

Abstract
In the present study the functional role of the C-terminus of the human ether-à-go-go-related gene K+ channel HERG was investigated using a series of C-terminal deletion constructs expressed in Xenopus oocytes.
Constructs with deletions of 311 or more amino acid residues failed to form functional channels. Truncation by 215 amino acid residues or fewer had no discernable effects on channel activity. Truncation by 236 or 278 amino acid residues accelerated deactivation, and caused a faster recovery from inactivation.
In high extracellular K+, channel deactivation of HERG results from the binding of the N-terminus to a site within the pore. This slows channel deactivation by a knock-off mechanism. Here, it was shown that C-terminal deletions also abolished this effect of high extracellular K+. Mutants containing deletions in both the N- and C-termini deactivated with rates similar to those observed in individual deletion mutants.
In contrast, experiments with double-deletion constructs showed additive effects of the N- and C-termini on the voltage dependence of activation, and on the kinetics of inactivation and recovery from inactivation. The reduction of inactivation in these mutants contributed to an increase in peak current amplitude.
These results indicate that residues within the C-terminus of HERG play a role in channel expression as well as in most aspects of channel gating. The regulation of channel deactivation is likely to be mediated by an interaction with the N-terminus, but the regulation of the voltage dependence of activation, and of rate processes associated with inactivation, does not require the N-terminus.
The human eag (ether-à-go-go)-related gene (HERG) encodes a component of cardiac delayed rectifier potassium current IKr (Sanguinetti et al. 1995; Trudeau et al. 1995) and is critical for the maintenance of normal rhythmicity in the human heart (Curran et al. 1995). IKr mediates the terminal repolarization of the ventricular action potential (Sanguinetti & Jurkewicz, 1990), and the loss of this current due to inherited mutations in HERG or drug block leads to a prolongation of the action potential and an associated susceptibility to arrhythmia.
Being a member of the eag family of voltage-gated K+ channels, HERG shares the structural characteristics of this family (Warmke & Ganetzky, 1994). Overall sequence similarity between Eag- and Shaker-type channels is quite low except for the pore (H5) region. A much higher degree of homology is found between eag-type channels and plant inwardly rectifying K+ channels (AKT1 and KAT1) (Anderson et al. 1992; Sentenac et al. 1992), as well as cyclic nucleotide-gated (CNG) channels. In addition, eag channels have a region of unknown function within the C-terminus that is homologous to the cyclic nucleotide-binding domain (CNBD) of CNG channels. It is presumed that eag-type channels are tetramers like members of the CNG family of ion channels (Kaupp et al. 1989; Liu et al. 1996). Ludwig et al. (1997) have identified a C-terminal domain (CAD, residues 897-937) that is important for rat eag (r-eag) subunit interactions. Similarly, Daram et al. (1997) have identified two regions in the AKT1 channel that are important for subunit interaction. The first of these regions is located in the putative CNBD of the channel and interacts with the region between amino acid residues 371 and 516. The second region is between the last 81 amino acid residues of the protein and a region lying between the channel hydrophobic core and the putative CNBD.
The functional role of the C-terminus of HERG and other members of the eag family of channels is poorly understood. Studies of other voltage-gated K+ channels have demonstrated roles for the C-terminus in channel gating. For example, the N- and C-termini act in concert to mediate inactivation in mKv4.1 channels (mouse Shal) by a mechanism distinct from the ‘ball-and-chain’ mechanism first described for Shaker channels (Hoshi et al. 1991; Demo & Yellen, 1991; Perozo et al. 1992; Jerng & Covarrubias, 1997). In rat drk-1 channels, deletions of the C-terminus cause a hyperpolarizing shift of the voltage dependence of activation, but normal activation is either restored or compensated for by the additional deletion of the N-terminus (VanDongen et al. 1990). Miller & Aldrich (1996) showed that inward rectification could be induced in Shaker K+ channels by mutations that shift activation to more negative voltage ranges. This increases the inactivation at resting potentials and, when the membrane potential is stepped to more negative potentials, causes HERG-like tail currents that first recover from inactivation prior to deactivating. Mutations causing a more extreme shift of activation result in currents that recover from inactivation but do not deactivate, producing maintained inward currents like those of KAT-1 inward rectifier currents from plants. The authors suggested on the basis of these mutant phenotypes that the voltage-dependent ‘activation’ of KAT-1 channels is actually the recovery from inactivation as the channels enter the open state. Marten & Hoshi (1997) showed that the voltage dependence of this process is altered by deletions of the extreme C-terminus. Thus, sequences in the C-terminus may be important in regulating inactivation in KAT-1.
In the present study, C-terminal deletions of HERG have been constructed in order to: (i) delineate the minimum amount of C-terminus necessary to form a functional ion channel, (ii) investigate the possible contribution of the CNBD to channel function and (iii) identify other regions in the C-terminus of HERG that may contribute to HERG channel function. Additionally, deletions in the C-terminus were coupled with N-terminal deletions to distinguish between N- and C-terminal effects that are cooperative or independent.
METHODS
Construction of deletion mutations
HERG C-terminal deletion clones were constructed using a one-step PCR. The forward primer provided a Bgl II site. The reverse primers contained a stop codon followed by an EcoR I site, as illustrated in Fig. 1A. The N-terminal part of double deletions and N-terminal deletions was constructed as described in Wang et al. (1998). Constructs were confirmed by restriction enzyme analyses and DNA sequencing.
Figure 1. HERG C-terminal and HERG N- and C-terminal deletions.
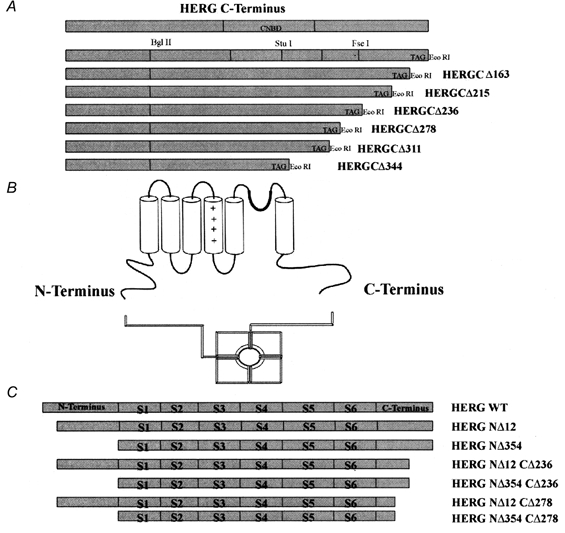
A, schematic representation of HERG C-terminal deletions. B, schematic representation of K+ channel (HERG) structure. C, schematic representation of HERG N- and C-terminal double-deletion constructs.
