

Abstract
Vascular endothelial growth factor (VEGF) increases hydraulic conductivity (Lp) in vivo. To determine the signal transduction cascade through which this is mediated, we measured the effect of inhibition of various signalling pathways on VEGF-mediated acute increases in Lp in individually perfused frog mesenteric microvessels.
VEGF receptors have previously been shown to activate phospholipase C-γ (PLCγ), protein kinase C (PKC) and MEK, the mitogen-activated and extracellular signal-related kinase (ERK) kinase. To determine the role of these signalling pathways we measured the effects of inhibitors of each on the VEGF-mediated increase in Lp.
VEGF-mediated increases in Lp were attenuated by pre-treatment with the PLC inhibitor U73122, but not affected by treatment with the inactive enantiomer U73343. The PLC inhibitor was also able to attenuate the increase in Lp mediated by the inflammatory mediator ATP.
Inhibition of either PKC or MEK activation using the selective inhibitors bisindolylmaleimide (BIM, 1 μm) and PD98059 (30 μm), respectively, did not change the VEGF-mediated increase in Lp. However, PD98059, BIM and U73122 all reduced phosphorylation of ERK1/2 determined by Western blot analysis with anti-phospho-ERK1/2 antibodies.
Furthermore, inhibition of the conversion of diacyl glycerol (DAG) to arachidonic acid, by perfusion with the DAG lipase inhibitor RHC80267 (50 μm), did not attenuate the increase in Lp brought about by VEGF.
These data suggest that VEGF acutely increases microvascular permeability in vivo through a mechanism that is dependent on PLC stimulation, but is independent of PKC or MEK activation or production of arachidonic acid from DAG. We therefore propose that VEGF acutely acts to increase Lp through the direct actions of DAG, independently of PKC or arachidonic acid.
Vascular endothelial growth factors (VEGFs) are a family of cytokines that act to increase the delivery of nutrients to tissue by three distinct mechanisms: new blood vessel growth (angiogenesis), increased blood flow (by vasodilatation) and increased vascular permeability (Bates et al. 1999). VEGF-A (generally referred to as VEGF) increases vascular permeability both acutely, over a period of a few minutes, and chronically, over a period of days. Overexpression of these growth factors has been demonstrated in a host of pathological conditions associated with increased angiogenesis and permeability, including all solid tumours so far studied, diabetic retinopathy, psoriasis and rheumatoid arthritis. VEGFs are currently being investigated as potential stimulators of new blood vessel growth in chronically underperfused tissue following myocardial infarction and peripheral ischaemia. Inhibitors of these growth factors or their receptors are being studied as putative anti-tumour agents. VEGFs have been shown to bind to three receptors: flt-1 (VEGFR-1), flk1/KDR (VEGFR-2) and flt-4 (VEGFR-2; found on lymphatic endothelial cells). Although some of the signalling pathways through which VEGFs act have been investigated in vivo (see Fig. 1), it is not known which of these are responsible for the increase in permeability. VEGF stimulation of flk1/KDR has been shown to result in tyrosine phosphorylation of phospholipase C-γ (PLC-γ) in vitro (Guo et al. 1995; Kroll & Waltenberger, 1997) and in vivo (Mukhopadhyay et al. 1998). VEGF has been shown to phosphorylate PLCγ in cultured human umbilical vein endothelial cells (HUVECs), an effect that was greatly attenuated by a monoclonal antibody directed against flk1/KDR and also by U73122, an inhibitor of PLC (Wu et al. 1999). Receptor tyrosine kinase-mediated PLCγ activation results in the production of inositol trisphosphate (IP3) and diacylglycerol (DAG) in large vessel endothelial cells in vitro (Xia et al. 1996). IP3, in turn, releases calcium from internal stores (Putney, 1990), which may then stimulate calcium influx by a capacitative entry pathway (Berridge, 1995). However, we have recently shown that VEGF acutely stimulates increased Lp and endothelial [Ca2+]i by a mechanism that is independent of the release of calcium from intracellular stores. Since the role of PLC in the VEGF-mediated increase in Lp is still unclear we have measured the effect of PLC inhibition in perfused intact capillaries of the frog mesentery.
