was inhibited by coincubation (8 h) of cells with 1 μm cycloheximide (Table 1). These results indicate that protein synthesis is required for the actions of this hormone on adenosine transport in non-diabetic smooth muscle. In contrast, the inhibitory effect of insulin on the increased Vmax for adenosine transport in diabetic smooth muscle cells was unaffected by cycloheximide (Table 1). Again, parallel results were seen for [3H]NBMPR binding (Table 2).
Figure 4. Immunoblot of inducible nitric oxide synthase in human umbilical artery smooth muscle cells after stimulation with insulin.
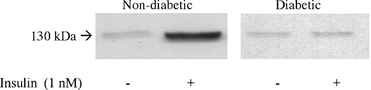
Smooth muscle cells isolated from non-diabetic or gestational diabetic pregnancies were deprived of serum for 24 h, washed and incubated with culture medium in the absence (-) or in the presence (+) of insulin (8 h). Data are representative of similar blots obtained in 3 different cell cultures.
Involvement of cAMP in the effect of insulin on adenosine transport
Incubation of non-diabetic cells with the adenylyl cyclase inhibitor SQ-22536 (Goldsmith & Abrams, 1992) led to a 3.1-fold increase in the Vmax for adenosine transport, with negligible changes in the apparent Km. The stimulatory effect of insulin on the Vmax for adenosine transport in non-diabetic cells was not altered in the presence of SQ-22536 (Table 1). However, the inhibitory action of insulin on the increased Vmax for adenosine transport in diabetic cells was blocked by SQ-22536. In parallel experiments, insulin increased the basal intracellular level of cAMP in diabetic, but not in non-diabetic smooth muscle cells (Fig. 5). This effect of insulin on cAMP levels was prevented by SQ-22536. The stimulatory effect of insulin on adenosine transport in non-diabetic smooth muscle cells was also inhibited by dibutyryl cAMP (dbcAMP, 0.3 ± 0.1 pmol (μg protein)−1 min−1, P < 0.05, n = 4). By contrast, the insulin-induced inhibition of stimulated rates of adenosine transport in diabetic cells was not affected by dbcAMP (0.2 ± 0.1 pmol (μg protein)−1 min−1, P 0.05, n = 4). When non-diabetic smooth muscle cells were incubated with SQ-22536, the Bmax for [3H]NBMPR binding was increased, with negligible changes in the apparent Kd for [3H]NBMPR (Table 2). In non-diabetic cells, insulin-stimulated binding of [3H]NBMPR was unaffected by SQ-22536, whereas the inhibitory action of insulin on the increased Bmax for [3H]NBMPR in diabetic cells was abolished (Table 2).
Figure 5. Effect of insulin on intracellular cAMP in human umbilical artery smooth muscle cells.
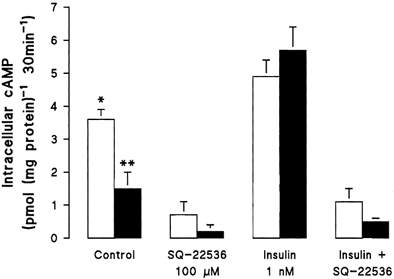
Cells were exposed for 8 h to insulin in the absence or presence of SQ-22536, and then intracellular cAMP (in the presence of 0.5 mm isobutylmethylxanthine, 30 min, 37 °C) was determined by radioimmunoassay in smooth muscle cells isolated from non-diabetic (□) or gestational diabetic (▪) pregnancies. Values are the means ±s.e.m. of experiments in 8 different cell cultures. *P < 0.05 vs. values in non-diabetic cells (except in the presence of insulin); **P < 0.05 vs. all values in diabetic cells, and Control and insulin in non-diabetic cells.
DISCUSSION
Effect of insulin on adenosine transport and NBMPR binding
This study has established that physiological concentrations of insulin modulate the activity of the equilibrative, NBMPR-sensitive adenosine transport (system es) in human umbilical artery smooth muscle cells (HUASMCs). Adenosine transport was increased by insulin in HUASMCs isolated from non-diabetic pregnancies. In contrast, the pre-existing enhanced transport capacity for adenosine in cells isolated from gestational diabetic pregnancies, which we have previously reported (Aguayo & Sobrevia, 2000), was reduced by insulin. The range of concentrations of insulin required to stimulate adenosine transport in non-diabetic HUASMCs (K1/2≈ 0.3 nm) was similar to that required to stimulate the Vmax for l-arginine transport, NO synthesis, and protein and DNA turnover in human umbilical vein endothelial cells (1 nm; Sobrevia et al. 1998). Diabetic cells appeared to be more sensitive to insulin, with the K1/2 value for reduction of adenosine transport being ≈ 0.01 nm.