Preparation of oocytes and RNA synthesis and injection
Oocytes were collected from female frogs (Xenopus laevis, Nasco, Fort Atkinson, WI, USA) anaesthetized by a 10-20 min exposure to 3-aminobenzoic acid ethyl ester (MS222; 0.5 g in 500 ml water). Frogs were humanely killed after the final collection. All procedures were approved by the University of Wisconsin Research Animals Resource Center and the NIH. The follicular membranes were removed from the oocytes by collagenase treatment (Collagenase B, Boehringer Mannheim) and, in some cases, by an osmotic shock procedure (Pajor et al. 1992). RNA was synthesized from linearized template DNA using the mMessage mMachine kit (Ambion, Austin, TX, USA). RNA was diluted in sterile water to different ratios to give upon expression an optimal 2-10 μA of outward current (evoked by steps to -10 or 0 mV). Oocytes were cultured at 18 °C in storage solution (96 mm NaCl, 2 mm KCl, 1 mm MgCl2, 1.8 mm CaCl2 and 5 mm Hepes, supplemented with 10 μg ml−1 gentamicin and 1 mg ml−1 BSA, pH 7.4).
Current recording and data analysis
Currents were recorded using two-electrode voltage clamp (OC-725A, Warner Equipment Corporation, Hamden, CT, USA) and pCLAMP 6.0.3 software (Axon Instruments, Foster City, CA, USA). pCLAMP and Origin 5.0 (Microcal Software Inc., MA, USA) were used for data analysis and plotting. The resistance of the electrodes was 0.5-1 MΩ in 2 m KCl. The normal (low K+ solution) bath solution contained 93 mm NaCl, 5 mm KCl, 1 mm MgCl2, 0.3 mm CaCl2 and 5 mm Hepes pH 7.4. In high K+ experiments the bath solution contained 300 mm KCl, 1 mm MgCl2, 0.3 mm CaCl2 and 5 mm Hepes pH 7.4 (Fig. 5). Solutions were kept isosmolar by addition of N-methyl-glutamine. No oocyte shrinkage was observed under these conditions and recordings were generally completed within 5 min. The holding potential was −80 mV and all recordings were done at room temperature, roughly 25 °C. Data were routinely discarded if the leak exceeded 10 % of the maximum conductance of the expressed currents, but for all the data in this study, the leak currents seen with steps from −80 to −100 mV were less than 0.2 μA, << 1 % of the maximum current of the expressed channel, and therefore no leak subtraction was utilized. Time constants (τ) for the transients were measured by curve fitting with Clampfit (Axon Instruments). Data were digitized at 2 or 4 kHz without filtering.
Figure 5. Effect of high K+ concentration on deactivation kinetics of WT, HERGCΔ236 and HERGCΔ278 channels.
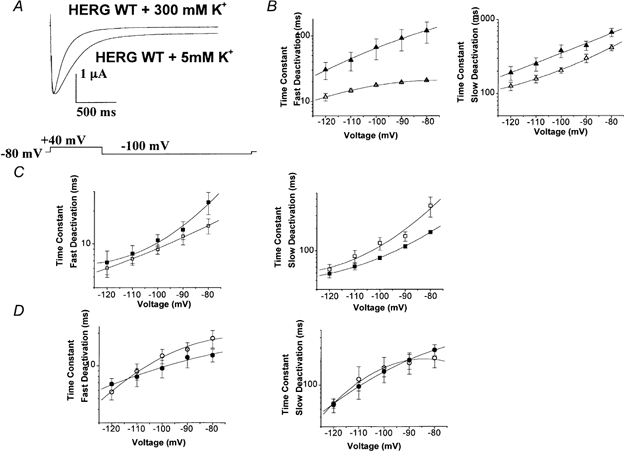
Deactivation kinetics were studied as in Fig. 3. A, high K+ accelerated the deactivation of WT channels. In this set of traces cells were held at −80 mV depolarized to +40 mV for 1 s and subsequently hyperpolarized to −100 mV for 3 s. B, fast and slow deactivation plots for WT HERG + 5 mm K+ (▴) and WT HERG + 300 mm K+ (▵), indicating that high K+ accelerated deactivation of WT HERG (n = 13, P = 0.011 for fast deactivation and P = 0.17 for slow deactivation). C, fast and slow deactivation plots for HERGCΔ236 + 300 mm K+ (□) and HERGCΔ236 + 5 mm K+ (□), indicating that high K+ did not alter the deactivation (n = 1, P = 0.41 for fast deactivation and P = 0.88 for slow deactivation). D, fast and slow deactivation plots for HERGCΔ278 + 300 mm K+ (^) and HERGCΔ278 + 5 mm K+ (•), indicating that high K+ did not alter deactivation (n = 11, P = 0.42 for fast deactivation and P = 0.33 for slow deactivation).
I-V relationships were determined by recording outward current in response to a standard voltage protocol with pulses from −80 to +70 mV for 1 s in steps of 10 mV from a holding potential of −80 mV. Each voltage command was followed by a repolarization to −100 mV, eliciting large inward tail currents. I-V plots were determined by plotting the current level at the end of the 1 s pulse versus command voltage.
Conductance (G) was calculated from the K+ current by dividing by the driving force for K+. The G-V relationship was determined in response to voltages from −100 to +70 mV in 10 mV steps from a holding potential of −80 mV. Tail currents were fitted to a double-exponential function (y = A1e-t/τ1+A2e-t/τ2) and extrapolated to the end of the command voltage. These values were plotted versus voltage and fitted with a single-power Boltzmann function of the form y= 1 − (1/(1 + e(V- V1/2)/k)). The midpoint is V1/2 and k is a slope factor.
The amplitudes of the outward currents (e.g. G-V, I-V) at each corresponding command voltage were normalized to the maximal tail current level evoked at −100 mV subsequent to a 1 s step to 70 mV (Figs 2C and D, and 7C). In activation curves (Fig. 4A, and Fig. 8A and B) all tail currents were normalized to maximum tail current measured at −100 mV in successive increments of 10 ms at +40 mV.
Figure 2. I-V and G-V relationships of HERG C-terminal deletion mutants.
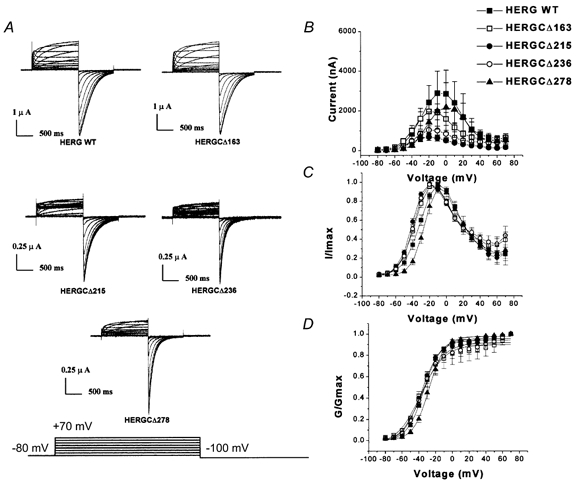
A, current traces recorded from HERG C-terminal deletion mutants. In these experiments cells were held at −80 mV and voltage commands were given from −80 to +70 mV for 1 s in steps of 10 mV and each step was followed by a step to −100 mV. B, peak amplitude I-V plots for HERGCΔ163 to HERGCΔ278 (symbols are indicated in figure). Deletion of amino acid residues from the C-terminus reduced the amplitude of HERG currents. One-way ANOVA showed that the mean currents of these constructs at each voltage were significantly different at the 0.05 level (n = 24, P = 0.0023). Deletion of more than 300 amino acid residues from the C-terminus resulted in no detectable current. C, normalized I-V plot for HERGCΔ163 to HERGCΔ278. D, normalized G-V plot for HERG C-terminal deletion mutants. Deletion of amino acid residues from the C-terminus of HERG had little effect on the slope of the steady-state activation curve. Slope (mV, mean ±s.e.m.) and V1/2 (mV, mean ±s.e.m.) values were 10.1 ± 0.7, −33.9 ± 0.8 for WT; 14.3 ± 2.1, −37.9 ± 2.6 for HERGCΔ163; 11.1 ± 1.0, −36.5 ± 1.2 for HERGCΔ215; 13.3 ± 1.8, −36.1 ± 2.1 for HERGCΔ236; and 9.5 ± 0.46, −27.7 ± 0.5 for HERGCΔ278. One-way ANOVA showed that the voltage dependences were not significantly different at the 0.05 level (n = 16 for each construct, P = 0.999).
