

Abstract
We have used giant patch-clamp recording to investigate the interaction between pH gating and K+-dependent gating in rat Kir1.1 (ROMK) channels heterologously expressed in Xenopus oocytes.
Gating by intracellular protons (pH gating) and extracellular K+ ions (K+-dependent gating) is a hallmark of Kir1.1 channels that mediate K+ secretion and control NaCl reabsorption in the kidney. pH gating is driven by protonation of an intracellular lysine residue (K80 in Kir1.1). K+-dependent gating occurs upon withdrawal of K+ ions from the extracellular side of the channel. Both gating mechanisms are thought to interact allosterically.
K+-dependent gating was shown to be strictly coupled to pH gating; it only occurred when channels were in the pH-inactivated closed state, but not in the open state. Moreover, K+-dependent gating was absent in the non-pH-gated mutant Kir1.1(K80M).
Channels inactivated by K+-dependent gating were reactivated upon addition of permeant ions to the extracellular side of the membrane, while impermeant ions failed to induce channel reactivation. Moreover, mutagenesis identified two residues in the P-helix (L136 and V140 in Kir1.1) that are crucial for K+-dependent gating. Replacement of these residues with the ones present in the non-K+-gated Kir2.1 abolished K+-dependent gating of Kir1.1 channels without affecting pH gating.
The results indicate that pH gating and K+-dependent gating are coupled to each other via structural rearrangements in the inner pore involving the P-helix.
Weak inward rectifier K+ channels (Kir) that allow for bidirectional K+ transport across secretory epithelia are essential for a number of physiological processes. Thus, in kidney, Kir channels are responsible for K+ secretion in the cortical collecting duct (CCD) and the distal convoluted tubule (DCT) and by recycling K+ into the lumen for reabsorption of NaCl via the Na+-K+-2Cl− cotransporter in the thick ascending loop of Henle (Wang et al. 1992; Hebert, 1998).
ROMK1 was the first Kir channel to be cloned (Ho et al. 1993); it shares the prototypic membrane topology of this family of K+ channels with intracellular N- and C-termini and two transmembrane domains (M1 and M2) flanking a well-conserved pore-forming region (P-region). The ROMK protein exists in three splice variations, designated ROMK1-3 (Kir1.1a-c; Kir1.1b shortened by 19 amino acids and Kir1.1c extended by seven amino acid residues with respect to Kir1.1a) (Shuck et al. 1994; Yano et al. 1994; Boim et al. 1995). Although differentially expressed in renal tubular cells (Boim et al. 1995), all splice variants encode weak inward-rectifier K+ channels with essentially identical functional properties. A number of Kir1.1 mutations leading to impairment or loss of channel activity have been identified in patients suffering from hyperprostaglandin E syndrome (the antenatal variant of Bartter syndrome; Seyberth et al. 1985) underlining the physiological importance of proper ROMK function (Simon et al. 1996; Derst et al. 1997).
As with their native counterparts, cloned Kir1.1 channels are gated by intracellular pH (pHi); they close in response to acidification and open upon alkalinization (Bleich et al. 1990; Wang et al. 1990; Tsai et al. 1995). The corresponding concentration-response curve displays a steep dependence of channel activity on pHi with a midpoint around neutral pH. As the sensor for intracellular protons, a lysine residue located N-terminal to M1 (K80 in Kir1.1a) and common to all pH-gated inward-rectifier channels (Kir1.1 and Kir4) has been identified (Fakler et al. 1996; Schulte et al. 1999). This residue displays a strong shift in its apparent pKa, which results from close electrostatic interaction with two highly conserved arginines in the N- and C-terminus (R41 and R311 in Kir1.1a); all three residues are arranged in a triad that seems well conserved among Kir channels (Schulte et al. 1999).
In addition to gating by pHi, activity of Kir1.1 channels critically depends on the presence of extracellular K+ (K+o) in the millimolar range (Doi et al. 1996). Removal of K+o leads to a decrease in channel activity, and reapplication of K+o restores channel activity in a concentration-dependent manner. The time course of this K+-dependent gating (or K+ gating) is markedly affected by pHi. While K+-dependent inactivation is slow around neutral pHi, it is speeded up considerably by intracellular acidification (Doi et al. 1996). Moreover, chimeras formed between Kir1.1 and non-K+-gated Kir2.1 suggest that K+-dependent gating is linked to the P-loop in the Kir1.1 sequence (Doi et al. 1996). Thus, gating by extracellular K+ and intracellular pH are thought to be coupled to each other via an allosteric mechanism.
Here, we studied the interdependence of gating by pHi and K+o in cloned Kir1.1a channels and investigated the structural determinants underlying the allosteric coupling of these gating mechanisms.
METHODS
Mutagenesis and cRNA synthesis
Site-directed mutagenesis with rat Kir1.1a was performed as described by Fakler et al. (1995) and verified by sequencing. For heterologous expression in Xenopus oocytes, all constructs were subcloned into the pBF expression vector (B. Fakler, unpublished work). Capped Kir wild-type and mutant cRNAs were synthesized in vitro using the mMESSAGE mMACHINE kit (Ambion, Austin, TX) and stored as stock aliquots at −70°C.
