

Abstract
The normal influence of heart rate (HR) on cardiac contraction and relaxation in the mouse remains uncertain despite its importance in interpreting many genetically engineered models. Prior in vivo data have repeatedly shown positive effects only at subphysiological heart rates, yet depressed basal conditions and use of load-dependent parameters probably have an impact on these results.
Open-chest mice of various strains (n = 16, etomidate/urethane anaesthesia) were instrumented with a miniaturized pressure-volume catheter employing absolute left ventricular (LV) volume calibration. HR was slowed (< 400 beats min−1) using ULFS-49, and atrial or ventricular pacing was achieved via an intra-oesophageal catheter. Pressure-volume data yielded cardiac-specific contractile indexes minimally altered by vascular load.
At a resting HR of 600 beats min−1, peak pressure-rise rate (dP/dtmax) was 16 871 ± 2941 mmHg s−1 (mean ±s.d.) and the relaxation time constant was 3.9 ± 0.8 ms, similar to values in conscious animals. Within the broad physiological range (500–850 beats min−1), load-insensitive contractile indexes and relaxation rate varied minimally, whereas dP/dtmax peaked at 600 ± 25 beats min−1 and decreased at higher rates due to preload sensitivity. Contraction and relaxation were enhanced modestly (13–15 %) at HRs of between 400 and 500 beats min−1.
The minimal force-frequency dependence was explained by rapid calcium cycling kinetics, with a mechanical restitution time constant of 9 ± 2.7 ms, and by dominant sarcoplasmic reticular buffering (recirculation fraction of 93 ± 1 %).
The mouse normally has a very limited force-frequency reserve at physiological HRs, unlike larger mammals and man. This is important to consider when studying disease evolution and survival of genetic models that alter calcium homeostasis and SR function.
The dependence of cardiac muscle contraction and relaxation on stimulation frequency plays an important role in modulating heart function. Increasing steady-state frequency under physiological conditions generally enhances contraction and relaxation of myocytes, muscle and intact chambers, with 2-fold increases in contractility occurring when heart rate (HR) is increased 2–3 times in humans and other mammals (Freeman et al. 1987; Liu et al. 1993; Bers, 1999; Layland & Kentish, 1999).
In studies performed in isolated tissue or cells from small rodents, however, negative force-frequency relations (FFRs) have often been observed (Rice et al. 2000), raising controversy over the role and nature of this mechanism in these mammals. Similar observations have been made in intact open- and closed-chest mice (Hoit et al. 1997; Palakodeti et al. 1997; Kadambi et al. 1999), with FFRs having an ascending portion between 300 and 400 beats min−1 but a descending limb at higher rates. Yet unrestrained conscious mice have resting HRs in the range 550–700 beats min−1 (Fewell et al. 1997; Uechi et al. 1998; Chruscinski et al. 1999; Gehrmann et al. 2000; Janssen et al. 2000; Mills et al. 2000) and maximal exercise or adrenergic stimulated rates of 800–850 beats min−1 (Vornanen, 1992; Desai et al. 1997; Uechi et al. 1998; Chruscinski et al. 1999; Janssen et al. 2000). Such results question the role of a FFR in mice, firstly because the heart rate range is limited, and secondly because it appears normally to reside in a descending limb.
There are two important limitations to existing data that probably play a role in these observations. One is that the reported basal heart rates and contractile function of intact murine hearts have been substantially depressed compared with conscious values, and this could be accompanied by altered calcium cycling kinetics (Yue et al. 1985; Wier & Yue, 1986). In isolated rat trabeculae where negative FFRs have often been observed, studies performed at physiological temperature and Ca2+ levels reveal positive relations (Layland & Kentish, 1999). Secondly, prior studies have relied solely on the maximal rate of LV pressure rise (dP/dtmax) to index systolic performance. This parameter is highly preload dependent particularly in the murine heart (Georgakopoulos et al. 1998; Kass et al. 1998), and faster HRs typically diminish preload volume by limiting diastolic filling time. This latter limitation can be obviated by use of pressure-volume relations (Freeman & Colston, 1990; Liu et al. 1993) which help separate contractile from loading effects; however, such analysis has not been reported in mice.
Several excitation-contraction coupling features of the murine heart suggest that while not negative, the FFR should be quite modest over physiological HRs. Frequency-dependent potentiation arises from increased net Ca2+ entry via sarcolemmal channels (Lewartowski & Pytkowski, 1987) and increased Ca2+ loading of the sarcoplasmic reticulum (SR) (Rice et al. 2000). Like rat, the mouse has a robust SR that appears to provide more than 90 % of cytosolic calcium during a twitch (Li et al. 1997). This would predict little further increase in net SR loading, limiting extrasystolic and steady-state frequency potentiation. The present study tested this hypothesis, employing a novel miniature pressure-volume catheter to derive cardiac-specific indexes that are minimally altered by preload or afterload change (Georgakopoulos et al. 1998; Georgakopoulos & Kass, 2000). We report that the murine force-frequency relation is minimal over physiological heart rates consistent with rapid beat-to-beat force-interval behaviour, and that the apparent reported descending limb largely results from loading influences on commonly measured contraction parameters.
METHODS
Animal preparation and instrumentation
Mice (n = 16), 25–40 g (31.2 ± 4.6 g) body weight, consisting of FVB, Black Swiss and C57BL/6 strains were housed under diurnal lighting, and allowed food and water ad libitum. The study was approved and all animals were handled in accordance to the guidelines of the Animal Care and Use Committee of the Johns Hopkins University.
Animals were induced with methoxyflurane, anaesthetized with an intraperitoneal injection of urethane (750–1000 mg kg−1), etomidate (10 mg kg−1) and morphine (1–2 mg kg−1), and intubated with a blunt 19 G needle inserted via tracheostomy. Supplemental doses (0.05 ml) were given if increases in heart rate or blood pressure were observed in response to tail pinch with forceps. Ventilation was initiated with 100 % oxygen using a custom-designed, constant flow ventilator delivering a tidal volume of 6.7 μl g−1 at 120 breaths min−1. The left external jugular vein was exposed by blunt dissection and directly cannulated with a 30 G needle connected to an infusion pump. Modest volume expansion was provided (100–150 μl of 12.5 % human albumin) at 30 μl min−1.