Figure 1. Diagram to show potential signalling pathways for increased vascular permeability.
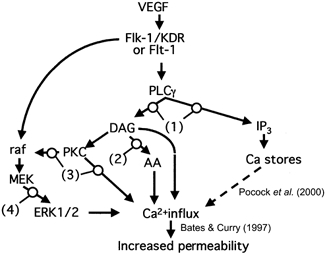
We have previously shown that VEGF increases intracellular calcium and vascular permeability through a store-independent calcium influx (Pocock et al. 2000; dashed line indicates that this pathway is known not to be the mechanism). In this set of experiments we determined whether inhibiting the action of PLC (1), DAG lipase (2), PKC (3) or MEK (4) would prevent the VEGF-mediated increase in vascular permeability. AA, arachidonic acid.
VEGF has also been shown to activate protein kinase C (PKC) both in cultured cells (Shen et al. 1999) and in vivo (Aiello et al. 1997). Activation of PKC leads, via raf activation, to phosphorylation of MEK, which in turn activates mitogen-activated protein (MAP) kinases (ERK1/2) (Mukhopadhyay et al. 1998). This pathway has been demonstrated for both VEGFR-1 (Sawano et al. 1997) and VEGFR-2 (Takahashi & Shibuya, 1997) in transfected cells. ERK1/2 activates phospholipase A2, which results in the production of prostaglandins such as prostacyclin (PGI2) (Wheeler-Jones et al. 1997). Prostaglandins cause both vasodilatation (Dusting et al. 1978) and increased solute flux (Williams & Morley, 1973). VEGF has been shown to both activate ERK1/2 and increase PGI2 synthesis in HUVECs. Both of these effects were abolished by the MEK inhibitor PD98059 (Wheeler-Jones et al. 1997).
Although much is known about the signalling pathways of VEGF in vitro, there is less evidence to establish how VEGF signals to increase microvascular permeability in vivo. We have previously shown that a short perfusion of a microvessel with VEGF increases microvascular permeability in two distinct but related fashions: a transient acute increase that lasts for under 3 min, and a sustained chronic increase that occurs from minutes to hours after VEGF perfusion and resolves between 24 and 48 h after exposure to VEGF (Bates & Curry, 1996). The acute increase in permeability has been shown to be associated with an increase in the endothelial intracellular calcium concentration in vivo, by a mechanism that involves calcium influx across the plasma membrane (Bates & Curry, 1997). However, a recent study in our laboratory suggested that the acute VEGF-induced calcium influx is not mediated through calcium store depletion, since the calcium increase is not attenuated by the sarco-endoplasmic reticulum Ca2+-ATPase inhibitor thapsigargin (Pocock et al. 2000). This leads to the suggestion that, in vivo, this acute calcium influx may be independent of IP3, and therefore possibly PLC. Since VEGF is thought to signal through the activation of ERK1/2, we considered that this pathway may be the most likely one for VEGF-mediated permeability increases. The aims of the current study were to determine whether this VEGF-mediated acute increase in microvascular permeability was dependent on the activation of PLC, PKC or ERK1/2.
METHODS
Materials
All experiments were carried out on male leopard frogs, Rana temporaria (20–35 g, supplied by Blades, UK), following local and national guidelines, and under licence. At the end of the experiments, frogs were killed by destruction of the brain. All chemicals were purchased from Sigma (unless otherwise specified), Calbiochem (PD98059, bisindolylmaleimide (BIM), U73122 and U73343), or Alexis (RHC80267). Chemicals were made up in water, except PD98059 (19.11 mm in ethanol), BIM (10 mm in DMSO), U73122 and U73343 (10 mm in ethanol), and RHC80267 (50 mm in ethanol). VEGF was a generous gift of Genentech Inc.