Heterologous expression in Xenopus oocytes
Oocytes were surgically removed under anaesthesia from adult females (which were humanely killed after the final oocyte collection) and manually dissected. Dumont stage VI oocytes were injected with the appropriate cRNA solution (approximately 50 nl each), treated with collagenase type II (Sigma, 0.5 mg ml−1 for 15 min) and incubated at 19 °C for 2-5 days prior to use. Animal care and experiments followed approved institutional guidelines at the University of Tübingen.
Electrophysiology
Two-electrode voltage-clamp measurements were done at room temperature as described by Doi et al. (1996). Briefly, electrodes were pulled from thick-wall borosilicate glass, and filled with 3 m KCl; they had resistances of 0.1-0.8 MΩ. Currents were recorded with a TurboTec 01C amplifier (npi electronic GmbH, Tamm, Germany) and sampled at 1 kHz with an ITC16 AD board (HEKA Elektronik, Lambrecht/Pfalz, Germany). The bath chamber was continuously perfused with solutions composed of (mm): 10 Hepes, 1.8 CaCl2 and either 90 KCl + 30 NaCl (90 K+o) or 120 NaCl (0 K+o), pH 7.3.
Giant-patch recordings in inside-out configuration were performed as previously described (Fakler et al. 1995). Patch pipettes pulled from thick-wall borosilicate glass had resistances of 0.3-0.5 MΩ when filled with pipette solution (120 K+o: 120 mm KCl, 10 mm Hepes, 0.18 mm CaCl2, pH 7.3; 120 Na+o, 120 Rb+o, 120 Cs+o and 120 NMDG+o were prepared by equimolar replacement of K+ with the respective ion). Currents recorded in response to voltage ramps or steps were sampled at 1 kHz with an EPC9 amplifier (HEKA Elektronik) with analog filter set to 3 kHz. For single-channel experiments, small patch pipettes were pulled from quartz glass capillaries and filled with standard 120 K+o or 120 Rb+o solution (resistance 3-7 MΩ). Currents were sampled at 10 kHz and filtered at 1 kHz.
Giant outside-out patches were obtained as follows: after establishing cell-attached configuration (seal resistances of > 3 GΩ) with a giant patch pipette (pulled as described above, filled with K+i (see below) + 10 μm PIP2 to prevent channel rundown), the patch was ruptured and the pipette was slowly withdrawn from the oocyte allowing the membrane to form a bell-shaped extension which eventually cut off as a giant outside-out patch. Intra- and extracellular solutions were composed as follows (mm): K+i/Rb+i/NMDG+i: 10 Hepes, 10 EGTA, 0.1 DTT and 120 KCl/120 RbCl/120 NMDG, adjusted to the given pH; 0 K+o/100 K+o: 10 Hepes, 1.44 MgCl2, 10 EGTA and 100 NaCl/100 KCl, pH 7.2. Solutions were applied to excised patches via a multibarrel pipette as described by Fakler et al. (1995).
Data evaluation
Relative permeability of Kir1.1 wild-type and mutant channels for K+, Rb+ and Cs+ was determined by linear regression according to a linearized form of the Goldman-Hodgkin-Katz equation: Pion1/Pion2=[ion 1]/[ion 2]exp{Vrev(zF/RT)}. Reversal potentials (Vrev) were determined in excised inside-out patches at zero current with voltage ramps from −100 to 100 mV; the patch pipette contained 30 mm Rb+; concentrations of K+ and Cs+ were varied on the intracellular side (10, 30 and 100 mm for Cs+, 3, 10, 30 and 100 mm for K+).
Relative conductance for Cs+ (gCs : gK) was determined from the asymptotic slope conductance of macroscopic currents (100 mm K+/100 mm Cs+) in excised patches. The single channel conductance was calculated from linear fits to single channel currents (≥ 50 ms periods of channel open state) and given as means ±s.d. of n patches.
Parameters characterizing pH gating of Kir1.1 channels were determined as described by Schulte et al. (1999). The apparent affinity of Kir1.1 channels for K+o was determined in inside-out patches by measuring relative current recovery from pH-induced inactivation with varying K+o (1, 3, 10, 30 and 100 mm) in the pipette; data points were plotted as means ±s.d. (of 5 experiments) and fitted with a logistic function.
All computational work was done on a Macintosh G3 using commercial software (IGOR, WaveMetrics, Lake Oswego, OR, USA) for fitting.