Figure 7. I-V and G-V relationships of HERG double-deletion mutants.
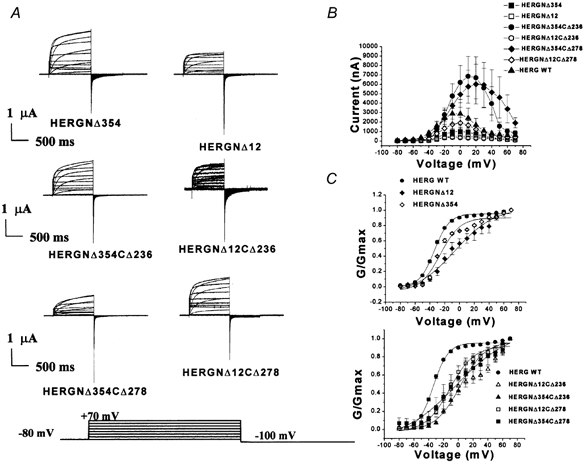
A, family of current traces recorded from HERG N-terminal deletion and N- and C-terminal double-deletion mutants with different test potentials. Outward currents were recorded in response to a voltage protocol in which commands were given from −80 to +70 mV for 1 s in steps of 10 mV from a holding potential of −80 mV; repolarization to −100 mV elicited inward tail currents. B, I-V relationships indicate that two double-deletion mutants, HERGNΔ354CΔ236 and HERGNΔ354CΔ278, had larger outward currents than WT channels (n = 16, P = 0.000003 for HERGNΔ354CΔ236 and HERGNΔ354CΔ278, and P = 0.000001 for the others). C, G-V relationships indicate that deletion of residues from the N-terminus and both the N- and C-termini of HERG shifted the activation to more positive voltages (n = 13, F= 0.747, P = 0.6). Slope (mV, mean ±s.e.m.) and V1/2 (mV, mean ±s.e.m.) values were 10.1 ± 0.7, −33.9 ± 0.8 for WT HERG; 16.1 ± 2.5, −22.1 ± 2.4 for HERGNΔ12CΔ236; 17.0 ± 1.7, +2.6 ± 1.7 for HERGNΔ354CΔ236; 16.5 ± 1.9, −11.6 ± 1.9 for HERGNΔ12CΔ278; 36.4 ± 10.7, −2.6 ± 7 for HERGNΔ354CΔ278; 26.7 ± 4.8, −5.9 ± 3.9 for HERGNΔ12; and 13.3 ± 2.7, −26 ± 3 for HERGNΔ354.
Figure 4. Activation and inactivation of HERG C-terminal deletion mutants.
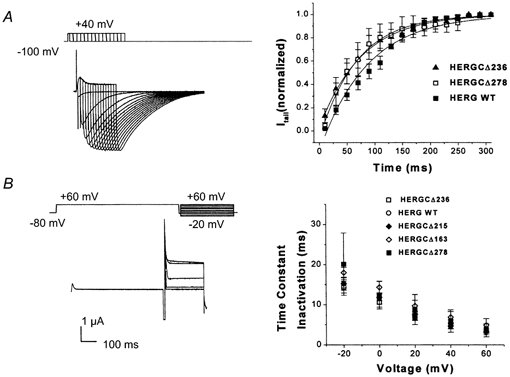
A, activation kinetics. Normalized tail currents were plotted versus activation pulse duration for WT HERG, HERGCΔ236 and HERGCΔ278 at +40 mV (only two fast deactivation phenotypes were measured). This protocol consisted of depolarizing steps, each followed by repolarization to −100 mV, given in successive increments of 10 ms at +40 mV. Slope (ms, mean ±s.e.m.) and time constant (ms, mean ±s.e.m.) values were −1.17 ± 0.05, 87.1 ± 5.9 for WT; −0.97 ± 0.02, 76.9 ± 4.9 for HERGCΔ236; and −1.06 ± 0.03, 69.0 ± 3.0 for HERGCΔ278. Activation time constants were not significantly different at the 0.05 level (n = 14, P = 0.764). B, in these experiments, channels were fully activated (and inactivated) by a pulse to +60 mV for 5 s. During a subsequent pulse to −80 mV channels recovered from inactivation. Then, prior to deactivation, a range of depolarizing potentials from +60 to −20 mV were given and channels re-entered the inactivated state. Inactivation time constants of HERG C-terminal deletion mutants were plotted versus voltage and were not different from those of WT channels at the 0.05 level (n = 12, P = 0.901).
Figure 8. Activation kinetics of double-deletion mutants.
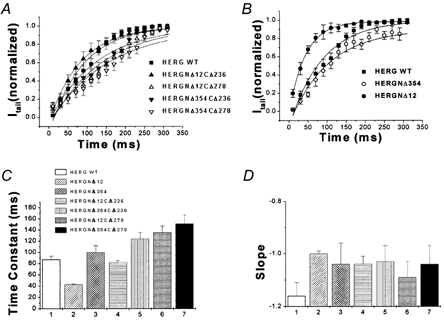
Activation rates of double-deletion mutants were significantly slower than those of WT channels and those of N- and C-terminal single-deletion mutants. A, normalized tail currents were plotted versus activation pulse duration for WT HERG, HERGNΔ12CΔ236, HERGNΔ12CΔ278, HERGNΔ354CΔ236 and HERGNΔ354CΔ278 at +40 mV (symbols indicated in figure). Activation time constants were fitted with a single-exponential function (see Fig. 4 for details; n = 17, P = 0.408). B, activation time constants at +40 mV for WT HERG, HERGNΔ12 and HERGNΔ354 are shown (n = 17, P = 0.044). Time constants were calculated from exponential fits (symbols indicated in figure). C, activation time constants of WT HERG, HERGNΔ12CΔ236, HERGNΔ12CΔ278, HERGNΔ354CΔ236, HERGNΔ354CΔ236, HERGNΔ12 and HERGNΔ354 are summarized in this bar graph (n = 17). D, slope values of activation curves at +40 mV for the constructs above (n = 17).
Deactivation curves were determined by activating channels from rest (-80 mV) with a 1 s pulse to +20 mV and measuring the closure of the channels during a range of pulses from 0 to −120 mV for 3 s. Deactivation τ values were derived from a Chebyshev fit to the deactivating current of the equation y=A0+A1e-t/τ1+A2e-t/τ2). Fitting began within 5 ms of the peak of the tail current, with the first cursor of the fitting window advanced to the first point in time that did not force a fit to the recovery phase; the second cursor was at the end of the 1.5 s pulse to −120 mV. The deactivation traces were best fitted with two exponentials.