Measurement of hydraulic conductivity (Lp)
Frogs were anaesthetised by immersion in 1 mg ml−1 MS222 (3-aminobenzoic acid ethyl ester) in water. The animal was then laid supine and the limbs secured lightly. A small incision (8–10 mm) was made in the right lateral skin and muscular body wall. The distal ileum was floated out and carefully draped over a 1 cm diameter transparent Perspex pillar. The ileum was supported with cotton wool soaked in frog Ringer solution (111 mm NaCl, 2.4 mm KCl, 1 mm MgSO4, 1.1 mm CaCl2, 0.20 mm NaHCO3, 2.63 mm Hepes acid and 2.37 mm Hepes sodium salt) if necessary. The pH of this solution was 7.40 ± 0.02 at room temperature. Anaesthesia was maintained by superfusion of the gut with 0.25 mg ml−1 MS222 in frog Ringer solution. The microvessels in the mesentery could be easily visualised under a Leitz inverted microscope (Leitz DMIL). A video camera (Panasonic WVBP32, 8 mm) was attached to the top of the microscope to allow binocular visualisation and simultaneous recording of a 270 μm segment of the vessel (out of a total length of 800–2000 μm). Capillaries were selected that had flowing blood, no white cells adhering to or rolling along the wall, a length of at least 800 μm with no side branches and a baseline Lp of less than 10 × 10−7 cm s−1 cmH2O−1. These vessels were true capillaries (divergent flow at one end and convergent at the other), and had a diameter of 15–35 μm. The video camera was connected through an electronic timer (ForA VT33) to a video cassette recorder (Panasonic AG7350, Panasonic, Bracknell, UK). The upper surface of the mesentery was kept continuously superfused with frog Ringer solution during the entire time that it was exposed. All experiments were carried out at room temperature (20–22 oC).
Measurement of baseline Lp
The Lp was measured using the Landis micro-occlusion method previously described (Michel et al. 1974), which has been extensively discussed in the literature (Curry et al. 1983), and adapted to measure rapid and chronic changes in Lp (Bates & Curry, 1996). Baseline Lp was defined as the conductivity during perfusion with 1 % bovine serum albumin (BSA) in frog Ringer solution. The vessel was cannulated with a glass micropipette filled with 1 % BSA in frog Ringer solution, with rat red blood cells as flow markers. The rat red cells were collected by direct cardiac puncture of 9-week-old halothane-anaesthetised rats (5 % halothane), and washed three times in frog Ringer solution before use. Rats were subsequently killed by cervical dislocation. The micropipette was refilled as previously described (Hillman et al. 2001). Baseline Lp was measured by occluding the vessel with a glass rod for 3–7 s, whilst perfusing it with 1 % BSA at a pressure of 30 cmH2O. Free flow was then allowed in the vessel for at least 7 s before another occlusion was made.
Calculation of Lp
The transcapillary water flow per unit area of capillary wall (Jv/S) was calculated from the initial velocity of the red cells (dl/dt) after occlusion, the capillary radius (r) and the length between the marker cell and the point of occlusion (l), all of which were measured off-line from the videotape:
| 1 |
Lp was calculated from the Starling equation:
| 2 |
where ΔP is the hydrostatic pressure difference and σΔΠ the effective oncotic pressure difference between the capillary and the interstitium. σΔΠ is assumed to be 3.6 cmH2O for a 1 % albumin solution.
Experimental protocol for measurement of Lp during perfusion with VEGF or ATP
After baseline Lp measurement the micropipette was refilled with a solution containing 1 nm VEGF or 30 μm ATP and the vessel occluded for 3–5 s as soon as possible to measure Lp. The occlusion was released and Lp measured approximately every 10 s over the next 2–3 min. The pipette was then refilled with 1 % BSA and the vessel perfused for at least 5 min to wash out the agonist, followed by perfusion with a modifying agent (10 min for U73122, U73343, BIM and RHC80267). Twenty minutes after perfusion with ATP or VEGF, Lp measurements were made and the pipette was then refilled with a combination of the same concentration of agonist as used previously and the modifying agent. Lp was measured over the next 2–3 min as described above. The vessel was washed out with 1 % BSA for a further 20 min, more Lp measurements were performed and then the vessel was perfused for a third time with the agonist used before. Previous work has shown that a 20 min recovery period is necessary for a second response to VEGF to be measured (Bates & Curry, 1996). All perfusates were made up in 1 % BSA in frog Ringer solution and contained rat red blood cells as flow markers. The time courses of the responses to VEGF or ATP (time-averaged Lp) were calculated by grouping each measurement of Lp, or Lp relative to baseline, within 15 s intervals (0–14.9 s, 15–29.9 s, etc.) for each vessel and then taking the mean and standard deviation of the group. This protocol was used to determine the effects of 1 μm BIM and 50 μm RHC80267 on VEGF-mediated increases in Lp, and 10 μm U73122 and 10 μm U73343 on VEGF- and ATP-mediated increases in Lp.