RESULTS
K+-dependent gating occurs in the pH-inactivated state
Figure 1 illustrates K+-dependent gating as observed in Xenopus oocytes expressing Kir1.1 channels. When oocytes were incubated in K+-free solution for > 2 h, no currents were recorded in response to voltage ramps (from −120 to 50 mV) in K+-free solution (0 K+o). However, upon application of 90 mm K+ to the extracellular side (90 K+o), Kir1.1-mediated currents appeared that increased over a period of minutes (Fig. 1A, upper recording; Doi et al. 1996). This slow K+-dependent increase and decrease in current was also observed in giant outside-out patches excised from such oocytes (Fig. 1B, upper recording). While at 0 K+o Kir1.1 currents (measured in response to voltage ramps from −100 to 50 mV) were small, they increased with time to more than 10-fold (13.2 ± 3.3, n = 3) after switching K+o to 100 mm (the instantaneous increase in current amplitude following switch to the 100 K+o solution is due to the respective shift in the K+ reversal potential, EK). Conversely, withdrawal of K+o from the patch decreased the current amplitude to the level observed at the beginning of the experiment (Fig. 1B, upper recording). In such experiments, the amplitude of single Kir1.1 channels remained unchanged indicating that the increase in current was due to a change in channel activity (data not shown; Sackin et al. 2001).
Figure 1. K+-dependent gating of Kir1.1 channels.
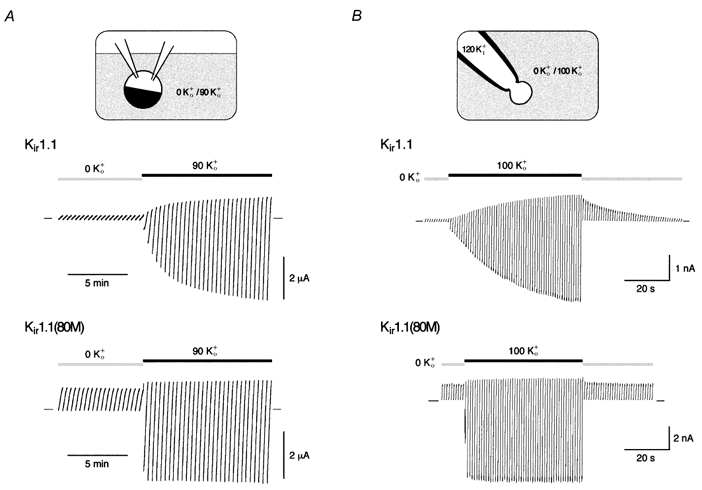
A, currents mediated by Kir1.1 (upper trace) or Kir1.1(K80M) channels (lower trace) recorded in oocytes preincubated in K+-free solution for more than 2 h. Change in bath solution from 0 K+o to 90 K+o as indicated by horizontal bars; currents were recorded in response to voltage ramps from −120 to 50 mV (in 20 s), and zero current is indicated by small bars. B, currents recorded in giant outside-out patches from oocytes expressing Kir1.1 (upper trace) or Kir1.1(K80M) channels (lower trace) and treated as in A. Currents are response to voltage ramps from −100 to 50 mV (in 400 ms) at pHi 7.1; application of extracellular solutions as indicated. Note the difference between slow increase in Kir1.1 currents due to recovery from K+-dependent inactivation and rapid increase in current amplitude due to the shift in EK resulting from the change in [K+]o.
Different from wild-type, K+-dependent gating was not observed in Kir1.1(K80M) channels where pH gating is abolished (Fakler et al. 1996; Schulte et al. 1999). As shown in Fig. 1A and B (lower recordings), mutant channels exhibited robust currents in 0 K+o (outward directed over the whole voltage range due to the negative EK). The switch to high K+o did not change channel activity, but rather led to an instantaneous increase in current amplitude resulting from the shift in EK (Fig. 1B, lower recording).
The lack of K+ gating in Kir1.1(K80M) channels suggested that K+-dependent gating is linked to pH gating. This was further investigated in a series of patch experiments, where 0 K+o was used as an extracellular solution and pH and K+ concentration of the intracellular solution were varied; currents were recorded at a potential of −80 mV that was stepped to 40 mV for 40 ms every second. In an initial experiment, the channel pore was depleted of K+ ions at a constant pHi of 8.0 by switching the intracellular solution from K+i to K+-free NMDG+i. As shown in Fig. 2A, Kir1.1-mediated outward currents were constant at pHi 8.0 and K+ depeletion did not induce K+-dependent inactivation as envisaged by the instantaneous recovery of currents upon switching the intracellular solution back to K+i. In a second set of experiments, channels were pH inactivated by intracellular acidification (application of K+i at pHi 6.0). Different from K+-depletion alone, Kir1.1 channels failed to recover in the absence of K+o (Fig. 2B) whereas in the presence of high K+o, pH-induced inactivation was fully reversible (Schulte et al. 1998; Figs. 2D and 4A). Furthermore, in Kir1.1(K80M) channels intracellular acidification together with K+ depletion failed to induce K+-dependent inactivation (Fig. 2C).
Figure 2. K+-dependent gating is linked to pH gating.