The stimulatory action of insulin on adenosine transport in non-diabetic cells was associated with a higher Vmax. This result complements previous reports that insulin stimulates synthesis and increases expression of Na+-dependent uridine transporters in rat liver parenchymal cells (Gómez-Angelats et al. 1996) and the human astrocytoma cell line U-373 MG (Kum et al. 1989), and describing Na+-independent, NBMPR-sensitive adenosine uptake in rat hippocampal slices (Morrison et al. 1992). It has also been reported that insulin increased mRNA levels for Na+-dependent adenosine transporters in cultures of IEC-6 rat intestinal epithelial cells (Jakobs et al. 1990), and enhanced gene expression of the high-affinity cationic amino acid transport system y+/CAT-1 in rat hepatocytes (Wu et al. 1994).
The stimulatory action of insulin on the Vmax for adenosine transport in non-diabetic HUASMCs was blocked by cycloheximide. Maximal binding of the es transport inhibitor [3H]NBMPR was also significantly increased, indicating that insulin increases the number of adenosine transporter proteins in the plasma membrane (insulin: 303 000 ± 27 000 vs. control: 182 000 ± 18 000 transporters cell−1, P < 0.05, n = 5), via a protein synthesis-dependent mechanism. The increase in membrane transporters induced by insulin was paralleled by an increase in the turnover number (i.e. Vmax/number of transporters per cell) for adenosine (503 ± 42 vs. 787 ± 67 adenosine molecules transporter−1 s−1, for control and insulin, respectively, P < 0.05, n = 5). Insulin did not alter significantly the apparent Km or Kd for adenosine transport or [3H]NBMPR binding, respectively. Thus, changes in adenosine transport are not due to alterations in the intrinsic properties of es transporters. However, an increase in the number of transporters and in their activity seems necessary to account for the ≈3-fold increase in adenosine transport induced by insulin in non-diabetic cells. In contrast, in HUASMCs from diabetic pregnancies, insulin reduced the diabetes-stimulated Vmax for adenosine transport via a protein synthesis-independent mechanism. Incubation of cells with insulin similarly reversed the diabetes-induced increase in Bmax for [3H]NBMPR binding (from 450 000 ± 45 000 to 165 000 ± 34 000 transporters cell−1, P < 0.05, n = 5).
Possible mechanisms for up-regulation of adenosine transport and NBMPR binding in non-diabetic cells exposed to insulin include an increase in the synthesis or a decreased internalization of the nucleoside transporters (see review, Baldwin et al. 1999). Recycling of NBMPR-sensitive nucleoside transporters has been demonstrated in cultured chromaffin cells (Torres et al. 1992) and is similar to the modulation of insulin-dependent GLUT-4 transporters in several other cell types (see Czech & Corvera, 1999). Similar mechanisms could account for the adaptive up-regulation of adenosine transport and NBMPR binding seen in diabetic cells.