To determine the effects of PD98059 on Lp, the vessel was perfused with 30 μm PD98059 (dissolved in ethanol, and then diluted 1:637 in Ringer solution containing 1 % BSA for 20 min), and Lp measurements made. The vessel was then perfused with 1 nm VEGF and 30 μm PD98059 and Lp measured. Lp values were compared to Lp values of vessels not pre-perfused with PD98059. We have previously shown that 20 min incubation of frog tissue with 30 μm PD98059 completely abolishes VEGF-mediated ERK1/2 phosphorylation (Hillman et al. 2001).
Western blot analysis
In order to ensure that PD98059, U73122 and BIM block MAP kinase phosphorylation in frog tissue, frog lungs were processed for Western blot analysis with an antibody to phosphorylated p44/p42 MAP kinase. Lungs were removed from frogs pithed by cervical dislocation and destruction of the brain. Parasites were removed from the lung if present. An equivalent volume of phosphate-buffered saline (PBS) alone or containing 1 nm VEGF, 30 μm PD98059, 30 μm PD98059 + 1 nm VEGF, 10 μm U73122, 10 μm U73122 + 1 nm VEGF, 1 μm BIM, or 1 nm VEGF + 1 μm BIM was added to each sample. The samples were chopped up crudely on ice using a sterile scalpel blade and then incubated for 30 min at room temperature. After this they were snap-frozen in liquid nitogen. An equivalent volume of protease inhibitor cocktail (1 μg ml−1 leupeptin, 1 μg ml−1 pepstatin A, 400 μg ml−1 AEBSF (4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride) and 5 mg ml−1 EDTA (sodium salt in PBS)) was added. After thawing, the samples were mixed gently and then spun for 2 min at 2000 g at 4 oC. The supernatant was removed and discarded. Protein was then extracted by resuspending the pellet in an equal volume of non-denaturing extraction buffer (300 mm NaCl, 20 mm Tris and 10 mm EDTA, containing 2 % Triton X-100, 0.2 % sodium dodecyl sulfate (SDS), 2 mm sodium orthovanadate (Na3VO4) and 1 mm phenyl methyl sulfonyl fluoride) and homogenising on ice. Homogenates were centrifuged for 10 min at 2000 g at 4 oC to separate the protein from cell debris. Aliquots (10 μl) were removed to determine protein concentration by the Bradford method (BioRad). For each sample, 100 μg of protein was used for SDS-PAGE. The gel was electroblotted onto a polyvinylidine difluoride membrane at 250 mA for 3 h. The membrane was washed in 1 × PBS−0.1 % Tween 20 (PBS-T), then blocked in 10 % reconstituted non-fat dried milk (Marvel) in PBS-T for 1 h, and probed for 1 h with the anti-phosphorylated p44/p42 MAP kinase antibody at 1:200 in 5 % Marvel in PBS-T. The membrane was washed in PBS-T, incubated with horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody (DAKO 1:1000) for 1 h and then washed 4 times for 5 min in PBS-T. Protein was detected with enhanced chemiluminescence Western blotting detection reagent (Roche, UK) and exposed to photo-sensitive film for 1 min and developed. Bands present were measured against coloured molecular mass markers (BioRad) also loaded onto the gel. Further samples of frog lung were incubated in 10 μm U73122 and 1 μm BIM and combinations of these agents with VEGF, and run on a gel as described above. Relative intensities of bands were calculated by importing the scanned images into NIH Image and measuring the mean density for an area covering the largest band. Figures are representative of three gels run for each condition.