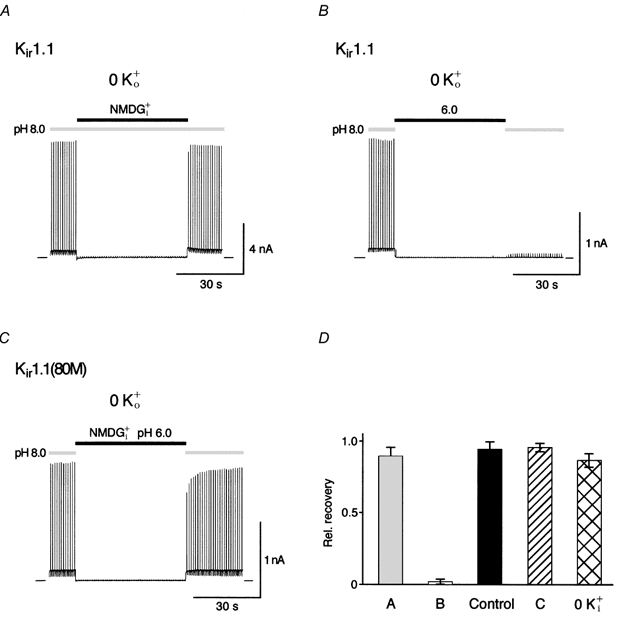
A, Kir1.1 currents recorded in the absence of extracellular K+ (0 K+o) from a giant inside-out patch with intracellular K+ switched from 120 mm (K+i) to 0 mm (NMDG+i) and back at a constant pHi of 8.0. Current was recorded at a membrane potential of −80 mV stepped to 40 mV for 40 ms every second. Note the complete recovery of currents after switching K+ back to 120 mm. B, recovery experiment as in A but with pHi switched from 8.0 to 6.0 and back at constant intracellular K+ of 120 mm. Note the lack of current recovery after pH-induced inactivation in the absence of K+o. C, currents mediated by Kir1.1(K80M) channels recorded in an experiment as in A but with simultaneously changing intracellular K+ and pHi (NMDG+i at pHi of 6.0). D, relative recovery of currents (measured at 40 mV) from experiments as in A-C given as means ±s.d. of ≥ 6 experiments; control refers to experiments as in B, but with 120 mm K+o; 0 K+i refers to experiments as in B but with 120 mm K+o and 0 mm K+i.
Figure 4. K+-dependent gating is selective for permeant ions on the extracellular side.
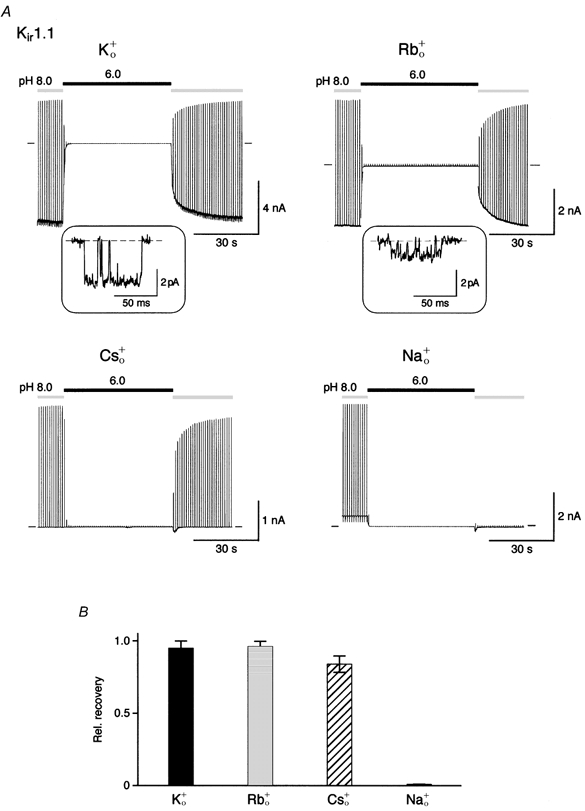
A, recovery experiments as in Fig. 2B with 120 mm K+ on the intracellular side (K+i) and 120 mm K+ (K+o), Rb+ (Rb+o), Cs+ (Cs+o) or Na+ (Na+o) in the recording pipette. Insets: Kir1.1 single channel currents recorded from inside-out patches at −100 mV in symmetrical 120 mm K+ (left) and 120 mm Rb+ (right); single channel conductances were 32 ± 0.5 pS (n = 5) and 11 ± 0.7 pS (n = 6), respectively; closed level indicated by the dashed line. B, relative recovery of currents from experiments as in A (acidification periods of 50 s); bars represent means ±s.d. of 5-12 experiments.
These results suggest that K+-dependent inactivation only occurs when channels are in the pH-inactivated closed state, while channels in the open state are not susceptible to this process.
K+-dependent gating exhibits selectivity for permeant ions
K+ gating was further characterized by determining the time course of K+-dependent inactivation. This was done by recording currents in the absence of K+o at pHi 8.0 before and after acidification of the channels for an increasing period of time; inactivation was quantified by normalizing the recovered current with respect to that observed prior to acidification. For acidification a pHi of 5.0 was used, where all channels were pH inactivated within less than 0.5 s (data not shown). As shown in Fig. 3, the time course of K+-dependent inactivation could be approximated by a monoexponential yielding a time constant of 8.2 s.
Figure 3. Time course of K+-dependent inactivation.