Involvement of nitric oxide in the action of insulin on adenosine transport
Insulin can attenuate the contraction of human vascular smooth muscle by increasing cGMP levels and the expression of iNOS (Kahn et al. 1997; Begum et al. 1998; Trovati et al. 1999). Moreover, NO can modulate the transport of nucleosides (Redzic et al. 1997; Soler et al. 2000; Montecinos et al. 2000; Aguayo & Sobrevia, 2000) and amino acids (Li et al. 1999; Ogonowski et al. 2000). In the present study, the protein level and activity of iNOS in non-diabetic smooth muscle cells was increased by insulin, in parallel with the increase in adenosine transport and NBMPR binding. This latter action of insulin was prevented by the NO synthase inhibitor l-NAME, showing that stimulation of adenosine transport in non-diabetic cells is mediated at least in part by NO. Begum et al. (1998) showed that insulin-stimulated NO production in human vascular smooth muscle cells led to rapid activation of mitogen-activated protein kinase phosphatase-1 (MKP-1) activity, which could therefore be one of the signals involved in regulating adenosine transport. Insulin-stimulated rates of adenosine transport in non-diabetic cells were not further augmented by dbcGMP or SNAP-derived NO, suggesting that the activation of soluble guanylyl cyclase by NO in response to insulin was sufficient to maximize effects on adenosine transport. Insulin has recently been reported to have a rapid stimulatory effect (5 min) on a constitutive (endothelial-like) NOS in human vascular smooth muscle cells derived from microarterioles (Trovati et al. 1999). Thus, insulin-induced vasodilatation in vivo may not be entirely endothelium dependent, as rapid activation of constitutive NOS followed by synthesis of the inducible NOS in vascular smooth muscle may maintain elevated NO levels in response to insulin in non-diabetic smooth muscle cells from microarterioles. However, the latter is unlikely in human umbilical artery smooth muscle cells, since in parallel studies we have amplified iNOS, but not endothelial NOS (eNOS) mRNAs by reverse transcription polymerase chain reaction (data not shown).
In HUASMCs, gestational diabetes led to pre-existing increased basal intracellular cGMP and l-citrulline production, which were not associated with a detectable increase in iNOS protein, and were not further enhanced by incubation of cells with insulin. These results suggest that smooth muscle cells from diabetic pregnancies are adaptively modified in a manner that elevates basal iNOS activity (perhaps by altered levels of substrate or cofactors), but additionally are less able to respond to insulin with increased synthesis of iNOS. Though we have not investigated the mechanisms involved, the latter finding is consistent with the recent demonstration by Begum & Ragolia (2000) that 24 h treatment of human vascular smooth muscle cells with insulin in the presence of high d-glucose, in contrast to the effects of treatment with insulin alone noted above (Begum et al. 1998), reduced iNOS expression (and consequently MKP-1 expression) by a pathway involving p38 mitogen-activated protein kinase.
Signalling cascades activated by insulin
In human vascular smooth muscle cells, as in many other cell types, insulin activates the PI3-kinase signalling pathway (Begum et al. 1998). Since the inhibitor of PI3-kinase wortmannin blocked the stimulatory action of insulin on adenosine transport in non-diabetic smooth muscle cells, it is likely that insulin modulates adenosine transport via activation of PI3-kinase. Begum et al. (1998) showed that insulin-stimulated NO production in these cells is blocked by wortmannin, which together with our results implies that PI3-kinase acts via NO synthase to upregulate adenosine transport. In addition, wortmannin blocked the stimulatory effect of diabetes on adenosine transport, implicating PI3-kinase in the effect of diabetes. The effect of diabetes is again presumably via NO synthase since this was elevated in the diabetic HUASMCs, its inhibition downregulated adenosine transport, and the inhibitory effect of insulin on adenosine transport in diabetic cells was blocked when cells were exposed to SNAP-derived NO.
Incubation of non-diabetic cells with the adenylyl cyclase inhibitor SQ-22536 increased adenosine transport and NBMPR binding but had no effect on the stimulation of transport and NBMPR binding by insulin (Tables 1 and 2), suggesting that the basal activity of adenylyl cyclase downregulates the availability of adenosine transporters in non-diabetic HUASMCs. This agrees with our previous results on non-diabetic HUASMCs (Aguayo & Sobrevia, 2000), where we found that adenosine transport was downregulated following direct activation of adenylyl cyclase by forskolin. It is also consistent with other reports showing that cAMP acts as a downregulator of adenosine transport in S49 lymphoma cells (Nagy et al. 1990), NG108-15 neuroblastoma × glioma cells (Coe et al. 1996), and undifferentiated Neuro-2A cells (Sen et al. 1999).
Trovati et al. (1999) noted a transient (within 30 min) increase in cAMP levels in response to insulin in non-diabetic human arteriolar smooth muscle cells, although levels returned to basal values after 3 h. In our study, insulin (8 h) increased intracellular levels of cAMP only in diabetic HUASMCs, suggesting that its effect on cAMP is enhanced and prolonged in diabetic cells by comparison with non-diabetic cells. SQ-22536 blocked this rise in cAMP and the inhibitory effect of insulin on adenosine transport and NBMPR binding, demonstrating that cAMP is one of the signalling molecules stimulated by insulin to downregulate adenosine transporters in diabetic smooth muscle cells.