Following stabilization, topical lidocaine gel was applied to the chest, and a lateral incision made at the xyphoid cartilage to expose the left ventricular (LV) apex. The 1.4F pressure-volume catheter (SPR-719, Millar) was inserted via an apical needle puncture, and advanced along the cardiac long axis. Proper catheter placement was verified by direct visualization of the electrodes at the apex, and of pressure-volume loop morphology.
A 2F pacing catheter (NuMed, Nicholville, NY, USA) consisting of four platinum electrodes 0.5 mm apart and a smooth tip was placed in the oesophagus, dorsal to the left atrium. Selection of specific electrode pairs enabled either atrial or ventricular capture using 5–7 V, 2 ms pulses (SD25, Grass Instruments, Quincy, MA, USA). Atrial pacing was used for all protocols except mechanical restitution (see below). Electrocardiograms (ECG) were recorded by needle electrodes placed on each limb. To control HR, we induced sinus bradycardia with the If inhibitor ULFS-49 (1,3,4,5-tetrahydro-7,8-dimethoxy-3-[3][2-(3,4-dimethoxyphenyl)ethyl]methylimino[propyl]-2H-3-benzazepin-2-on hydrochloride, Boehringer Ingelheim; 15–20 mg kg−1, i.p.) This agent has been shown to be free of inotropic effects (Kobinger & Lillie, 1984; Riley et al. 1987) and does not alter the time constant of mechanical restitution (Neumann et al. 1998).
Calibration of the volume signal was performed using a 5–10 μl bolus of 30 % hypertonic saline injected into the jugular vein (to determine the signal offset), and an ultrasound flow probe (1RB, Transonic) placed around the thoracic aorta (to determine signal gain). Data were digitized at 2 kHz and stored to disk for off-line analysis. At the end of the experiment, animals were killed with an overdose injection of urethane.
Protocol
Atrial pacing was initiated at 600 beats min−1(n = 11). Following collection of baseline data, pacing rate was reduced to 400 beats min−1, then increased in 50 beats min−1 increments to 850 beats min−1. Steady-state data were collected after 2 min at each frequency. Pacing was then initiated at the highest achievable rate, usually 850–950 beats min−1, and then abruptly terminated (returning to < 300 beats min−1) to evaluate potentiation and potentiation decay. The latter data was used to calculate the recirculation fraction, a measure of the amount of calcium cycled through the SR with each beat (Wohlfart, 1982; Seed et al. 1984).
Mechanical restitution curves (MRCs) were determined in five animals. The MRC relates contractile response of a single test beat to the time interval of this beat. It reflects net calcium cycling kinetics (Burkhoff et al. 1984; Wier & Yue, 1986; Schouten et al. 1987) related primarily to properties of the ryanodine release channel (Banijamali et al. 1991; Marx et al. 2000). To determine the MRC, hearts were ventricularly paced at 550 beats min−1, test beats were introduced every 10 cycles, and the test interval was varied from 35 to 250 ms. The same data were also used to determine the kinetics of restitution of LV relaxation (Prabhu & Freeman, 1992). Ventricular pacing was used to bypass the atrio-ventricular node which blocked or markedly delayed short test intervals. There was no significant functional difference between ventricular and atrial pacing (e.g. dP/dtmax: 16 059 ± 3151 for atrial pacing, 15 314 ± 2084 for ventricular pacing, P = not significant, n.s.).
Data analysis
Steady-state systolic and diastolic indexes were derived from 10–20 consecutive averaged beats. Systolic function was indexed by dP/dtmax, dP/dtmax normalized to instantaneous developed pressure (dP/dtmax/IP), ventricular end-systolic elastance (Ees), and preload-adjusted maximal power. The latter three measures are all minimally load dependent. Ees reflects chamber end-systolic stiffness, and is the slope of the end-systolic pressure-volume relation. For the present studies, Ees was approximated by the ratio of end-systolic pressure/volume (Suga et al. 1984). In addition, the end-systolic pressure-volume relation itself was examined in a subset of animals. Maximal ventricular power was obtained from the peak product of LV pressure times flow (derivative of LV volume signal), and normalized to end-diastolic volume to minimize preload dependence (Kass & Beyar, 1991). Isovolumic relaxation of LV pressure decline was assessed assuming a mono-exponential decay (Weiss et al. 1976) and by the pressure decay half-time (P1/2) (Mirsky, 1984).
The recirculation fraction (RF) was derived as previously described (Liu et al. 1993) using sequential cycles following abrupt termination of rapid atrial pacing. The amount of potentiation of any given beat (n) relative to steady-state (ss) (Rn= (dP/dtmax(n)/dP/dtmax(ss)) − 1) was plotted versus the value obtained for beat n+ 1; the slope of the relation was equal to the RF.
Mechanical restitution curves were constructed using Ees as the mechanical index (Freeman et al. 1987; Liu et al. 1993). This was chosen to avoid the influence of marked loading changes associated with varying test-stimulus intervals (TSI). TSI was measured from the ECG, and Ees for each test beat was normalized to the steady-state value (550 beats min−1). The resulting data were then fitted to a mono-exponential in the form:
where TSIo was the longest stimulus duration at which no mechanical force developed, τc was the time constant of mechanical restitution, and CRmax the plateau. For relaxation restitution, the portion of isovolumic pressure decay between dP/dtmin and 5 mmHg above EDP was used to derive the time constant of relaxation (τ) based on a logistic model (Matsubara et al. 1995). Each curve was normalized to the respective steady-state relaxation rate, and results fitted to a mono-exponential model: τ(TSI)/τ(550)=Ko+A[e-TSI/τCR], where Ko was the plateau asymptote, and τCR the time constant of isovolumic relaxation.
Statistical analysis
All data in the text and figures are presented as means ±s.d. The influence of heart rate on a given parameter was determined by repeated measures ANOVA, with post hoc analysis using Dunnett's multiple comparisons against the control heart rate of 600 beats min−1. Statistical significance was accepted at the P < 0.05 level.
RESULTS
Baseline haemodynamics
Baseline steady-state data were measured at an atrial pacing 16 871 ± 2941 mmHg s−1, and the pressure relaxation time constant was 3.9 ± 0.8 ms. LV systolic pressure was 115.4 ± 12.1 mmHg, LV diastolic pressure 6.0 ± 1.8 mmHg, ejection fraction 80.7 ± 4.4 % and cardiac output 12.5 ± 2.7 ml min−1. The values for systolic pressure, ejection fraction and cardiac output were close to those reported in conscious mice (Smits, 1998; Yang et al. 1999; Mills et al. 2000). Basal dP/dtmax was considerably higher than in many murine studies, but similar to that reported in awake animals (Palakodeti et al. 1997).