The inactivation in this study was measured using a three-pulse voltage protocol. First, channels were fully activated (and inactivated) by a pulse to +60 mV for 5 s. During a subsequent pulse to −80 mV channels recovered from inactivation. Then, prior to deactivation, a range of depolarizing potentials from +60 to −20 mV were given and channels re-entered the inactivated state. The resulting decay traces represent the kinetics of channel inactivation. The inactivation time constant was similarly derived from a fit of the equation y=Ae-t/τ to the inactivating current. In this case the fitting window began as soon as the clamp settled, after approximately 2 ms, and continued to the end of the 300 ms pulse.
The activation time constant was derived using an envelope of tails protocol (Trudeau et al. 1995; Liu et al. 1996; Wang et al. 1998). This protocol consists of depolarizing steps, each followed by repolarization to −100 mV, given in successive increments of 10 ms at +40 mV. The deactivating phase of tail currents elicited for each trace was fitted with a double-exponential function and back extrapolated to the initial time point of repolarization. The current measured at t = 0 is assumed to be proportional to the total number of channels that have undergone the transition from the closed to the open state. The resulting values were plotted against the duration of depolarization and fitted to a single-exponential function to obtain the time constant of activation. These activating currents are actually sigmoidal in nature and so the fit to a single exponential represents a simplifying approximation.
The kinetics of recovery from inactivation were determined after a 1 s pulse to +60 mV maximally activated channels. Pulses to −10 to −100 mV then inactivated the channels and these currents were best fitted with a single-exponential function. Time constants derived from these fits were voltage dependent.
Data are expressed as means ±s.e.m. (n = number of oocytes used for each construct). Statistical comparisons of multiple means in plots of variables versus voltage were performed using one-way ANOVA.
RESULTS
Expression of HERG C-terminal deletion mutants
A schematic representation of the HERG C-terminal deletions is shown in Fig. 1A. A putative K+ channel structure is presented in Fig. 1B. The functional channel is formed from four α subunit polypeptides (MacKinnon, 1991; Liman et al. 1992). Each α subunit is thought to consist of six transmembrane segments (S1-S6), a pore-forming (H5) region and a cytoplasmic C-terminus and N-terminus (Ranganathan et al. 1996; Jan & Jan, 1997). Sequential deletion analyses indicate that at least 881 residues are necessary for channel expression since deletion of 311 or 344 residues from the C-terminal end resulted in non-functional channels (HERGCΔ311 and HERGCΔ344). These results dictated our construction of N-terminal and C-terminal double-deletion mutants, and these constructs are summarized in Fig. 1C.
Deletion of 163 and 215 residues from the C-terminal end resulted in authentic HERG-like currents. The kinetic properties were indistinguishable from those of the WT channel (G-V, inactivation, deactivation and activation curves). Figure 2A shows families of currents from WT HERG and four functional C-terminal deletion mutants (HERGCΔ163 to HERGCΔ278). Comparison of the fitted G-V curves of these HERG C-terminal deletion mutants with those of WT HERG indicated that there were no significant shifts (Fig. 2C and D). In contrast, I-V curves of C-terminal deletion mutants revealed that the total amplitudes of HERG currents were significantly reduced (Fig. 2B). Percentage reductions in amplitude for all voltages tested (-80 to +70 mV) were 27 % for HERGCΔ163, 75 % for HERGCΔ215, 59 % for HERGCΔ236 and 28 % for HERGCΔ278. Percentage reductions in amplitudes at −80 mV were 40.5 % for HERGCΔ163, 77.9 % for HERGCΔ215, 38.6 % for HERGCΔ236 and 45.2 % for HERGCΔ278. This finding supported earlier work suggesting that removal of amino acid residues from the C-terminus via missense mutations or frame shifts in arrhythmia patients may cause the reduction of total HERG current in association with LQT2 syndrome (Satler et al. 1996; Itoh et al. 1998; Berthet et al. 1999).
The reduction of the current amplitude increased with shortening of the HERG C-terminus (27 % for HERGCΔ163 and 75 % for HERGCΔ215), but decreased again with further shortening (59 % for HERGCΔ236 and 28 % for HERGCΔ278). Similar results have previously been reported for Shaker channels (Hopkins et al. 1994) and drk1 channels (VanDongen et al. 1990).
C-terminal deletions accelerate deactivation
Sequential deletion of 236 and 278 residues from the HERG C-terminus resulted in currents with faster deactivation when compared to the WT HERG channel (Fig. 3A). Fast deactivation time constants were significantly different for HERGCΔ236 and HERGCΔ278 whereas they were not significantly different for HERGCΔ163 and HERGCΔ215 (Fig. 3B). Slow deactivation time constants were significantly different for HERGCΔ236 and HERGCΔ278 whereas they were not significantly different for HERGCΔ163 and HERGCΔ215 (Fig. 3C). Additionally, the relative contributions of the fast and slow components of deactivation for the two shortest mutants (HERGCΔ236, HERGCΔ278) and WT were determined by calculating the fraction of fast deactivating component in the tail currents (data not shown). There were no statistical differences between the fractions of fast and slow components of deactivation between these mutants and WT (P = 0.93 for HERGCΔ236 and P = 0.1 for HERGCΔ278). The G-V relationship (Fig. 2D), and time constants for activation (Fig. 4A) and inactivation (Fig. 4B) were also unchanged. Smaller deletions failed to alter current from that seen in WT channels (Fig. 3A).
Figure 3. Deactivation kinetics of HERG C-terminal deletion mutants.
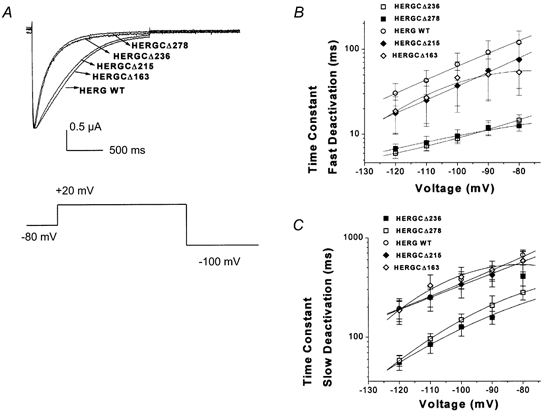
A, deactivation traces recorded from HERG C-terminal deletion mutants, indicating that the two shortest mutants, HERGCΔ236 and HERGCΔ278, had significantly faster deactivation rates than those of WT channels. In this experiment cells were depolarized by activating channels from rest (-80 mV) with a 1 s pulse to +20 mV and measuring the closure of the channels during a range of pulses to 100 mV for 3 s. B, plot of fast deactivation time constants versus voltage for C-terminal deletion mutants (at −120 mV they were 30.4 ± 9 ms for WT, 18.8 ± 9 ms for HERGCΔ163, 17.8 ± 7 ms for HERGCΔ215, 6.01 ± 0.8 ms for HERGCΔ236 and 7 ± 2 ms HERGCΔ278. One-way ANOVA showed that the mean currents of HERGCΔ236 and HERGCΔ278 at each voltage were significantly different from WT at the 0.05 level (n = 13, P = 0.0008) whereas they were not significantly different for HERGCΔ163 and HERGCΔ215 (n = 13, P = 0.166). C, plot of slow deactivation time constants versus voltage for C-terminal deletion mutants. One-way ANOVA showed that the mean currents of HERGCΔ236 and HERGCΔ278 at each voltage were significantly different from WT at the 0.05 level (n = 13, P = 0.045) whereas they were not significantly different for HERGCΔ163 and HERGCΔ215 (n = 13, P = 0.93). Both time constants were significantly shorter at the 0.05 level in the deletion mutants (at −120 mV they were 192 ± 40 ms for WT, 188 ± 54 ms for HERGCΔ163, 185 ± 42.5 ms for HERGCΔ215, 58 ± 16 ms for HERGCΔ236 and 56 ± 9 ms for HERGCΔ278).