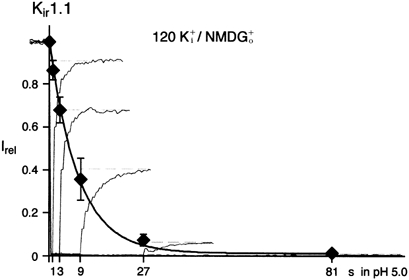
Time course of K+-dependent inactivation monitored by the current recovery as obtained from experiments as in Fig. 2B. K+i at pH 5.0 was applied for periods of 1, 3, 9, 27 and 81 s. Overlaid are current traces from such experiments normalized to the initial outward current amplitude at pH 8.0 (only currents at 40 mV are shown); data points are means ±s.d. of 5 experiments. Line is monoexponential fit to the data that yielded a time constant of 8.2 s.
Next, the sensitivity of K+-dependent gating to changes in ion species was tested. This was done in recovery experiments where K+ was replaced by various alkali ions on the extracellular side of the membrane. As shown in Fig. 4, recovery of Kir1.1 channels from pH-induced inactivation was almost complete in 120 mm K+o, Rb+o and Cs+o, whereas no recovery was observed in 120 mm Na+o. This pattern roughly paralleled the permeability sequence of Kir1.1. While K+, Rb+ and Cs+ were permeant (relative permeabilities of PK : PRb : PCs= 1 : 0.6 : 0.039, for details see Methods), Na+ did not penetrate through the channel. The extent of recovery was independent of the absolute permeance, since Cs+ induced full recovery although it was more than 160-fold less permeant than K+ (data not shown).
These findings suggest that K+-dependent gating may be governed by occupancy of an ion binding site located in the channel's selectivity filter.
K+-dependent gating is determined by the P-helix
Among Kir channels, K+ gating was only observed with Kir1.1 and Kir4 channels, but not with other subtypes including Kir2.1 (Doi et al. 1996; Pearson et al. 1999). Comparison of primary sequence uncovered three positions within the highly conserved stretch around the selectivity filter that were different between Kir1.1 and Kir2.1 (Fig. 5). Residues V140 and L136 in the P-helix of Kir1.1 were tested for their ability to affect K+-dependent gating after replacing them with the respective residues of the Kir2.1 sequence. As shown in Fig. 6, both mutants, Kir1.1(L136I) and Kir1.1(V140T), expressed K+-selective channels (PK : PRb : PCs= 1 : 0.63 : 0.053 and 1 : 0.67 : 0.013 for Kir1.1(L136I) and Kir1.1(V140T), respectively) that were gated by intracellular pH indistinguishable from wild-type channels (Fig. 6A and B). In contrast to wild-type, however, these channels exhibited complete recovery from pH-induced inactivation independent of the presence of extracellular K+ (Fig. 6). Thus, in either case the mutation in the P-helix abolished K+-dependent gating.
Figure 5. Amino acid sequences of a P-loop stretch from Kir1.1 and Kir2.1 channels aligned to the respective region from the KcsA channel.
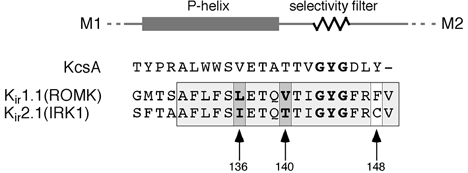
Box shaded in grey marks a sequence stretch highly conserved in the Kir family; arrows mark differences between Kir1.1 and Kir2.1.
Figure 6. Mutations in the Kir1.1 P-helix abolish K+-dependent gating.
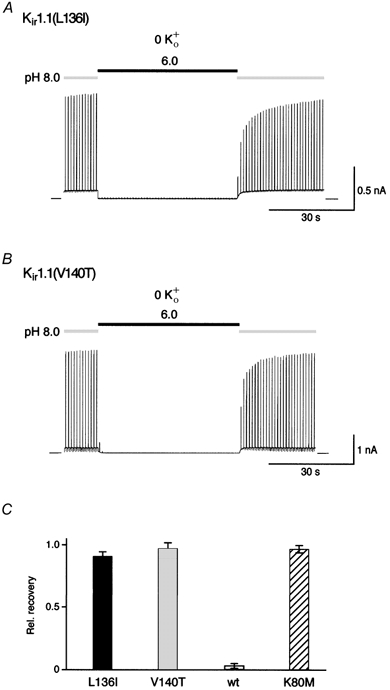
A and B, complete recovery of currents in Kir1.1(L136I) and Kir1.1(V140T) channels. Experimental conditions as in Fig. 2B. C, relative recovery of currents from experiments as in A and B (acidification periods of 50 s); bars represent means ±s.d. of 5 (Kir(L136I)) and 12 (Kir1.1(V140T)) experiments. Data for Kir1.1 and Kir1.1(K80M) were added for better comparison.
Together with the results shown above these findings suggest that the P-helix is involved in the conformational changes underlying K+-dependent gating and its coupling to pH gating.