Force-frequency relations
Example steady-state pressure-volume loops at varying HRs are shown in Fig. 1A. These demonstrate the minimal decline in end-systolic volume with increasing heart rate, but a marked decline in chamber end-diastolic (preload) volume. When systolic function was assessed using dP/dtmax (Fig. 1B), we observed a biphasic relationship similar to that previously reported (Hoit et al. 1997; Palakodeti et al. 1997; Kadambi et al. 1999), although the peak frequency was at 600 beats min−1, higher than in prior studies. In contrast, dP/dtmax/IP, which adjusted for preload change, did not display the descending limb at faster rates (Fig. 1C), and only a modest 16.3 % rise between 400 and 600 beats min−1 (P < 0.0001). The extent of preload (EDV) change with HR was also directly examined (Fig. 1D) and revealed a gradual decline over the full measured range (P < 0.0001).
Figure 1.
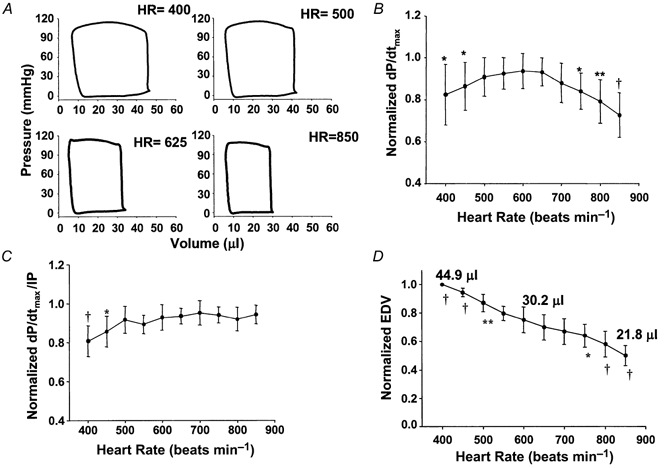
A, the effects of increasing atrial pacing rate on steady-state pressure-volume loops. There was a progressive decline in end-diastolic volume but little change in end-systolic volume, as heart rate increased. The loops maintained a normal box shape even at high rates. B, effect of increasing heart rate on dP/dtmax. Significantly lower values were observed at rates exceeding 600 beats min−1 and at reduced rates. C, the biphasic change in dP/dtmax is absent when the parameter is normalized to instantaneous developed pressure to diminish preload dependence (dP/dtmax/IP). In particular, this eliminated the descending limb at higher rates. D, effect of heart rate on end-diastolic volume shows marked preload dependence that can influence common contractile parameters such as dP/dtmax. *P < 0.05, **P < 0.001, †P < 0.0001 vs. 600 beats min−1.
To further test the importance of concomitant loading changes to the observed FFR, we examined pressure-volume relations. Figure 2 shows example pressure-volume loops measured during transient inferior vena caval occlusion. The upper left corners define the end-systolic pressure-volume relation. This relation shifted upward to the left (increased contractility) between heart rates of 420 and 600 beats min−1, but there was very little further change at faster rates (shown in the subsequent panels). Group pressure-volume analysis is provided for the end-systolic elastance and preload-adjusted maximal power in Fig. 3A. Both parameters displayed responses similar to that for dP/dtmax/IP. Ees rose 35 % from 400 to 600 beats min−1; however, in the physiological range there was minimal change. Virtually identical results were observed with the power index.
Figure 2.
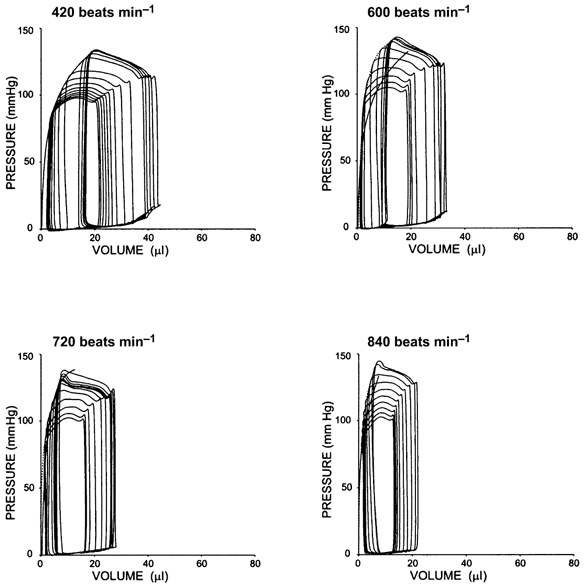
Example pressure-volume relations in an atrially paced murine heart over a broad range of heart rate. Data were recorded during transient preload reduction to generate these sets of relations. The upper left relation is the end-systolic pressure-volume relation (ESPVR) and reflects contractile function. Between 420 and 600 beats min−1, the relation shifted upward to the left (increased contractility). However, at heart rates above this (e.g. in the physiological range) there was very little further change. Similar data were obtained in four animals.
Figure 3.
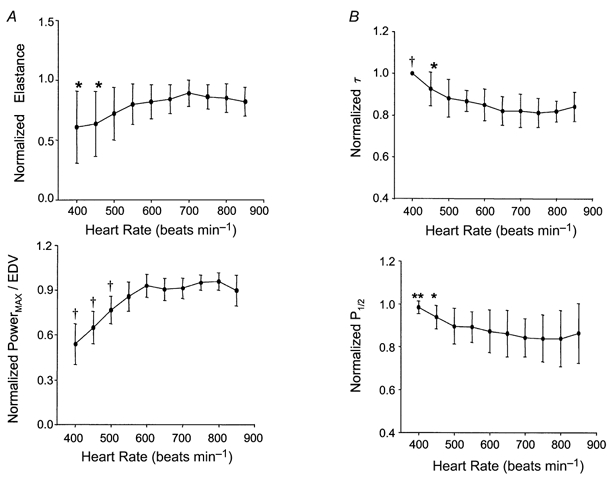
A, assessment of heart rate effects on contraction indexed by load-independent measures: end-systolic elastance (Ees) and preload-adjusted maximal power (powermax). Consistent with the ESPVR example in Fig. 2, neither index displayed a descending limb at fast heart rates, and both revealed only modest positive dependence on rate in subphysiological ranges (i.e. < 500 beats min−1). *P < 0.05, **P < 0.001, †P < 0.0001 vs. 600 beats min−1. B, similar analysis for ventricular relaxation parameters as assessed by a logistic relaxation time constant (τ) and pressure half-time (P1/2). Relaxation rate was significantly shortened with increasing rate only at subphysiological levels, with no further changes above 500 beats min−1. *P < 0.05, †P < 0.0001 vs. 600 beats min−1.