Modulation of deactivation by the C-terminus is sensitive to elevated K+
The N-terminus in HERG slows deactivation by binding to the mouth of the pore. Elevation of external K+ concentration results in the removal of the N-terminus from the pore (‘knock off’ effect), and this increases the rate of channel deactivation (Armstrong, 1966; MacKinnon & Miller, 1988; Wang et al. 1998). The effect of high K+ concentration was lost when the N-terminus was deleted, supporting the interpretation that elevated K+ destabilizes the binding of the N-terminus (Wang et al. 1998).
Here, we used a high K+ concentration to probe the relationship between the C-terminus and channel blockade by the N-terminus. Elevation of external K+ concentration to 300 mm caused a significant acceleration of both slow and fast deactivation time constants in WT channels (Fig. 5A and B). In contrast, 300 mm K+ did not produce a significant change in the fast and slow deactivation time constants for HERGCΔ236 and HERGCΔ278 (Fig. 5C and D). This supports our hypothesis that the N- and C-termini of HERG could cooperate in regulating HERG deactivation kinetics. These results suggest that in C-terminal deletion mutants the affinity of the N-terminus for the mouth of the pore is reduced, but it is also possible that in HERG channels N-terminal deletions modify the function of the C-terminus.
Deactivation kinetics in N- and C-terminal double-deletion mutants
As described above, deactivation is accelerated by either N- or C-terminal deletion. In order to investigate the functional interactions of N- and C-termini further, we generated constructs simply by combining the two deletion mutants using a restriction site central to the N- and C-termini. If the double-deletion mutant exhibited the same fast deactivation kinetics as the individual deletions, this would suggest that these two domains share a common mechanistic pathway in the control of this process. Alternatively, if the kinetics were faster than in either single-deletion construct, then independent effects on deactivation would be implicated.
The double-deletion constructs HERGNΔ12CΔ236, HERGNΔ354CΔ236, HERGNΔ12CΔ278 and HERGNΔ354CΔ278 were able to form functional channels (Fig. 7A) and the fast deactivation time constants were the same as those of the mutants with individual deletions in either the C- or N-terminus alone (in the single-deletion constructs the fast deactivation time constants were similar; Fig. 6A and B).
Figure 6. Deactivation kinetics of double-deletion mutants.
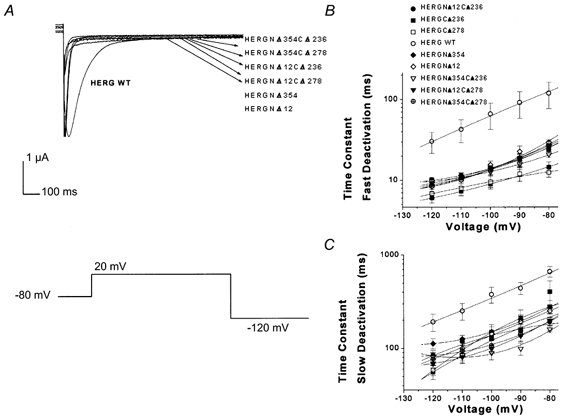
Deactivation in double-deletion mutants was not faster than in individual N-terminal and C-terminal deletion mutants. A, deactivation current traces from HERG C-terminal, N-terminal and double-deletion mutants. In this set of traces cells were held at −80 mV, depolarized to +20 mV for 1 s and subsequently hyperpolarized to −120 mV for 3 s. B, plot of fast deactivation time constants versus voltage for C-terminal, N-terminal and double-deletion mutants. Symbols for plots are indicated in the figure (n = 16, P = 0.383). C, plot of slow deactivation time constants versus voltage for C-terminal, N-terminal and double-deletion mutants. Symbols as in B (n= 16, P = 0.773).
Additional effects of double deletions
I-V relationships of these mutants are shown in Fig. 7B. This plot indicates that the two double-deletion mutants with the largest deletions (HERGNΔ354CΔ236 and HERGNΔ354CΔ278) had significantly larger currents than those of WT channels as well as the corresponding individual N- and C-terminal deletion channels. This was due to the slower inactivation kinetics of these constructs compared to WT channels. Thus the greater current measured for these constructs can be accounted for by alterations in gating behaviour.
The steady-state dependence of conductance on voltage for double-deletion mutants was determined from tail currents following long command pulses. The G-V relationship was determined in response to voltages from −100 to +70 mV in 10 mV steps from a holding potential of −80 mV. The plots were fitted by a single Boltzmann function (see Methods). In these constructs, the G-V relationship was shifted in the depolarizing direction, as indicated by the more positive V1/2 values. The steepness (slope factor, k) of the G-V curves was also reduced (Fig. 7C).
Activation plots for double-deletion and individual deletion mutants are shown in Fig. 8A and B (see Methods). Activation time constants and slope values of WT HERG, HERGNΔ12CΔ236, HERGNΔ12CΔ278, HERGNΔ354CΔ236, HERGNΔ354CΔ278, HERGNΔ12 and HERGNΔ354 are shown in Fig. 8C and D. All the double-deletion mutants showed slower time constants than WT channels. Slope values for double-deletion mutants were greater than for WT channels, indicating that the effects of the N- and C-termini on activation kinetics were additive.
Inactivation in double-deletion mutants was significantly slower than in the individual deletion mutants (either C-terminus or N-terminus deleted; Fig. 9A and B). This indicates that part of the C-terminus could contribute to the fast inactivation of HERG channels.
Figure 9. Inactivation kinetics of double-deletion mutants.
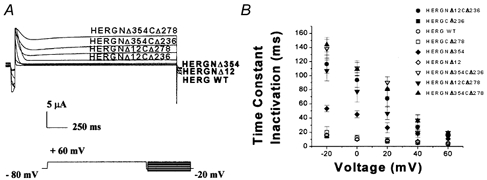
A, family of current traces recorded from WT HERG, HERGNΔ12CΔ236, HERGNΔ12CΔ278, HERGNΔ354CΔ236, HERGNΔ354CΔ278, HERGNΔ12 and HERGNΔ354 are shown. Inactivation pulse protocol consisted of a pulse to +60 mV for 5 s with a subsequent pulse to −80 mV and a range of depolarizing potentials from +60 to −20 mV. B, inactivation time constants of WT HERG, HERGNΔ12CΔ236, HERGNΔ12CΔ278, HERGNΔ354CΔ236, HERGNΔ354CΔ278, HERGNΔ12 and HERGNΔ354 were plotted against test voltage (n = 20, P = 0.0004). Symbols are defined in the figure. Inactivation was slower in deletion mutants than in WT channels.