Statistics
Measurements of Lp during perfusion with VEGF were not normally distributed, therefore non-parametric statistics were used to compare and contrast Lp values (medians are shown for actual Lp values). Error ranges are demonstrated by interquartile range (IQR) values. Wilcoxon tests were used for paired statistical comparison, Mann-Whitney U tests for unpaired statistical comparison. A probability value of P < 0.05 was accepted as significant. The normalized changes in Lp are given as means ± standard error of the mean (s.e.m.) and differences were tested using Student's t test or ANOVA as appropriate.
RESULTS
Effects of MEK, PLC and PKC inhibitors on VEGF-induced ERK1/2 phosphorylation
In order to determine whether the inhibitors were able to affect their targets in Rana temporaria, the effect of VEGF on ERK phosphorylation was measured. Incubation of frog lungs with VEGF resulted in a significant increase in the amount of phosphorylated ERK detected using phospho-ERK-specific antibodies in the frog (Fig. 2), as we have previously described. This VEGF-dependent phosphorylation was significantly reduced in the presence of 10 μm U73122, 30 μm PD98059 and 1 μm BIM. These studies showed that VEGF-mediated ERK activation is dependent on PKC and PLC activation, and that the inhibitors could effectively block their respective targets.
Figure 2. Representative Western blots of frog lung tissue treated with saline or inhibitor, and with VEGF or VEGF and inhibitor.
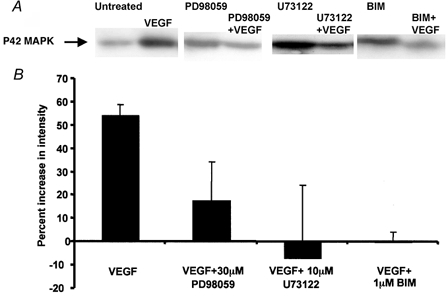
A, blots were probed with mouse anti-phospho-ERK1/2 primary antibody, and HRP-conjugated goat anti-mouse secondary antibody. B, mean ±s.e.m. relative densities, calculated from four independent experiments.
Effects of MEK inhibiton on VEGF-mediated increases in Lp
Measurements were made on 10 vessels perfused with PD98059 for 20 min and then perfused with 1 nm VEGF. This resulted in a rapid and significant 8.9 (± 3.3)-fold increase in Lp from 2.6 (± 0.9) × 10−7 to 10.6 (± 3.7) × 10−7 cm s−1 cmH2O−1(n = 10). The magnitude and time course were no different from the changes in Lp brought about by perfusion with 1 nm VEGF alone that we have previously published. The time-averaged mean changes in Lp for perfusion with PD98059 are shown in Fig. 3, and compared with 18 vessels perfused with VEGF alone (data from vessels used in previously published work; Bates & Curry, 1996, 1997). Perfusion with PD98059 therefore appeared to have no effect on the acute increase in Lp.
Figure 3. The effect of MEK inhibition on the VEGF-mediated increase in permeability.
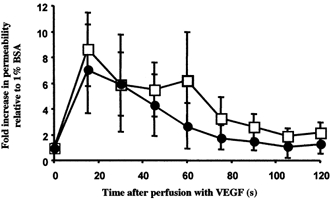
Time-averaged Lp measurements during 1 nm VEGF perfusion on 10 vessels with (•) and 18 vessels without (□) 20 min pre-treatment with 30 μm PD98059. Values are means ±s.e.m.
Effect of BIM on VEGF-induced changes in Lp
Measurements of Lp were made on 10 vessels perfused with 1 nm VEGF and then 1 nm VEGF with BIM (see Fig. 4A). Perfusion with 1 nm VEGF caused a 7.8 (± 2.6)-fold increase in Lp, from a median ± IQR baseline of 1.4 (± 0.2) × 10−7 to a peak of 6.8 (± 1.1) × 10−7 cm s−1 cmH2O−1 (P < 0.01). Perfusion with 1 μm BIM did not affect the Lp. After perfusion with 1 μm BIM, subsequent perfusion with 1 nm VEGF caused a 14.0 (± 5.9)-fold increase in Lp, from 1.4 (± 0.4) × 10−7 to 7.7 (± 5.8) × 10−7 cm s−1 cmH2O−1 (P < 0.01). This increase was not significantly different from that observed before perfusion with the inhibitor. In nine of these vessels, Lp measurements were also made after 20 min washout of BIM with 1 % BSA. Perfusion of 1 nm VEGF caused a 14.5 (± 9.6)-fold (n = 9) increase in Lp from a baseline of 1.4 (± 0.6) × 10−7 to a peak of 9.4 (± 7.5) × 10−7 cm s−1 cmH2O−1(P < 0.01). This increase was not significantly different from either the first or second responses. The overall increase in Lp with BIM (14 (± 5.9)-fold, n = 10) was not significantly different from that without BIM (11 (± 4.7)-fold, n = 19, ANOVA). The time course of the increase in Lp stimulated by VEGF was also similar between the vessels perfused with and without BIM.