Figure 3B displays effects of HR on relaxation. Relaxation time constants declined modestly in the subphysiological HR range (−15.1 %, P < 0.0001; −11.3 %, P < 0.001; for τ and P1/2, respectively). However, rates remained static when HR was further varied over the physiological range. Thus, both contractile and relaxation enhancement with frequency was minimal in the normal operating HR range, with only modest effects observed at bradycardic rates.
Short-term force-interval relations
Figure 4 displays an example of the data used to determine the recirculation fraction. Following rapid pacing termination (Fig. 4A, arrow), dP/dtmax increased and then declined over ensuing beats. Heart rate and end-diastolic volume were unchanged during this period (see Fig. 4A, top), so dP/dtmax could be used to index contractile function. Plotting the data from beat n+ 1 versus beat n yielded a linear relationship (Fig. 4B, mean r2= 0.97) the slope of which averaged 93 ± 1 %(n = 10 mice). This reflects a high proportion of internally recycled calcium in the murine heart.
Figure 4.
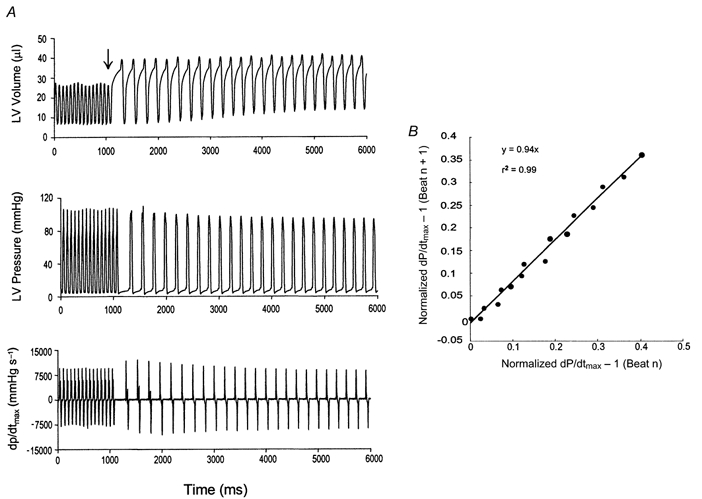
A, example of recirculation fraction determination in mice. Steady-state LV volume (top), pressure (middle) and dP/dtmax (bottom) are shown at rapid pacing rate. Following pacing cessation (arrow) there is initial potentiation in dP/dtmax followed by a gradual decline. End-diastolic volume and subsequent cycle length are minimally unaltered for each post-paced beat. B, geometric decay of potentiation demonstrated by linear plot of difference equation relation dP/dtmax for given beat to prior beat. The slope of this regression is the recirculation fraction (0.94 for this example).
Figure 5A shows typical data used to generate the mechanical restitution curve. The curves derived from group data are shown in Fig. 5B and C. Systolic function at short test intervals (∼65 ms) was remarkably close to that present at steady state. The mechanical restitution time constant based on Ees analysis averaged 9.0 ± 2.7 ms, with a plateau near 100 %. The shortest interval to elicit a mechanical response (TSIo) was 40 ± 4.7 ms. In contrast, when similar relations were derived using dP/dtmax (which is more affected by TSI-dependent loading changes than Ees), a longer time constant was derived (21.4 ms) and the MRC relation appeared to decline at longer TSIs. Relaxation restitution mirrored the rapid kinetics observed for systolic restitution with a time constant, τCR, of 10.68 ± 1.36 ms and a plateau of 100.9 ± 2.4 % (Fig. 5C).
Figure 5.
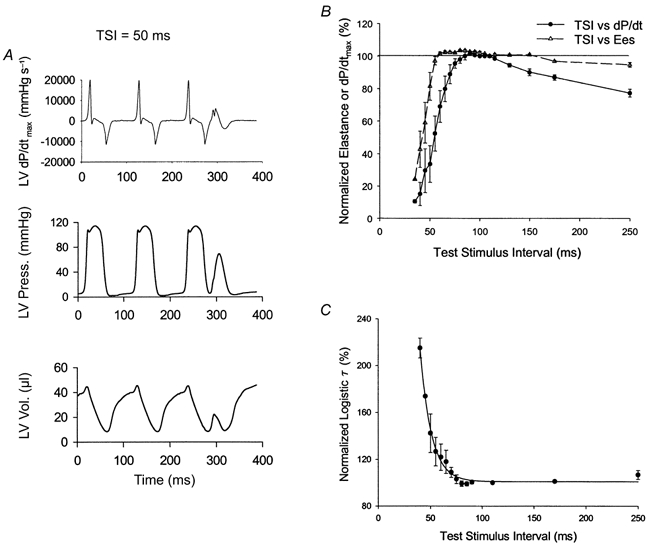
A, raw data tracings for left ventricular pressure, volume and dP/dtmax during determination of mechanical restitution. Data for a single test stimulus interval (TSI) at 50 ms is shown. Despite this very short interval, contraction is clearly observed – with nearly 50 % of the systolic pressure developed, and a small systolic ejection. B, mechanical restitution curves derived either from Ees (▵) or dP/dtmax (•) data. Data are combined by normalizing each relation to 100 % at the steady-state cycle length (100 ms). Restitution is very rapid - particulary as determined from less load-independent measures such as Ees. C, relaxation restitution curves for τ reveal little change in relaxation rate over a broad range of intervals (down to 75 ms).
DISCUSSION
The present data differ from previously published results with respect to several key findings. First, the murine force-frequency relation was flat above 600 beats min−1, with a modest positive relation observed only at lower rates. Secondly, there was no descending limb of the FFR at high frequencies. Lastly, and in concert with the minimal steady-state FFR, short-term force-interval data indicated a high percentage of Ca2+ recycling via the SR and very fast systolic and relaxation restitution processes.