Recovery from inactivation can be observed directly as the rising phase of the tail current upon repolarization. To measure the recovery from inactivation, families of tail currents were generated by first inactivating channels using a pulse to +60 mV and then repolarizing to a range of voltages from −10 to −100 mV. Single-exponential fits to the rising phases of the tail current yielded voltage-dependent time constants. Recovery from inactivation in all deletion mutants, including double-deletion mutants, was significantly faster than in WT channels (Fig. 10A–C). This indicates that both termini can contribute to the recovery from inactivation in HERG channels.
Figure 10. Recovery from inactivation in all deletion mutants.
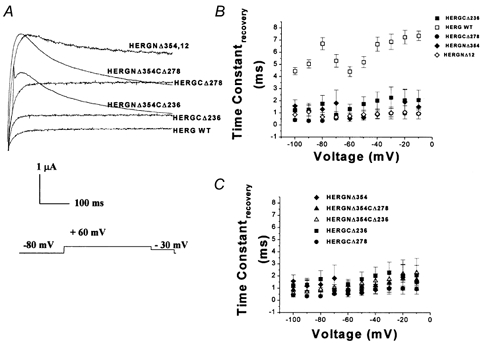
A, recovery from inactivation current traces. Pulse protocol consisted of a 1 s pulse to +60 mV followed by a pulse to −30 mV. B, time constants of recovery plotted versus test voltage for WT HERG, C-terminal deletion mutants HERGCΔ236 and HERGCΔ278, and N-terminal deletion mutants HERGNΔ12 and HERGNΔ354. Symbols are indicated in the figure (n = 24, P = 0). C, recovery plots for HERGNΔ354CΔ236, HERGNΔ354CΔ278, HERGCΔ236, HERGCΔ278 and HERGNΔ354 (n = 24, P = 0). All deletion mutants had significantly faster recovery than WT channels.
Activation of double-deletion mutants was significantly slower than in WT channels and in individual deletion mutants (Fig. 10A and B). Thus the two termini can contribute independently to the activation kinetics of the channels.
DISCUSSION
The present work explored the function of the C-terminus of HERG. Sequential deletions indicated that at least 881 residues are necessary to reconstruct HERG channel properties, since deletion of 311 and 344 residues from the C-terminal end eliminated functional expression in oocytes. Deletion of 163 and 215 residues from the C-terminal end resulted in authentic HERG currents, essentially indistinguishable from those of the WT channel in terms of the voltage dependence of conductance, inactivation, deactivation and activation kinetics. These results are consistent with the finding that the HERG C-terminus splice variant HERGUSO, which lacks 462 residues (splice from CNBD), did not form functional channels in Xenopus oocytes (Kupershmidt et al. 1998). Ligation of 104 residues at the C-terminal end of HERG rescued HERGUSO current. Our finding that deletion of 163 and 215 residues from the C-terminal end did not prevent the expression of a HERG-like current indicates that these residues are not necessary for the expression of functional channels, although these constructs showed reduced current amplitude. Thus, rescue of HERGUSO by ligation of the C-terminal 104 residues may reflect a robust assembly process which does not require a unique C-terminal sequence. Indeed, sequences capable of promoting assembly may be distributed broadly throughout the C-terminus. Some residues in the C-terminus may also make assembly more difficult, so that truncations of 236 to 278 residues actually aid in assembly and increase expression.
Similarly, Zhou et al. (1998) found that some LQT2 mutations in the C-terminus of HERG result in protein processing defects that block normal transport to the cell surface. These mutant channels are retained in the endoplasmic reticulum, where they are rapidly degraded. The V822M mutation (335 residues from the C-terminus, within the CNBD) possesses this phenotype, and several lines of evidence suggest that this mutant channel is retained in an intracellular compartment (Zhou et al. 1998). This mutation is a HERG C-terminus point mutation located within the same region as our non-functional deletion constructs (HERGCΔ311, 344), suggesting that even point mutations in this particular region affect the formation of functional channels.
The present study is also consistent with findings that truncation of the C-terminus via missense or frame-shift mutations identified in arrhythmia patients reduces the cell-surface expression of HERG channels, leading to LQT2 syndrome (Satler et al. 1996; Itoh et al. 1998; Berthet et al. 1999).
C- and N-terminal cooperation in deactivation
Wang et al. (1998) showed that the HERG N-terminus slows deactivation by a mechanism similar to that seen in N-type inactivation in Shaker channels, namely by binding to the internal mouth of the pore and blocking channel closure. These results suggest that the N-terminus stabilizes the open state and, by a separate mechanism, promotes C-type inactivation in HERG.
In this study, sequential deletion of 236 and 278 residues from the HERG C-terminus resulted in HERG-like currents with faster deactivation than WT HERG currents. Both high K+ and double-deletion experiments suggested that the C-terminus of HERG regulates deactivation kinetics by a mechanism that involves the N-terminus. Presumably, truncation of 236 or 278 amino acid residues from the HERG C-terminus disrupts a functional interaction between these domains and accelerates HERG deactivation.
Double-deletion mutants have different steady-state activation, and different activation, inactivation, and recovery of inactivation kinetics
All double-deletion mutants (HERGNΔ12CΔ236, HERGNΔ354CΔ236, HERGNΔ12CΔ278 and HERGNΔ354CΔ278) possessed G-V curves that were shifted to positive voltages relative to WT and individual deletion mutant channels. The activation kinetics were significantly slower than in the WT channels and the individual deletion mutants.
Most of the predicted phosphorylation sites in the HERG protein are located in the C-terminus. Previous studies showed that deletions in the HERG N-terminus do not alter the activation kinetics (Wang et al. 1998). A recent study showed that mutation of a protein kinase A (PKA) phosphorylation site altered HERG activation kinetics (Thomas et al. 1999). Cui et al. (2000) demonstrated that stimulation of PKA reduces HERG currents in transfected cells. In isolated guinea-pig cardiomyocytes, the rapid component of the delayed rectifier potassium current, IKr, which is produced by the HERG channel, was decreased by stimulating protein kinases, presumably PKA (Kiehn et al. 1998).
In this study we have shown that HERG C-terminal truncations (HERGCΔ236, HERGCΔ278) alter the activation kinetics of the channel, but double deletions produce dramatic reductions in the kinetics of channel activation. Thus the two termini can contribute independently to the activation kinetics of the HERG channel.
Inactivation in double-deletion mutants was significantly slower than in the individual deletion mutants, indicating that the C- and N-termini make additive contributions to the inactivation kinetics of HERG channels. As a consequence of the slow inactivation kinetics of double-deletion channels, outwardly rectifying currents were significantly increased (Fig. 7B). HERGNΔ354 was previously reported to have slower inactivation than WT channels, whereas HERGNΔ12 has the same inactivation rate as WT channels (Wang et al. 1998). Additionally Chen et al. (1999) demonstrated that mutation of a single residue (i.e. R56Q) altered the gating of both deactivation and inactivation. Thus, the N-terminus and the C-terminus of HERG both contribute to inactivation, and may cooperate with each other to maintain the N-terminal inactivation gate near the inner mouth of the channel as described by Jerng & Covarrubias (1997) for the mKv4.1 channel.