DISCUSSION
Regulation of channel activity by extracellular K+ has been described for native (Giebisch, 1998) and cloned (Doi et al. 1996) ROMK (Kir1.1) channels as well as for Kir4.1 (Pearson et al. 1999). The mechanism underlying this gating process, however, has remained poorly understood. The results presented here demonstrate that K+-dependent gating is strictly coupled to pH gating, only occuring when channels are pH inactivated. Mechanistically, K+ gating is thought to result from dissociation of K+ ions from the channel and conformational changes in the inner pore involving the P-helix.
K+-dependent gating of Kir1.1 was observed in both whole oocytes and excised outside-out patches (Fig. 1). The kinetics of K+-dependent inactivation/reactivation as determined in excised patches, however, were significantly faster than those measured in whole oocytes with the two-electrode voltage-clamp technique. These differences are most likely to be due to the large differences in solution exchange observed between both experimental configurations.
Basically, K+ gating describes a gating reaction that occludes the channel pore as a result of dissociation of K+ ions from the channel (Doi et al. 1996). Different from most other gating processes, however, K+-dependent gating did not act on the open channel, but rather affected Kir1.1 channels when they were in the pH-inactivated closed state. Thus, K+ gating was neither observed at pHi 8.0, even when the pore was completely depleted from K+ (Fig. 2A), nor present in Kir1.1(K80M) mutant channels in which pH gating was abolished by disruption of the pH sensor (Fig. 1 and Fig. 2C). Consequently, the conformational change(s) associated with pH gating may be regarded a prerequisite for K+-dependent gating.
A further characteristic feature of K+ gating is its ion selectivity. Thus, while the permeant monovalents Rb+, NH4+ or Cs+ were equally as potent as K+ in preventing K+-dependent inactivation, the impermeant Na+ and NMDG+ failed to keep the pore open (Fig. 4; Doi et al. 1996). As permeation is governed by binding site(s) in the channel's selectivity filter (Doyle et al. 1998; Stampe et al. 1998), it seemed likely that K+ gating may be accompanied by conformational changes in the channel pore similar to what was reported as a ‘pore collapse’ for C-type inactivation of voltage-gated K+ channels (reviewed by Yellen, 1998). In line with this interpretation, a number of residues (F148C, Q152E and E151D) located in the extracellular portion of the Kir1.1 pore were recently suggested to affect K+-dependent gating (Sackin et al. 2001). Additionally, we identified two residues in the P-helix, I136 and V140, that markedly affected K+-dependent gating. Replacement of either site with the respective residue from non-K+-gated Kir2.1 eliminated K+-dependent gating without any effect on pH gating (Fig. 6). Kir1.1(V140T), which enhances block by extracellular Ba2+ (Zhou et al. 1996), showed a higher single channel conductance (data not shown; Zhou et al. 1996), increased relative permeability for K+ (see above; Choe et al. 2000) and altered single channel kinetics (data not shown; H. Sackin, personal communication), whereas in Kir1.1(L136I) these parameters were almost identical to Kir1.1 wild-type. Thus, the presence or absence of K+-dependent gating could not be linked to any of these single channel parameters.
Interestingly, the P-helix was recently shown to move during pH gating in KcsA, a two segment-type K+ channel from Streptomyces lividans (Perozo et al. 1998; Heginbotham et al. 1999). When viewed in the structural context of the KcsA crystal (Doyle et al. 1998), side chains of L136 and V140 (positions 70 and 74 in KcsA) appeared to interact with neighbouring subunits rather than with the ion binding sites (Fig. 7A). This is consistent with the idea that P-helix mutations act by preventing the slow gating reaction observed upon K+ depletion of the pore (Fig. 3).
Figure 7. Structural model of the KcsA pore and sequential model of Kir1.1 gating by pHi and K+o.
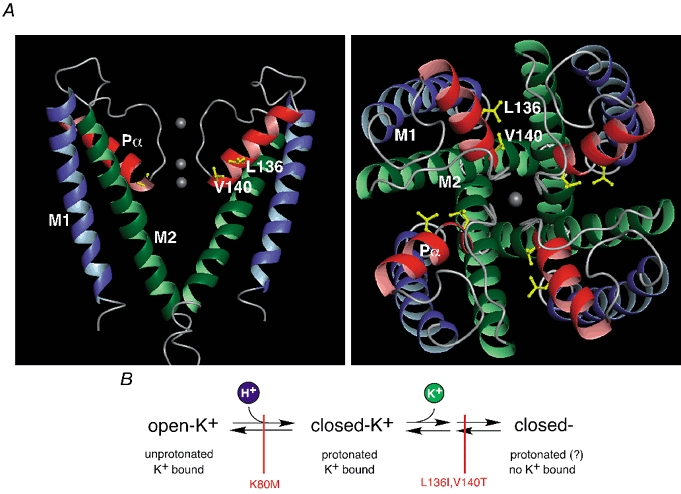
A, structural model of the KcsA pore as determined by X-ray crystallography (Doyle et al. 1998) viewed from the side (left panel, only two subunits shown for clarity) and the top (right panel); transmembrane segments in green (M1) and blue (M2) and the P-helices (Pα) in red; K+ binding sites are indicated by grey spheres. Side chains of residues 70 and 74 homologous to residues 136 and 140 in Kir1.1 were replaced by I and V and highlighted. Note that the side chains are oriented towards the neighbouring subunits rather than to the ion conducting pathway. B, sequential model of Kir1.1 gating by pHi and K+o as deduced from the experiments presented here. ‘open-K+’ represents active channels; ‘closed-K+’ is pH-inactivated closed state with K+ bound to the pore binding site(s); ‘closed-’ is K+-inactivated closed state; possible intermediate states are omitted for simplicity. These channel conformations are linked through gating transitions (pH gating and K+-dependent gating, symbolized by arrows) which can be blocked independently by point mutations (indicated in red).