Earlier studies of the FFR (Hoit et al. 1997; Palakodeti et al. 1997; Kadambi et al. 1999) in in situ murine hearts suggested heart rate as a potent modulator of cardiac function. However, heart rate was typically varied between 100 and 600 beats min−1, with a peak response observed at 250–400 beats min−1. Our data support a positive rate effect on both contraction and relaxation only over this lower frequency range. However, these rates are subphysiological for intact mice, and while this may reflect mechanisms that can enhance net intracellular Ca2+ loading, such as sarcolemmal exchange currents or SR leakage currents, it is less likely to be relevant to the intact animal.
Similar to prior studies, we found dP/dtmax displayed a biphasic response to increasing heart rate in the mouse. Such data have led to suggestions that the frequency at optimal dP/dtmax can serve to the index inotropic state (Kadambi et al. 1999). However, the biphasic response did not reflect actual contractile decline at higher frequencies, but rather the influence of preload decline. dP/dtmax varies directly with end-systolic elastance and peak rate of elastance rise, inversely with the time to end-systole (Tes), but also directly with end-diastolic volume (Little, 1985). At the lower range, increasing HR elevated Ees while Tes declined −19 ± 6 % (from 400–600 beats min−1), and thus despite concomitant EDV reduction dP/dtmax increased. However, Ees was little altered at faster rates (Fig. 2A) and Tes declined only an additional −5 %, so in this range, the decrease in preload dominated and dP/dtmax decreased. The lack of a true descending limb of contractile function in mice is consistent with recent data measured in rat trabeculae (Layland & Kentish, 1999), a species long considered to possess a descending limb of the FFR (Bers, 1999). This study performed at physiological temperature, calcium buffering and heart rate failed to find a descending limb, and suggested that its presence was probably dependent on experimental conditions or metabolic limitations as previously suggested (Schouten & ter Keurs, 1986).
The minimal steady-state force-frequency response in mice differs from that of larger mammals, such as dog and human, where heart rate typically varies over a 3-fold range, and contractile function more than doubles over the same range (Freeman & Colston, 1990; Liu et al. 1993). These results, however, are compatible with observed rapid calcium cycling and enhanced SR buffering as reflected in the mechanical restitution time constant and recirculation fraction. The recirculation fraction first reported by Wohlfart (1982) and Seed et al. (1984) derives from the geometric decay of contractile potentiation, reflecting the relative amount of Ca2+ recycled via the SR rather than extruded across the sarcolemmal membrane. One assumes that net gain in Ca2+ via sarcolemmal influx is negligible and that the relation between SR calcium release and peak mechanical performance is linear. The former assumption may be particularly valid in the mouse, which has an extremely short action potential, with no discernible calcium plateau (Nuss & Marban, 1994).
The present study reports the first in vivo RF in mice, and the value of 93 % is similar to the 90.3 % value reported for isolated murine myocytes (Li et al. 1997). Such values imply a dominant role for SR Ca2+ recycling, with ∼7 % of activator Ca2+ being extruded via the Na+-Ca2+ exchanger and other mechanisms. This also implies that a much smaller fraction (∼7 %) of activator Ca2+ enters the cell via the L-type Ca2+ channels or forward mode Na+-Ca2+ exchanger with each action potential, since steady-state Ca2+ influx effectively equals net Ca2+ efflux per cardiac cycle (Delbridge et al. 1997). This contrasts to 50 % values reported in canine and human heart (Seed et al. 1984; Liu et al. 1993; Bers, 1999) which leaves far more room to enhance SR uptake via Ca2+ entry through sarcolemmal channels.
The mechanical restitution time constant measures the net time required for total calcium cycling, and stems principally from the restitution time of the SR calcium release channel (Banijamali et al. 1991). This assumes a linear relation between calcium concentration and mechanical function; clearly an over-simplification. As in many studies performed in isolated intact mammalian hearts including human (Burkhoff et al. 1984; Yue et al. 1985; Wier & Yue, 1986; Freeman & Colston, 1990; Liu et al. 1993) the MRC is nearly mono-exponential, with a plateau at long test intervals. The very short time constant of < 10 ms is 5 times less than that previously published for wild-type mouse heart (Hoit et al. 2000). However, this value is consistent with data from conscious dogs or humans, which reported approximately 8-fold higher time constants at 8-fold slower heart rates compared with mice (Freeman & Colston, 1990; Liu et al. 1993). The disparity is probably due to basal cardiodepression (pacing rate of 300 beats min−1) as well as the use of dP/dtmax to index systolic function in these earlier studies. When dP/dtmax was employed as the systolic index, we also observed a longer time constant. This was related to loading changes, since at short test intervals dP/dtmax declines due to preload dependence, while at long intervals it decreases due to the reduction in arterial pressure. Both effects have been previously observed in isolated canine hearts (Burkhoff et al. 1984). The short time constant predicts that only heart rates faster than ∼30 ms cycle length (2000 beats min−1) would be expected to reduce contractile force due to inadequate diastolic time for full mechanical restitution. This heart rate is physiologically impossible, making it unlikely that a true descending limb would be observed in mice.
The present results revealing rapid cycling kinetics in the murine heart are supported by observations in isolated mouse myocytes showing marked resistance to Ca2+ overload even at supraphysiological levels of external Ca2+ (Gao et al. 1998). These authors concluded that the mouse must possess extremely efficient Ca2+ removal mechanisms. From the above results on recirculation fraction, the source of this removal is most probably the SR, which has been shown to have an initial maximal calcium uptake capability that is 2-fold faster than other species (McCollum et al. 1972; Levitsky et al. 1981; Luo et al. 1994).
One important contributor to rapid Ca2+ cycling kinetics and minimal FFR in mice may be enhanced basal sympathetic tone. Heart rate variability studies in conscious mice have revealed a predominant influence of sympathetic innervation, with minimal parasympathetic input (Mansier et al. 1998; Gehrmann et al. 2000; Janssen et al. 2000). While we did not directly measure plasma noradrenaline (norepinephrine) levels in the present study, recent data (Dash et al. 2001; M. De Biasi, personal communication) reports values between 3.6 and 6 ng ml−1. This is more than an order of magnitude above values reported in humans (Francis et al. 1990). Loss of sympathetic stimulation may explain commonly observed elevations of diastolic pressure in isolated mouse hearts when paced at fast stimulation rates. This can be reversed by isoproterenol infusion (Lim et al. 1999). Another potential factor is greater co-operativity of myofilament interaction following Ca2+ binding to troponin C. Gao et al. (1998) reported a Hill coefficient for the force-pCa relationship at near 10, allowing for large changes in force with relatively small changes in Ca2+ concentration. The anticipated effect would be to restore near-maximal mechanical function even with moderate increases in released calcium, further shortening the MRC time constant (Rice et al. 2000).