In conclusion, our study has established that adenosine transport in human umbilical artery smooth muscle cells in vitro is upregulated by insulin in non-diabetic smooth muscle cells through a mechanism that involves increased iNOS expression, NO synthesis and cGMP. However, insulin downregulates diabetes-stimulated adenosine transport via activation of adenylyl cyclase and elevated levels of cAMP (see Fig. 6). Modulation of adenosine transport by insulin was associated with changes in the number of nucleoside transporters in the plasma membrane of smooth muscle cells. Adenosine transport is thus regulated differentially by insulin in arterial smooth muscle cells derived from non-diabetic or uncontrolled gestational diabetic pregnancies. It is worth noting that umbilical artery smooth muscle cells isolated from gestational diabetics maintain their phenotypic difference in adenosine transport under identical culture conditions to the non-diabetic cells. While we do not yet understand the mechanisms involved, we presume that it represents an adaptive response to components of the altered extracellular milieu to which the cells are exposed in utero, which may include periodic episodes of severe hyperglycaemia as well as hyperinsulinaemia.
Figure 6. Postulated cell signalling mechanisms by which insulin modulates adenosine transport in smooth muscle cells isolated from umbilical arteries obtained from non-diabetic and gestational diabetic pregnancies.
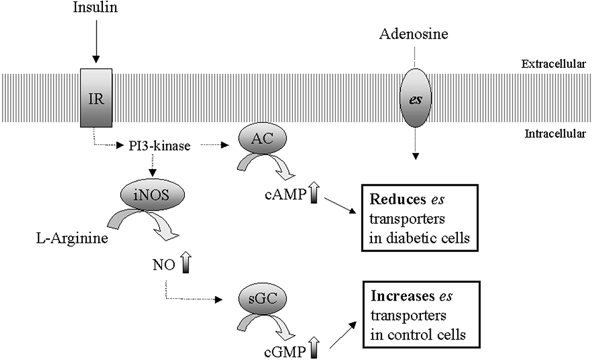
Adenosine is incorporated into human umbilical artery smooth muscle cells by equilibrative, Na+-independent nitrobenzylthioinosine (NBMPR)-sensitive nucleoside transporters (system es or hENT-1). In non-diabetic and diabetic cells the effect of insulin on es transport involves signalling through PI3-kinase. Thereafter, the main control mechanisms diverge between non-diabetic and diabetic cells. In non-diabetic cells, system es is increased by insulin by a pathway involving increased expression and activity of the inducible isoform of nitric oxide synthase (iNOS), leading to increased intracellular cGMP levels. Inhibition of adenylyl cyclase (AC) increases basal es levels in these cells, but has no effect on insulin-stimulated es levels. By contrast, the tonically elevated levels of es in diabetic cells are reduced by insulin through a pathway involving AC; inhibition of the NO/cGMP pathway also reduces es levels, but does not affect the ability of insulin to reduce es levels in these cells.
Adenosine is produced and released from cells during periods of relative hypoxia, and is an important contributor to the compensatory hyperaemia, due to its interaction with specific receptors on vascular smooth muscle cells (Ralevic & Burnstock, 1998). Although we are not aware of studies directly addressing the question, it is reasonable to expect that the ability of locally released adenosine to cause vasodilatation is modulated by its uptake by the smooth muscle cells. Gestational diabetes-induced adenosine transport in smooth muscle cells (Aguayo & Sobrevia, 2000) may represent an adaptive response to the reported gestational diabetes-induced (Sobrevia et al. 1994) or hyperglycaemia-induced (Montecinos et al. 2000) inhibition of adenosine transport in human endothelium. Thus, the actions of insulin in non-diabetic and diabetic vascular smooth muscle may represent an important cellular mechanism involved in the modulation of extracellular adenosine concentrations in diabetes mellitus.
Acknowledgments
This study was supported by Fondo Nacional de Ciencia y Tecnología (FONDECYT 1000354 and 7000354), Dirección de Investigación (9733871-ID and 9933931-ID)-Universidad de Concepción (Chile), and the Wellcome Trust (UK). C. Aguayo holds a CONICYT (Chile) PhD fellowship. C. Flores and J. Parodi hold Beca Docente University of Concepción MSc fellowships. R. Rojas is a recipient of a University of Concepción fellowship for undergraduate pharmacy studies. We thank the midwives of Clínica Francesa and Hospital Regional-Concepción (Chile) labour wards for the generous supply of umbilical cords, Mrs Susana Rojas for her expert technical assistance, and Miss Isabel Jara for excellent secretarial assistance.