Figure 7B summarizes the results in a kinetic model of Kir1.1 gating by pHi and K+o. Thus, channels undergo at least two consecutive conformational changes. In a first step, reflecting pH gating and triggered by protonation of K80 (pKa= 6.8; Schulte et al. 1999), channels enter a closed state with K+ ions present at the respective binding sites (closed-K+). Upon dissociation of these ions, K+-dependent gating associated with structural rearrangements involving the P-helix drives channels into a second closed state (closed-). Equilibrium of this second transition is defined by the apparent affinity of closed channels for K+o (KD= 1.6 mm; see Methods). As result of this sequential mechanism, the apparent pKa of ROMK should be shifted to higher values at physiological K+o, which has indeed been reported (Sackin et al. 2001). This model also explains the pHi dependence of K+ gating kinetics (Doi et al. 1996). K+-dependent inactivation becomes very slow at alkaline pHi as the fraction of channels susceptible to this reaction (closed-K+) is reduced, whereas recovery is governed entirely by the slow transition from (closed-) to (closed-K+), independent of pHi.
Thus, the interaction of gating by K+o and pHi may play an important role in the regulation of ROMK activity under physiological conditions.
Acknowledgments
We would like to thank D. Bentrop for the illustration of the KcsA structural model and D. Oliver and T. Baukrowitz for reading and helpful discussion of the manuscript.
References
- Bleich M, Schlatter E, Greger R. The luminal K+ channel of the thick ascending limb of Henle's loop. Pflügers Archiv. 1990;415:449–460. doi: 10.1007/BF00373623. [DOI] [PubMed] [Google Scholar]
- Boim MA, Ho K, Shuck ME, Bienkowski MJ, Block JH, Slightom JL, Yang Y, Brenner BM, Hebert SC. ROMK inwardly rectifying ATP-sensitive K+ channel: II. Cloning and distribution of alternative forms. American Journal of Physiology. 1995;268:F1132–1140. doi: 10.1152/ajprenal.1995.268.6.F1132. [DOI] [PubMed] [Google Scholar]
- Choe H, Sackin H, Palmer LG. Permeation properties of inward-rectifier potassium channels and their molecular determinants. Journal of General Physiology. 2000;115:391–404. doi: 10.1085/jgp.115.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derst C, Konrad M, Köckerling A. Mutations in the ROMK gene in antenatal Bartter syndrome are associated with impaired K+ channel function. Biochemical and Biophysical Research Communications. 1997;203:641–645. doi: 10.1006/bbrc.1996.6024. [DOI] [PubMed] [Google Scholar]
- Doi T, Fakler B, Schultz JH, Schulte U, Brändle U, Weidemann S, Zenner HP, Lang F, Ruppersberg JP. Extracellular K+ and intracellular pH allosterically regulate renal Kir1. 1 channels. Journal of Biological Chemistry. 1996;271:17261–17266. doi: 10.1074/jbc.271.29.17261. [DOI] [PubMed] [Google Scholar]
- Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, Mackinnon R. The structure of the potassium channel: molecular basis of K+ conductivity and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Fakler B, Brändle U, Glowatzki E, Weidemann S, Zenner HP, Ruppersberg JP. Strong voltage-dependent inward-rectification of inward rectifier K+ channels is caused by intracellular spermine. Cell. 1995;80:149–154. doi: 10.1016/0092-8674(95)90459-x. [DOI] [PubMed] [Google Scholar]
- Fakler B, Schultz JH, Yang J, Schulte U, Brändle U, Zenner HP, Jan LY, Ruppersberg JP. Identification of a titratable lysine residue that determines sensitivity of kidney potassium channels (ROMK) to intracellular pH. EMBO Journal. 1996;16:4093–4099. [PMC free article] [PubMed] [Google Scholar]
- Giebisch G. Renal potassium transport: mechanism and regulation. American Journal of Physiology. 1998;274:F817–833. doi: 10.1152/ajprenal.1998.274.5.F817. [DOI] [PubMed] [Google Scholar]
- Hebert SC. Roles of Na-K-2Cl and NaCl cotransporters and ROMK potassium channels in urinary concentration mechanism. American Journal of Physiology. 1998;275:F325–327. doi: 10.1152/ajprenal.1998.275.3.F325. [DOI] [PubMed] [Google Scholar]