There are several limitations to this study. Although our basal haemodynamics were similar to those reported in conscious mice, the data were obtained under anaesthesia, and effects of this anaesthesia cannot be ruled out. However, urethane anaesthesia was chosen to minimize changes in autonomic neuromodulation (Billman et al. 1989), and should not have tipped the balance to favour sympathetic stimulation. Furthermore, it is unlikely that increased HR itself activated sympathetic tone since systolic pressure and cardiac output were minimally altered over the range of HRs studied. The volume catheter signal is linear over the normal range of mouse heart volumes, but does display a decline in gain at higher volumes (Georgakopoulos & Kass, 2000) as encountered with some post-pacing delays. This would result in overestimation of Ees at low rates. However, qualitatively similar results were observed with the power index and dP/dtmax/IP, both volume gain independent.
Conclusions
Over the physiological operating range of murine heart rate (550–850 beats min−1), the mouse heart displays minimal force-frequency dependence, which is only positive at rates below this range. Short-term force-interval analysis supports a dominant role for SR recycling and rapid recycling kinetics that largely underlie this behaviour. These features, which bear some similarities to other small rodents, such as rat, may have important practical implications regarding the interpretation of phenotypic changes associated with altered calcium handling in genetically mutated animals. Chronic assessment of cardiac phenotype and genotype, i.e. hypertrophy, failure evolution, expression of fetal response genes, are based on the behaviour existing in the intact conscious animal. Genetic mutations or alterations in adrenergic signalling or calcium handling proteins that would augment SR calcium loading might appear far more influential when studied in isolated or depressed hearts and muscle preparations than in the intact heart. In contrast, alterations depressing SR function and cycling are anticipated to be far more detrimental in mice than perhaps in humans or larger mammals. This suggests caution is warranted when extrapolating findings in mouse to human disease, especially as a basis for therapeutic strategies.
Acknowledgments
This study was supported by a grant from National Institutes of Health (NHLBI: PO1-HL59408).
References
- Banijamali HS, Gao WD, Macintosh BR, Ter Keurs HE. Force-interval relations of twitches and cold contractures in rat cardiac trabeculae. Effect of ryanodine. Circulation Research. 1991;69:937–948. doi: 10.1161/01.res.69.4.937. [DOI] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Boston, MA, USA: Kluwer Academic; 1999. [Google Scholar]
- Billman GE, Hoskins RS, Randall DC, Randall WC, Hamlin RL, Lin YC. Selective vagal postganglionic innervation of the sinoatrial and atrioventricular nodes in the non-human primate. Journal of the Autonomic Nervous System. 1989;26:27–36. doi: 10.1016/0165-1838(89)90104-5. [DOI] [PubMed] [Google Scholar]
- Burkhoff D, Yue DT, Franz MR, Hunter WC, Sagawa K. Mechanical restitution of isolated perfused canine left ventricles. American Journal of Physiology. 1984;246:H8–16. doi: 10.1152/ajpheart.1984.246.1.H8. [DOI] [PubMed] [Google Scholar]
- Chruscinski AJ, Rohrer DK, Schauble E, Desai KH, Bernstein D, Kobilka BK. Targeted disruption of the beta2 adrenergic receptor gene. Journal of Biological Chemistry. 1999;274:16694–16700. doi: 10.1074/jbc.274.24.16694. [DOI] [PubMed] [Google Scholar]
- Dash R, Kadambi VJ, Schmidt AG, Tepe NM, Biniakiewicz D, Gerst MJ, Canning AM, Abraham WT, Hoit BD, Liggett SB, Lorenz JN, Dorn GW, Kranias EG. Interactions between phospholamban and beta-adrenergic drive may lead to cardiomyopathy and early mortality. Circulation. 2001;103:889–896. doi: 10.1161/01.cir.103.6.889. [DOI] [PubMed] [Google Scholar]
- Delbridge LM, Satoh H, Yuan W, Bassani JW, Qi M, Ginsburg KS, Samarel AM, Bers DM. Cardiac myocyte volume, Ca2+ fluxes, and sarcoplasmic reticulum loading in pressure-overload hypertrophy. American Journal of Physiology. 1997;272:H2425–2435. doi: 10.1152/ajpheart.1997.272.5.H2425. [DOI] [PubMed] [Google Scholar]
- Desai KH, Sato R, Schauble E, Barsh GS, Kobilka BK, Bernstein D. Cardiovascular indexes in the mouse at rest and with exercise: new tools to study models of cardiac disease. American Journal of Physiology. 1997;272:H1053–1061. doi: 10.1152/ajpheart.1997.272.2.H1053. [DOI] [PubMed] [Google Scholar]
- Fewell JG, Osinska H, Klevitsky R, Ng W, Sfyris G, Bahrehmand F, Robbins J. A treadmill exercise regimen for identifying cardiovascular phenotypes in transgenic mice. American Journal of Physiology. 1997;273:H1595–1605. doi: 10.1152/ajpheart.1997.273.3.H1595. [DOI] [PubMed] [Google Scholar]
- Francis GS, Benedict C, Johnstone DE, Kirlin PC, Nicklas J, Liang CS, Kubo SH, Ridin-Toretsky E, Yusuf S. Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. A substudy of the Studies of Left Ventricular Dysfunction (SOLVD) Circulation. 1990;82:1724–1729. doi: 10.1161/01.cir.82.5.1724. [DOI] [PubMed] [Google Scholar]
- Freeman GL, Colston JT. Evaluation of left ventricular mechanical restitution in closed-chest dogs based on single-beat elastance. Circulation Research. 1990;67:1437–1445. doi: 10.1161/01.res.67.6.1437. [DOI] [PubMed] [Google Scholar]