Figure 4. The effect of BIM on the VEGF-mediated increase in Lp.
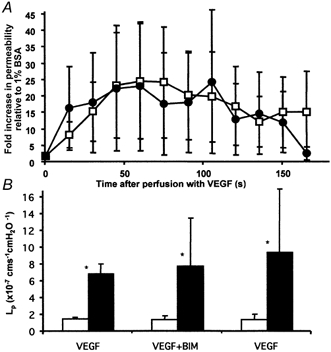
A, time-averaged Lp measurements on 10 vessels during 1 nm VEGF perfusion with (•) and without (□) 20 min pre-treatment with 1 μm BIM. Values are means ±s.e.m. B, median baseline (□) and peak (▪) Lp before, during and after perfusion with 1 μm BIM. * Significantly increased compared to baseline (P < 0.01). The peak increases are not significantly different from each other.
Effects of u73122 and u73343 on VEGF- and ATP-induced changes in Lp
Having established that neither BIM nor PD98059 was capable of reducing the increase in permeability, we were concerned to confirm our assumption that PLC was in fact involved in the VEGF-mediated increase in Lp. Measurements of Lp were made on 10 vessels perfused with 1 nm VEGF and then reperfused with VEGF and the PLC inhibitor U73122.
Perfusion with 1 nm VEGF caused an immediate and transient 5.9 (± 2.1)-fold increase in Lp, from a baseline of 3.4 (± 2.4) × 10−7 to a peak of 20.6 (± 10.1) × 10−7 cm s−1 cmH2O−1 (see Fig. 5). Perfusion with 10 μm U73122 did not significantly increase the Lp. After perfusion with U73122, subsequent perfusion with 1 nm VEGF caused a 2.7 (± 0.4)-fold increase in Lp, from 6.1 (± 6.6) × 10−7 to 13.2 (± 4.6) × 10−7 cm s−1 cmH2O−1. The increase in Lp was significantly smaller than with VEGF before U73122 perfusion (P < 0.05). After a 20 min washout of U73122 with 1 % BSA, perfusion of 1 nm VEGF caused an 8.8 (± 3.8)-fold (n = 8) increase in Lp from a baseline of 5.3 (± 5.6) × 10−7 to a peak of 21.4 (± 4.5) × 10−7 cm s−1 cmH2O−1. This increase was not significantly different from the increase observed before U73122 treatment. U73343, the inactive enantiomer of U73122, did not reduce the VEGF-mediated increase in permeability. Perfusion of seven vessels with VEGF following U73343 treatment resulted in an 8.3 (± 4.5)-fold increase in Lp, which was not significantly different from that with VEGF alone (6.2 (± 2.9)-fold, see Fig. 5 and Fig. 6).
Figure 5. The effect of PLC inhibitors on the VEGF-mediated increase in Lp.
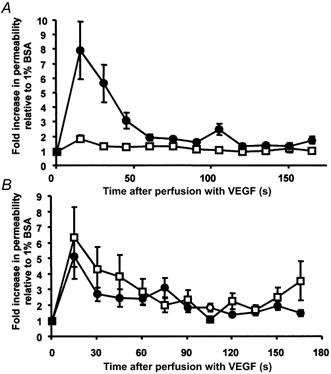
A, time-averaged Lp measurements on 10 vessels during 1 nm VEGF perfusion with (□) and without (•) 20 min pre-treatment with 10 μm U73122. B, time-averaged Lp measurements on seven vessels during 1 nm VEGF perfusion with (□) and without (•) 20 min pre-treatment with 10 μm of the inactive enantiomer of U73122, U73343. Values are means ±s.e.m.