The present study also demonstrates that for HERG both the N- and C-termini can contribute to fast C-type inactivation. Deletion of both N- and C-termini slowed C-type inactivation. Recovery from inactivation for C- or N-terminal deletion mutants was significantly faster than in WT channels (HERGCΔ236, HERGCΔ278 and HERGNΔ12, HERGNΔ354). Double-deletion mutants exhibited fast recovery from inactivation (recovery from inactivation time constants were the same as those for individual N- or C-terminal deletion mutants). Thus, the effect on recovery from inactivation in double-deletion constructs was non-additive. This indicates that the N- and C-termini may functionally interact to regulate the slow recovery from inactivation in HERG channels.
Final remarks and conclusions
The reduction of the current amplitude of the HERG C-terminal deletion mutants supports the hypothesis that the C-terminus plays a role in the formation of functional channels, as suggested for other K+ channels (Hopkins et al. 1994; Kim et al. 1995; Ludwig et al. 1997; Wood & Vogeli, 1997; Arnold & Clapham, 1999; Bentley et al. 1999). In contrast, removal of the entire N-terminus still results in functional channels (Spector et al. 1996; Schonherr & Heinemann, 1996; Terlau et al. 1997; Wang et al. 1998). This contrasts sharply with Shaker-type channels where the N-terminus is necessary for assembly (Gulbis et al. 2000; Minor et al. 2000), and the recently crystallized T1 domain (Kobertz et al. 2000) plays an important role. Thus, in HERG channels assembly is more dependent on the C-terminus.
Although the functional characteristics of C-terminal deletion and the double-deletion mutants are clear, the mechanisms of all the effects remain obscure. However, some of the functional characteristics of the recently cloned Mirp1 channel gene, which has been shown to assemble with HERG channels and is proposed to form IKr currents in human heart, resemble those of our C-terminal deletion mutants. For example Mirp1-HERG complex channels show faster deactivation kinetics and their amplitudes are smaller than thoes of WT HERG channels (Abbott et al. 1999). Similar results were obtained with the C-terminal deletion mutants (Fig. 2B and Fig. 3). However this channel complex showed slow activation kinetics, whereas HERG C-terminal deletions did not alter the activation kinetics. Based on the overall similarities, we would speculate that Mirp1 interacts with the HERG C-terminus.
Our findings suggest that (a) the HERG C-terminus regulates channel deactivation by interacting with the N-terminus, (b) N- and C-termini make additive contributions to activation and inactivation kinetics of the channel, and (c) truncation of both the N- and C-termini causes dramatic increases in the HERG channel inactivation time constants. Thus, the N-terminus and the C-terminus of HERG are important determinants of inactivation, and may functionally interact with each other to maintain the inactivation gate near the inner mouth of the channel.
Acknowledgments
We would like thank Professor Gail Robertson for supporting this research (NIH grant NIH R02 HL55973), Jinling Wang for providing HERG N-terminus constructs and Professor Meyer Jackson for help in preparing the manuscript.
References
- Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- Anderson JA, Huprikar SS, Kochian LV, Lucas WJ, Gaber RF. Functional expression of a probable Arabidopsis thaliana potassium channel in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the USA. 1992;89:3736–3740. doi: 10.1073/pnas.89.9.3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM. Time course of TEA induced anomalous rectification in the squid giant axons. Journal of General Physiology. 1966;50:491–503. doi: 10.1085/jgp.50.2.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold DB, Clapham DE. Molecular determinants for subcellular localization of PSD-95 with an interacting K+ channel. Neuron. 1999;23:149–157. doi: 10.1016/s0896-6273(00)80761-8. [DOI] [PubMed] [Google Scholar]
- Bentley GN, Brooks MA, O'Neill CA, Findlay JB. Determinants of potassium channel assembly localised within the cytoplasmic C-terminal domain of Kv2. 1. Biochimica et Biophysica Acta. 1999;1418:176–184. doi: 10.1016/s0005-2736(99)00021-8. [DOI] [PubMed] [Google Scholar]
- Berthet M, Denjoy I, Donger C, Demay L, Hammoude H, Klug D, Schulze-Bahr E, Richard P, Funke H, Schwartz K, Coumel P, Hainque B, Guicheney P. C-terminal HERG mutations: the role of hypokalemia and a KCNQ1-associated mutation in cardiac event occurrence. Circulation. 1999;99:1464–1470. doi: 10.1161/01.cir.99.11.1464. [DOI] [PubMed] [Google Scholar]
- Chen J, Zou A, Splawski I, Keating MT, Sanguinetti MC. Long QT syndrome-associated mutations in the Per-Arnt-Sim (PAS) domain of HERG potassium channels accelerate channel deactivation. Journal of Biological Chemistry. 1999;274:10113–10118. doi: 10.1074/jbc.274.15.10113. [DOI] [PubMed] [Google Scholar]
- Cui J, Melman Y, Palma E, Fishman GI, McDonald TV. Cyclic AMP regulates the HERG K+ channel by dual pathways. Current Biology. 2000;10:671–674. doi: 10.1016/s0960-9822(00)00516-9. [DOI] [PubMed] [Google Scholar]
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- Daram P, Urbach S, Gaymard F, Sentenac H, Cherel I. Tetramerization of the AKT1 plant potassium channel involves its C-terminal cytoplasmic domain. EMBO Journal. 1997;16:3455–3463. doi: 10.1093/emboj/16.12.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demo SD, Yellen G. The inactivation gate of the Shaker K+ channel behaves like an open-channel blocker. Neuron. 1991;7:743–753. doi: 10.1016/0896-6273(91)90277-7. [DOI] [PubMed] [Google Scholar]
- Gulbis JM, Zhou M, Mann S, MacKinnon R. Structure of the cytoplasmic beta subunit-T1 assembly of voltage-dependent K+ channels. Science. 2000;289:123–127. doi: 10.1126/science.289.5476.123. [DOI] [PubMed] [Google Scholar]
- Hopkins WF, Demas V, Tempel BL. Both N- and C-terminal regions contribute to the assembly and functional expression of homo- and heteromultimeric voltage-gated K+ channels. Journal of Neuroscience. 1994;14:1385–1393. doi: 10.1523/JNEUROSCI.14-03-01385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Itoh T, Tanaka T, Nagai R, Kamiya T, Sawayama T, Nakayama T, Tomoike H, Sakurada H, Yazaki Y, Nakamura Y. Genomic organization and mutational analysis of HERG, a gene responsible for familial long QT syndrome. Human Genetics. 1998;102:435–439. doi: 10.1007/s004390050717. [DOI] [PubMed] [Google Scholar]