- Freeman GL, Little, W C & O'Rourke RA. Influence of heart rate on left ventricular performance in conscious dogs. Circulation Research. 1987;61:455–464. doi: 10.1161/01.res.61.3.455. [DOI] [PubMed] [Google Scholar]
- Gao WD, Perez NG, Marban E. Calcium cycling and contractile activation in intact mouse cardiac muscle. Journal of Physiology. 1998;507:175–184. doi: 10.1111/j.1469-7793.1998.175bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrmann J, Hammer PE, Maguire CT, Wakimoto H, Triedman JK, Berul CI. Phenotypic screening for heart rate variability in the mouse. American Journal of Physiology. 2000;279:H733–740. doi: 10.1152/ajpheart.2000.279.2.H733. [DOI] [PubMed] [Google Scholar]
- Georgakopoulos D, Kass DA. Estimation of parallel conductance by dual-frequency conductance catheter in mice. American Journal of Physiology. 2000;279:H443–450. doi: 10.1152/ajpheart.2000.279.1.H443. [DOI] [PubMed] [Google Scholar]
- Georgakopoulos D, Mitzner WA, Chen CH, Byrne BJ, Millar HD, Hare JM, Kass DA. In vivo murine left ventricular pressure-volume relations by miniaturized conductance micromanometry. American Journal of Physiology. 1998;274:H1416–1422. doi: 10.1152/ajpheart.1998.274.4.H1416. [DOI] [PubMed] [Google Scholar]
- Hoit BD, Ball N, Walsh RA. Invasive hemodynamics and force-frequency relationships in open- versus closed-chest mice. American Journal of Physiology. 1997;273:H2528–2533. doi: 10.1152/ajpheart.1997.273.5.H2528. [DOI] [PubMed] [Google Scholar]
- Hoit BD, Kadambi VJ, Tramuta DA, Ball N, Kranias EG, Walsh RA. Influence of sarcoplasmic reticulum calcium loading on mechanical and relaxation restitution. American Journal of Physiology. 2000;278:H958–963. doi: 10.1152/ajpheart.2000.278.3.H958. [DOI] [PubMed] [Google Scholar]
- Janssen BJ, Leenders PJ, Smits JF. Short-term and long-term blood pressure and heart rate variability in the mouse. American Journal of Physiology. 2000;278:R215–225. doi: 10.1152/ajpregu.2000.278.1.R215. [DOI] [PubMed] [Google Scholar]
- Kadambi VJ, Ball N, Kranias EG, Walsh RA, Hoit BD. Modulation of force-frequency relation by phospholamban in genetically engineered mice. American Journal of Physiology. 1999;276:H2245–2250. doi: 10.1152/ajpheart.1999.276.6.H2245. [DOI] [PubMed] [Google Scholar]
- Kass DA, Beyar R. Evaluation of contractile state by maximal ventricular power divided by the square of end-diastolic volume. Circulation. 1991;84:1698–1708. doi: 10.1161/01.cir.84.4.1698. [DOI] [PubMed] [Google Scholar]
- Kass DA, Hare JM, Georgakopoulos D. Murine cardiac function: a cautionary tail. Circulation Research. 1998;82:519–522. doi: 10.1161/01.res.82.4.519. [DOI] [PubMed] [Google Scholar]
- Kobinger W, Lillie C. Cardiovascular characterization of UL-FS 49, 1,3,4,5-tetrahydro-7,8-dimethoxy-3-[3-][2-(3,4-dimethoxyphenyl)ethyl] methylimino]propyl]-2H-3-benzazepin-2-on hydrochloride, a new ‘specific bradycardic agent’. European Journal of Pharmacology. 1984;104:9–18. doi: 10.1016/0014-2999(84)90363-7. [DOI] [PubMed] [Google Scholar]
- Layland J, Kentish JC. Positive force- and [Ca2+]i-frequency relationships in rat ventricular trabeculae at physiological frequencies. American Journal of Physiology. 1999;276:H9–18. doi: 10.1152/ajpheart.1999.276.1.H9. [DOI] [PubMed] [Google Scholar]
- Levitsky DO, Benevolensky DS, Levchenko TS, Smirnov VN, Chazov EI. Calcium-binding rate and capacity of cardiac sarcoplasmic reticulum. Journal of Molecular and Cellular Cardiology. 1981;13:785–796. doi: 10.1016/0022-2828(81)90236-4. [DOI] [PubMed] [Google Scholar]
- Lewartowski B, Pytkowski B. Cellular mechanism of the relationship between myocardial force and frequency of contractions. Progress in Biophysics and Molecular Biology. 1987;50:97–120. doi: 10.1016/0079-6107(87)90005-8. [DOI] [PubMed] [Google Scholar]
- Li L, Satoh H, Ginsburg KS, Bers DM. The effect of Ca2+-calmodulin-dependent protein kinase II on cardiac excitation-contraction coupling in ferret ventricular myocytes. Journal of Physiology. 1997;501:17–31. doi: 10.1111/j.1469-7793.1997.017bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CC, Liao R, Varma N, Apstein CS. Impaired lusitropy-frequency in the aging mouse: role of Ca2+-handling proteins and effects of isoproterenol. American Journal of Physiology. 1999;277:H2083–2090. doi: 10.1152/ajpheart.1999.277.5.H2083. [DOI] [PubMed] [Google Scholar]
- Little WC. The left ventricular dP/dtmax-end-diastolic volume relation in closed-chest dogs. Circulation Research. 1985;56:808–815. doi: 10.1161/01.res.56.6.808. [DOI] [PubMed] [Google Scholar]
- Liu CP, Ting CT, Lawrence W, Maughan WL, Chang MS, Kass DA. Diminished contractile response to increased heart rate in intact human left ventricular hypertrophy: systolic versus diastolic determinants. Circulation. 1993;88:1893–1906. doi: 10.1161/01.cir.88.4.1893. [DOI] [PubMed] [Google Scholar]
- Luo W, Grupp IL, Harrer J, Ponniah S, Grupp G, Duffy JJ, Doetschman T, Kranias EG. Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circulation Research. 1994;75:401–409. doi: 10.1161/01.res.75.3.401. [DOI] [PubMed] [Google Scholar]
- McCollum WB, Besch HRJ, Entman ML, Schwartz A. Apparent initial binding rate of calcium by canine cardiac-relaxing system. American Journal of Physiology. 1972;223:608–614. doi: 10.1152/ajplegacy.1972.223.3.608. [DOI] [PubMed] [Google Scholar]