References
- Aguayo C, Flores C, Sobrevia L. Diabetes- and insulin-increased adenosine transport is associated with a higher number of nucleoside transporters in human umbilical artery smooth muscle cells. Journal of Physiology. 2000a;527 P. [Google Scholar]
- Aguayo C, Siow R C M, Mann GE, Pearson JD, Sobrevia L. Regulation of adenosine transport by insulin in human umbilical artery smooth muscle cells from diabetic pregnancies. Journal of Physiology. 2000b;525:17. doi: 10.1111/j.1469-7793.2001.00243.x. P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguayo C, Sobrevia L. Nitric oxide, cGMP and cAMP modulate nitrobenzylthioinosine-sensitive adenosine transport in human umbilical artery smooth muscle cells from gestational diabetes. Experimental Physiology. 2000;85:399–409. [PubMed] [Google Scholar]
- Baldwin SA, Mackey JR, Cass CE, Young JD. Nucleoside transporters: molecular biology and implications for therapeutic development. Molecular Medicine Today. 1999;5:216–224. doi: 10.1016/S1357-4310(99)01459-8. [DOI] [PubMed] [Google Scholar]
- Baron AD. Vascular reactivity. American Journal of Cardiology. 1999;84:25–27. doi: 10.1016/s0002-9149(99)00354-9. [DOI] [PubMed] [Google Scholar]
- Beck DW, Vinters HV, Moore SA, Hart MN, Cancilla PA. Uptake of adenosine by cultured cerebral vascular smooth muscle cells. Journal of Neurochemistry. 1983;41:939–941. doi: 10.1111/j.1471-4159.1983.tb09037.x. [DOI] [PubMed] [Google Scholar]
- Begum N, Ragolia L. High glucose and insulin inhibit VSMC MKP-1 expression by blocking iNOS via p38 MAPK activation. American Journal of Physiology − Cell Physiology. 2000;278:C81–91. doi: 10.1152/ajpcell.2000.278.1.C81. [DOI] [PubMed] [Google Scholar]
- Begum N, Ragolia L, Rienzie J, McCarthy M, Duddy N. Regulation of mitogen-activated protein kinase phosphatase-1 induction by insulin in vascular smooth muscle cells. Evaluation of the role of the nitric oxide signaling pathway and potential defects in hypertension. Journal of Biological Chemistry. 1998;273:25164–25170. doi: 10.1074/jbc.273.39.25164. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Parkinson FE. Effect of adenosine receptor agonists on release of the nucleoside analogue [3H]formycin B from cultured smooth muscle DDT1 MF-2 cells. European Journal of Pharmacology. 1998;346:339–344. doi: 10.1016/s0014-2999(98)00069-7. [DOI] [PubMed] [Google Scholar]
- Coe IR, Yao L, Diamond I, Gordon AS. The role of protein kinase C in cellular tolerance to ethanol. Journal of Biological Chemistry. 1996;271:29468–29472. doi: 10.1074/jbc.271.46.29468. [DOI] [PubMed] [Google Scholar]
- Contreras R, Fuentes O, Mann GE, Sobrevia L. Diabetes and insulin-induced stimulation of l-arginine transport and nitric oxide synthesis in rabbit isolated gastric glands. Journal of Physiology. 1997;498:787–796. doi: 10.1113/jphysiol.1997.sp021902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech MP, Corvera S. Signaling mechanisms that regulate glucose transport. Journal of Biological Chemistry. 1999;274:1865–1868. doi: 10.1074/jbc.274.4.1865. [DOI] [PubMed] [Google Scholar]
- Foga IO, Gaiger JD, Parkinson FE. Nucleoside transport-mediated uptake and release of [3H]adenosine in DDT1 MF-2 smooth muscle cells. European Journal of Physiology. 1996;318:455–460. doi: 10.1016/s0014-2999(96)00720-0. [DOI] [PubMed] [Google Scholar]