- Jan LY, Jan YN. Voltage-gated and inwardly rectifying potassium channels. Journal of Physiology. 1997;505:267–282. doi: 10.1111/j.1469-7793.1997.267bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng HH, Covarrubias M. K+ channel inactivation mediated by the concerted action of the cytoplasmic N- and C-terminal domains. Biophysical Journal. 1997;72:163–174. doi: 10.1016/S0006-3495(97)78655-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupp UB, Niidome T, Tanabe T, Terada S, Bonigk W, Stuhmer W, Cook NJ, Kangawa K, Matsuo H, Hirose T. Primary structure and functional expression from complementary DNA of the rod photoreceptor cyclic GMP-gated channel. Nature. 1989;342:762–766. doi: 10.1038/342762a0. [DOI] [PubMed] [Google Scholar]
- Kiehn J, Karle C, Thomas D, Yao X, Brachmann J, Kubler W. HERG potassium channel activation is shifted by phorbol esters via protein kinase A-dependent pathways. Journal of Biological Chemistry. 1998;273:25285–25291. doi: 10.1074/jbc.273.39.25285. [DOI] [PubMed] [Google Scholar]
- Kim E, Niethammer M, Rothschild A, Jan YN, Sheng M. Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature. 1995;378:85–88. doi: 10.1038/378085a0. [DOI] [PubMed] [Google Scholar]
- Kobertz WR, Williams C, Miller C. Hanging gondola structure of the T1 domain in a voltage-gated K+ channel. Biochemistry. 2000;39:10347–10352. doi: 10.1021/bi001292j. [DOI] [PubMed] [Google Scholar]
- Kupershmidt S, Snyders DJ, Raes A, Roden DM. A K+ channel splice variant common in human heart lacks a C-terminal domain required for expression of rapidly activating delayed rectifier current. Journal of Biological Chemistry. 1998;273:27231–27235. doi: 10.1074/jbc.273.42.27231. [DOI] [PubMed] [Google Scholar]
- Liman ER, Tytgat J, Hess P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- Liu DT, Tibbs GR, Siegelbaum SA. Subunit stoichiometry of cyclic nucleotide-gated channels and effects of subunit order on channel function. Neuron. 1996;16:983–990. doi: 10.1016/s0896-6273(00)80121-x. [DOI] [PubMed] [Google Scholar]
- Ludwig J, Owen D, Pongs O. Carboxy-terminal domain mediates assembly of the voltage-gated rat ether-à-go-go potassium channel. EMBO Journal. 1997;16:6337–6345. doi: 10.1093/emboj/16.21.6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon R. New insights into the structure and function of potassium channels. Current Opinion in Neurobiology. 1991;1:14–19. doi: 10.1016/0959-4388(91)90005-r. [DOI] [PubMed] [Google Scholar]
- MacKinnon R, Miller C. Mechanism of charybdotoxin block of the high conductance, Ca2+-activated K- channel. Journal of General Physiology. 1988;91:335–349. doi: 10.1085/jgp.91.3.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marten I, Hoshi T. Voltage-dependent gating characteristics of the K+ channel KAT1 depend on the N and C termini. Proceedings of the National Academy of Sciences of the USA. 1997;94:3448–3453. doi: 10.1073/pnas.94.7.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AG, Aldrich RW. Conversion of a delayed rectifier K+ channel to a voltage-gated inward rectifier K+ channel by three amino acid substitutions. Neuron. 1996;16:853–858. doi: 10.1016/s0896-6273(00)80105-1. [DOI] [PubMed] [Google Scholar]
- Minor DL, Lin YF, Mobley BC, Avelar A, Jan YN, Jan LY, Berger JM. The polar T1 interface is linked to conformational changes that open the voltage-gated potassium channel. Cell. 2000;102:657–670. doi: 10.1016/s0092-8674(00)00088-x. [DOI] [PubMed] [Google Scholar]
- Pajor AM, Hirayama BA, Wright EM. Molecular biology approaches to comparative study of Na+-glucose cotransport. American Journal of Physiology. 1992;263:R489–495. doi: 10.1152/ajpregu.1992.263.3.R489. [DOI] [PubMed] [Google Scholar]
- Perozo E, Papazian DM, Stefani E, Bezanilla F. Gating currents in Shaker K+ channels. Implications for activation and inactivation models. Biophysical Journal. 1992;62:160–168. 169–171. doi: 10.1016/S0006-3495(92)81802-7. discussion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan R, Lewis JH, MacKinnon R. Spatial localization of the K+ channel selectivity filter by mutant cycle-based structure analysis. Neuron. 1996;16:131–139. doi: 10.1016/s0896-6273(00)80030-6. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current. Journal of General Physiology. 1990;96:195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satler CA, Walsh EP, Vesely MR, Plummer MH, Ginsburg GS, Jacob HJ. Novel missense mutation in the cyclic nucleotide-binding domain of HERG causes long QT syndrome. American Journal of Medical Genetics. 1996;65:27–35. doi: 10.1002/(SICI)1096-8628(19961002)65:1<27::AID-AJMG4>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Sentenac H, Bonneaud N, Minet M, Lacroute F, Salmon JM, Gaymard F, Grignon C. Cloning and expression in yeast of a plant potassium ion transport system. Science. 1992;256:663–665. doi: 10.1126/science.1585180. [DOI] [PubMed] [Google Scholar]
- Schonherr R, Heinemann SH. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. Journal of Physiology. 1996;493:635–642. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector PS, Curran ME, Zou A, Keating MT, Sanguinetti MC. Fast inactivation causes rectification of the IKr channel. Journal of General Physiology. 1996;107:611–619. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Zhang W, Karle CA, Kathofer S, Schols W, Kubler W, Kiehn J. Deletion of protein kinase A phosphorylation sites in the HERG potassium channel inhibits activation shift by protein kinase A. Journal of Biological Chemistry. 1999;274:27457–27462. doi: 10.1074/jbc.274.39.27457. [DOI] [PubMed] [Google Scholar]
- Terlau H, Heinemann SH, Stuhmer W, Pongs O, Ludwig J. Amino terminal-dependent gating of the potassium channel rat eag is compensated by a mutation in the S4 segment. Journal of Physiology. 1997;502:537–543. doi: 10.1111/j.1469-7793.1997.537bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau MC, Warmke JW, Ganetzky B, Robertson G. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- VanDongen AM, Frech GC, Drewe JA, Joho RH, Brown AM. Alteration and restoration of K+ channel function by deletions at the N- and C-termini. Neuron. 1990;5:433–443. doi: 10.1016/0896-6273(90)90082-q. [DOI] [PubMed] [Google Scholar]
- Wang J, Trudeau MC, Zappia AM, Robertson GA. Regulation of deactivation by an amino terminal domain in human ether-a-go-go-related gene potassium channels. Journal of General Physiology. 1998;112:637–647. doi: 10.1085/jgp.112.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmke JW, Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proceedings of the National Academy of Sciences of the USA. 1994;91:3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood LS, Vogeli G. Mutations and deletions within the S8-S9 interdomain region abolish complementation of N- and C-terminal domains of Ca2+-activated K+ (BK) channels. Biochemical and Biophysical Research Communications. 1997;240:623–628. doi: 10.1006/bbrc.1997.7714. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Epstein ML, January CT. HERG channel dysfunction in human long QT syndrome. Intracellular transport and functional defects. Journal of Biological Chemistry. 1998;273:21061–21066. doi: 10.1074/jbc.273.33.21061. [DOI] [PubMed] [Google Scholar]