- Mansier D, Medigue C, Vardon G, Bestel J, Swynghedauw B. Lack of vagal tone in mice. FASEB Journal. 1998;12 Abstract. [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Matsubara H, Takaki M, Yasuhara S, Araki J, Suga H. Logistic time constant of isovolumic relaxation pressure-time curve in the canine left ventricle. Better alternative to exponential time constant. Circulation. 1995;92:2318–2326. doi: 10.1161/01.cir.92.8.2318. [DOI] [PubMed] [Google Scholar]
- Mills PA, Huetteman DA, Brockway BP, Zwiers LM, Gelsema AJ, Schwartz RS, Kramer K. A new method for measurement of blood pressure, heart rate, and activity in the mouse by radiotelemetry. Journal of Applied Physiology. 2000;88:1537–1544. doi: 10.1152/jappl.2000.88.5.1537. [DOI] [PubMed] [Google Scholar]
- Mirsky I. Assessment of diastolic function: suggested methods and future considerations. Circulation. 1984;69:836–841. doi: 10.1161/01.cir.69.4.836. [DOI] [PubMed] [Google Scholar]
- Neumann T, Ravens U, Heusch G. Characterization of excitation-contraction coupling in conscious dogs with pacing-induced heart failure. Cardiovascular Research. 1998;37:456–466. doi: 10.1016/s0008-6363(97)00246-0. [DOI] [PubMed] [Google Scholar]
- Nuss HB, Marban E. Electrophysiological properties of neonatal mouse cardiac myocytes in primary culture. Journal of Physiology. 1994;479:265–279. doi: 10.1113/jphysiol.1994.sp020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palakodeti V, Oh S, Oh B-H, Mao L, Hongo M, Peterson KL, Ross J., Jr Force-frequency effect is a powerful determinant of myocardial contractility in the mouse. American Journal of Physiology. 1997;273:H1283–1290. doi: 10.1152/ajpheart.1997.273.3.H1283. [DOI] [PubMed] [Google Scholar]
- Prabhu SD, Freeman GL. Kinetics of restitution of left ventricular relaxation. Circulation Research. 1992;70:29–38. doi: 10.1161/01.res.70.1.29. [DOI] [PubMed] [Google Scholar]
- Rice JJ, Jafri MS, Winslow RL. Modeling short-term interval-force relations in cardiac muscle. American Journal of Physiology. 2000;278:H913–931. doi: 10.1152/ajpheart.2000.278.3.H913. [DOI] [PubMed] [Google Scholar]
- Riley DC, Gross GJ, Kampine JP, Warltier DC. Specific bradycardic agents, a new therapeutic modality for anesthesiology: hemodynamic effects of UL-FS 49 and propranolol in conscious and isoflurane-anesthetized dogs. Anesthesiology. 1987;67:707–716. [PubMed] [Google Scholar]
- Schouten VJ, ter Keurs HEDJ. The force-frequency relationship in rat myocardium. The influence of muscle dimensions. Pflügers Archiv. 1986;407:14–17. doi: 10.1007/BF00580714. [DOI] [PubMed] [Google Scholar]
- Schouten VJ, Van Deen JK, De Tombe P, Verveen AA. Force-interval relationship in heart muscle of mammals. A calcium compartment model. Biophysics Journal. 1987;51:13–26. doi: 10.1016/S0006-3495(87)83307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seed WA, Noble MI, Walker JM, Miller GA, Pidgeon J, Redwood D, Wanless R, Franz MR, Schoettler M, Schaefer J. Relationships between beat-to-beat interval and the strength of contraction in the healthy and diseased human heart. Circulation. 1984;70:799–805. doi: 10.1161/01.cir.70.5.799. [DOI] [PubMed] [Google Scholar]
- Smits JF, Debets JJ, Janssen BJ. Variability of cardiac output in conscious mice. FASEB Journal. 1998;12 doi: 10.1152/ajpregu.00406.2001. Abstract. [DOI] [PubMed] [Google Scholar]
- SUGA H, SAGAWA K. Instantaneous pressure-volume relationships and their ratio in the excised, supported canine left ventricle. Circulation Research. 1984;35:117–126. doi: 10.1161/01.res.35.1.117. [DOI] [PubMed] [Google Scholar]
- Uechi M, Asai K, Osaka M, Smith A, Sato N, Wagner TE, Ishikawa Y, Hayakawa H, Vatner DE, Shannon RP, Homcy CJ, Vatner SF. Depressed heart rate variability and arterial baroreflex in conscious transgenic mice with overexpression of cardiac Gsalpha. Circulation Research. 1998;82:416–423. doi: 10.1161/01.res.82.4.416. [DOI] [PubMed] [Google Scholar]
- Vornanen M. Maximum heart rate of soricine shrews: correlation with contractile properties and myosin composition. American Journal of Physiology. 1992;262:R842–851. doi: 10.1152/ajpregu.1992.262.5.R842. [DOI] [PubMed] [Google Scholar]
- Weiss JL, Frederiksen JW, Weisfeldt ML. Hemodynamic determinants of the time-course of fall in canine left ventricular pressure. Journal of Clinical Investigation. 1976;58:751–760. doi: 10.1172/JCI108522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wier WG, Yue DT. Intracellular calcium transients underlying the short-term force-interval relationship in ferret ventricular myocardium. Journal of Physiology. 1986;376:507–530. doi: 10.1113/jphysiol.1986.sp016167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlfart B. Analysis of mechanical alternans in rabbit papillary muscle. Acta Physiologica Scandinavica. 1982;115:405–414. doi: 10.1111/j.1748-1716.1982.tb07098.x. [DOI] [PubMed] [Google Scholar]
- Yang XP, Liu YH, Rhaleb NE, Kurihara N, Kim HE, Carretero OA. Echocardiographic assessment of cardiac function in conscious and anesthetized mice. American Journal of Physiology. 1999;277:H1967–1974. doi: 10.1152/ajpheart.1999.277.5.H1967. [DOI] [PubMed] [Google Scholar]
- Yue DT, Burkhoff D, Franz MR, Hunter WC, Sagawa K. Postextrasystolic potentiation of the isolated canine left ventricle. Relationship to mechanical restitution. Circulation Research. 1985;56:340–350. doi: 10.1161/01.res.56.3.340. [DOI] [PubMed] [Google Scholar]