- Goldsmith BA, Abrams TW. cAMP modulates multiple K+ currents, increasing spike duration and excitability in Aplysia sensory neurons. Proceedings of the National Academy of Sciences of the USA. 1992;89:11481–11485. doi: 10.1073/pnas.89.23.11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Angelats M, DelSanto B, Mercader J, Ferrer-Martinez A, Felipe A, Casado J, Pastor-Anglada M. Hormonal regulation of concentrative nucleoside transport in liver parenchymal cells. Biochemical Journal. 1996;313:915–920. doi: 10.1042/bj3130915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JF. Peritz' F test: BASIC program of a robust multiple comparison test for statistical analysis of all differences among group means. Computing in Biology and Medicine. 1984;14:437–445. doi: 10.1016/0010-4825(84)90044-1. [DOI] [PubMed] [Google Scholar]
- Jakobs ES, VanOsCorby DJ, Paterson AR. Expression of sodium-linked nucleoside transport activity in monolayer cultures of IEC-6 intestinal epithelial cells. Journal of Biological Chemistry. 1990;265:22210–22216. [PubMed] [Google Scholar]
- Kahn AM, Husid A, Odebunmi T, Allen JC, Seidel CL, Song T. Insulin inhibits vascular smooth muscle contraction at a site distal to intracellular Ca2+ concentration. American Journal of Physiology. 1997;274:E885–892. doi: 10.1152/ajpendo.1998.274.5.E885. [DOI] [PubMed] [Google Scholar]
- Kum W, Cockram CS, Zhu SQ, Teoh R, Young JD. Insulin binding to astrocytoma cells and its effect on uridine incorporation into nucleic acid. Journal of Neurochemistry. 1989;52:242–247. doi: 10.1111/j.1471-4159.1989.tb10923.x. [DOI] [PubMed] [Google Scholar]
- Li H, Marshall ZM, Whorton AR. Stimulation of cystine uptake by nitric oxide: regulation of endothelial cell glutathione levels. American Journal of Physiology. 1999;276:C803–811. doi: 10.1152/ajpcell.1999.276.4.C803. [DOI] [PubMed] [Google Scholar]
- Montecinos VP, Aguayo C, Flores C, Wyatt AW, Pearson JD, Mann GE, Sobrevia L. Regulation of adenosine transport by d-glucose in human fetal endothelial cells: involvement of nitric oxide, protein kinase C and mitogen-activated protein kinase. Journal of Physiology. 2000;529:777–790. doi: 10.1111/j.1469-7793.2000.00777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison PD, MacKinnon MW, Bartrup JT, Skett PG, Stone TW. Changes in adenosine sensitivity in the hippocampus of rats with streptozotocin-induced diabetes. British Journal of Pharmacology. 1992;105:1004–1008. doi: 10.1111/j.1476-5381.1992.tb09092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz M, Sweiry JH, Mann GE. Insulin stimulates cationic amino acid transport activity in the isolated perfused rat pancreas. Experimental Physiology. 1995;80:745–753. doi: 10.1113/expphysiol.1995.sp003883. [DOI] [PubMed] [Google Scholar]
- Nagy LE, Diamond I, Casso DJ, Franklin C, Gordon AS. Ethanol increases extracellular adenosine by inhibiting adenosine uptake via the nucleoside transporter. Journal of Biological Chemistry. 1990;265:1946–1951. [PubMed] [Google Scholar]
- Obata T, Kashiwagi A, Maegawa H, Nishio Y, Ugi S, Hidaka H, Kikkawa R. Insulin signaling and its regulation of system A amino acid uptake in cultured rat vascular smooth muscle cells. Circulation Research. 1996;79:1167–1176. doi: 10.1161/01.res.79.6.1167. [DOI] [PubMed] [Google Scholar]
- Ogonowski AA, Kaesemeyer WH, Jin L, Ganapathy V, Leibach FH, Caldwell RW. Effects of NO donors and synthase agonists on endothelial cell uptake of l-Arg and superoxide production. American Journal of Physiology − Cell Physiology. 2000;278:C136–143. doi: 10.1152/ajpcell.2000.278.1.C136. [DOI] [PubMed] [Google Scholar]
- Olsson RA, Pearson JD. Cardiovascular purinoceptors. Physiological Reviews. 1990;70:761–845. doi: 10.1152/physrev.1990.70.3.761. [DOI] [PubMed] [Google Scholar]