- Heginbotham L, Lemasurier M, Kolmakova-Partensky L, Miller C. Single Streptomyces lividans K+ channels: functional asymmetries and sidedness of proton activation. Journal of General Physiology. 1999;114:551–560. doi: 10.1085/jgp.114.4.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, Kanazirska MV, Hebert SC. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature. 1993;362:31–38. doi: 10.1038/362031a0. [DOI] [PubMed] [Google Scholar]
- Pearson WL, Dourado M, Schreiber M, Salkoff L, Nichols CG. Expression of a functional Kir4 family inward rectifier K+ channel from a gene cloned from mouse liver. Journal of Physiology. 1999;514:639–653. doi: 10.1111/j.1469-7793.1999.639ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo E, Cortes DM, Cuello LG. Three-dimensional architecture and gating mechanism of a K+ channel studied by EPR spectroscopy. Nature Structural Biology. 1998;5:459–469. doi: 10.1038/nsb0698-459. [DOI] [PubMed] [Google Scholar]
- Sackin H, Syn S, Palmer LG, Choe H, Walters DE. Regulation of ROMK by extracellular cations. Biophysical Journal. 2001;80:683–697. doi: 10.1016/S0006-3495(01)76048-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte U, Hahn H, Konrad M, Jeck N, Derst C, Wild K, Weidemann S, Ruppersberg JP, Fakler B, Ludwig J. pH gating of ROMK (Kir1. 1) channels: Control by an Arg-Lys-Arg triad disrupted in antenatal Bartter syndrome. Proceedings of the National Academy of Sciences of the USA. 1999;96:15298–15303. doi: 10.1073/pnas.96.26.15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte U, Hahn H, Wiesinger H, Ruppersberg JP, Fakler B. pH-dependent gating of ROMK (Kir1. 1) channels involves conformational changes in both N and C termini. Journal of Biological Chemistry. 1998;273:34575–34579. doi: 10.1074/jbc.273.51.34575. [DOI] [PubMed] [Google Scholar]
- Seyberth HW, Rascher W, Schweer H, Kühl PG, Mehls O, Schärer K. Congenital hypokalemia with hypercalciuria in preterm infants: a hyperprostaglandinuric tubular syndrome different from Bartter syndrome. Journal of Pediatrics. 1985;107:694–701. doi: 10.1016/s0022-3476(85)80395-4. [DOI] [PubMed] [Google Scholar]
- Shuck ME, Bock JH, Benjamin CW, Tsai TD, Lee KS, Slightom JL, Bienkowski MJ. Cloning and characterization of multiple forms of the human kidney ROMK potassium channel. Journal of Biological Chemistry. 1994;269:24261–24270. [PubMed] [Google Scholar]
- Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, Dipietro A, Trachtman H, Sanjad SA, Lifton RP. Genetic heterogeneity of Bartter's syndrome revealed by mutations in the K+ channel, ROMK. Nature Genetics. 1996;14:152–156. doi: 10.1038/ng1096-152. [DOI] [PubMed] [Google Scholar]
- Stampe P, Arreola J, Perez-Cornejo P, Begenisich T. Nonindependent K+ movement through the pore in IRK1 potassium channels. Journal of General Physiology. 1998;112:475–484. doi: 10.1085/jgp.112.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai TD, Shuck ME, Thompson DP, Bienkowski MJ, Lee KS. Intracellular H+ inhibits a cloned rat kidney outer medulla K+ channel expressed in Xenopus oocytes. American Journal of Physiology. 1995;37:C1173–1178. doi: 10.1152/ajpcell.1995.268.5.C1173. [DOI] [PubMed] [Google Scholar]
- Wang W, Sackin H, Giebisch G. Renal potassium channels and their regulation. Annual Review of Physiology. 1992;54:81–96. doi: 10.1146/annurev.ph.54.030192.000501. [DOI] [PubMed] [Google Scholar]
- Wang W, Schwab A, Giebisch G. Regulation of small-conductance K+ channel in apical membrane of rat cortical collecting tubule. American Journal of Physiology. 1990;259:F494–502. doi: 10.1152/ajprenal.1990.259.3.F494. [DOI] [PubMed] [Google Scholar]
- Yano H, Philipson LH, Kugler JL, Tokuyama Y, Davis EM, LeBeau M, Nelson DJ, Bell GI, Takeda J. Alternative splicing of human inwardly rectifying K+ channel ROMK1 mRNA. Molecular Pharmacology. 1994;45:854–860. [PubMed] [Google Scholar]
- Yellen G. The moving parts of voltage-gated ion channels. Quarterly Reviews of Biophysics. 1998;31:239–295. doi: 10.1017/s0033583598003448. [DOI] [PubMed] [Google Scholar]
- Zhou H, Chepilko S, Schütt W, Choe H, Palmer LG, Sackin H. Mutations in the pore region of ROMK enhance Ba2+ block. American Journal of Physiology. 1996;271:C1949–1956. doi: 10.1152/ajpcell.1996.271.6.C1949. [DOI] [PubMed] [Google Scholar]