- Paterson A R P, Kolassa N, Cass CE. Transport of nucleoside drugs in animal cells. Pharmacology and Therapeutics. 1981;12:515–536. doi: 10.1016/0163-7258(81)90096-6. [DOI] [PubMed] [Google Scholar]
- Pearson JD, Carleton JS, Hutchings A, Gordon JL. Uptake and metabolism of adenosine by pig aortic and smooth-muscle cells in culture. Biochemical Journal. 1978;170:421–429. doi: 10.1042/bj1700265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poston L, Taylor PD. Endothelium-mediated vascular function in insulin-dependent diabetes mellitus. Clinical Science. 1995;88:245–255. doi: 10.1042/cs0880245. [DOI] [PubMed] [Google Scholar]
- Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacological Reviews. 1998;50:413–492. [PubMed] [Google Scholar]
- Redzic ZB, Segal MB, Markovic ID, Gasic JM, Vidovic V, Rakic LM. The characteristics of basolateral nucleoside transport in the perfused sheep choroid plexus and the effect of nitric oxide inhibition on these processes. Brain Research. 1997;767:26–33. doi: 10.1016/s0006-8993(97)00530-1. [DOI] [PubMed] [Google Scholar]
- Sen RP, Delicado EG, Miras-Portugal MT. Differential modulation of nucleoside transport types in neuroblastoma cells by protein kinase activation. Neuropharmacology. 1999;38:1009–1015. doi: 10.1016/s0028-3908(99)00029-5. [DOI] [PubMed] [Google Scholar]
- Sobrevia L, Cesare P, Yudilevich DL, Mann GE. Diabetes-induced activation of system y+ and nitric oxide synthase in human endothelial cells: association with membrane hyperpolarization. Journal of Physiology. 1995;489:183–192. doi: 10.1113/jphysiol.1995.sp021040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobrevia L, Jarvis SM, Yudilevich DL. Adenosine transport in cultured human umbilical vein endothelial cells is reduced in diabetes. American Journal of Physiology. 1994;267:C39–47. doi: 10.1152/ajpcell.1994.267.1.C39. [DOI] [PubMed] [Google Scholar]
- Sobrevia L, Mann GE. Dysfunction of the endothelial l-arginine-nitric oxide signalling pathway in diabetes and hyperglycaemia. Experimental Physiology. 1997;82:1–30. doi: 10.1113/expphysiol.1997.sp004038. [DOI] [PubMed] [Google Scholar]
- Sobrevia L, Yudilevich DL, Mann GE. Elevated d-glucose induces insulin insensitivity in human umbilical endothelial cells isolated from gestational diabetic pregnancies. Journal of Physiology. 1998;506:219–230. doi: 10.1111/j.1469-7793.1998.219bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler C, Felipe A, Casado FJ, Celada A, Pastor-Anglada M. Nitric oxide regulates nucleoside transport in activated B lymphocytes. Journal of Leukocyte Biology. 2000;67:345–349. doi: 10.1002/jlb.67.3.345. [DOI] [PubMed] [Google Scholar]
- Torres M, Delicado EG, Fideu MD, Miras-Portugal MT. Down-regulation and recycling of the nitrobenzylthioinosine-sensitive nucleoside transporter in cultured chromaffin cells. Biochimica et Biophysica Acta. 1992;1105:291–299. doi: 10.1016/0005-2736(92)90207-3. [DOI] [PubMed] [Google Scholar]
- Trovati M, Massucco P, Mattiello L, Costamagna C, Aldieri E, Cavalot F, Anfossi G, Bosia A, Ghigo D. Human vascular smooth muscle cells express a constitutive nitric oxide synthase that insulin rapidly activates, thus increasing guanosine 3′:5′-cyclic monophosphate and adenosine 3′:5′-cyclic monophosphate concentrations. Diabetologia. 1999;42:831–819. doi: 10.1007/s001250051234. [DOI] [PubMed] [Google Scholar]
- Wu JY, Robinson D, Kung HJ, Hatzoglou M. Hormonal regulation of the gene for the type C ecotropic retrovirus receptor in rat liver cells. Journal of Virology. 1994;68:1615–1623. doi: 10.1128/jvi.68.3.1615-1623.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]