Figure 6. Comparison of the effects of the PLC inhibitors on store-independent (VEGF) and store-mediated (ATP) increases in Lp.
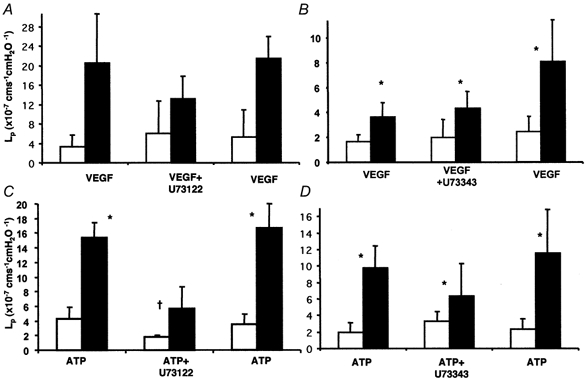
Median ± IQR for baseline (□) and peak (▪) measurements before, during and after perfusion with the inhibitors. A, effect of U73122 on the VEGF-mediated increase in Lp in 10 vessels. B, effect of U73343 on the VEGF-mediated increase in Lp in seven vessels. C, effect of U73122 on the ATP-mediated increase in Lp in seven vessels. D, effect of U73343 on the ATP-mediated increase in Lp in five vessels. *P < 0.05 compared to baseline; † significantly less than without inhibitor (P < 0.05).
In order to determine whether the dose of U73122 was able to abolish the increase in permeability brought about by other inflammatory mediators, the effect of U73122 on ATP-mediated increased permeability was investigated. Measurements were made in seven vessels perfused with ATP. In these vessels, 30 μm ATP stimulated a rapid and transient 4.7 (± 0.5)-fold increase in Lp from a mean baseline of 4.3 (± 1.5) × 10−7 cm s−1 cmH2O−1 to a peak of 15.4 (± 2.1) × 10−7 cm s−1 cmH2O−1. After perfusion with U73122 (10 μm), perfusion with ATP caused a 2.3 (± 0.4)-fold increase in Lp from 1.9 (± 0.2) × 10−7 to 5.7 (± 3.0) × 10−7 cm s−1 cmH2O−1. This increase was significantly smaller than the response to ATP before U73122 perfusion (P < 0.05). After a 20 min washout of U73122 with 1 % BSA, perfusion of 30 μm ATP caused a 4.8 (± 2.2)-fold (n = 4) increase in Lp from a baseline of 3.5 (± 1.5) × 10−7 to a peak of 16.7 (± 3.4) × 10−7 cm s−1 cmH2O−1. This increase was not significantly different from the increase observed before U73122 treatment (Student's t test). Treatment of five vessels with the inactive enantiomer U73343 did not affect the increase in Lp brought about by ATP (see Fig. 6).
Effect of DAG lipase inhibition on VEGF-induced changes in Lp
PLC is known to produce IP3 and DAG. We have previously shown that store-mediated calcium release (and therefore presumably IP3) is not necessary for VEGF to increase vascular Lp. Since PKC does not appear to be involved either, we tried to determine whether the increase in VEGF may be mediated by the breakdown of DAG by DAG lipase to arachidonic acid. Prostacyclin has already been shown to be involved with VEGF signalling in endothelial cells in culture, so this was a likely possibility. Measurements of Lp were made on seven vessels perfused with 50 μm RHC80267 (an inhibitor of DAG lipase) and 1 nm VEGF. VEGF caused an 8.1 (± 4.0)-fold increase in Lp, from a median baseline of 1.8 (± 0.4) × 10−7 to a peak of 8.8 (± 3.6) × 10−7 cm s−1 cmH2O−1. The vessels were subsequently perfused with 50 μm RHC80267 for 20 min and then perfused with 1 nm VEGF and RHC80267. This resulted in a 9.0 (± 5.3)-fold increase in Lp from 2.7 (± 1.0) × 10−7 to 10.6 (± 3.5) × 10−7 cm s−1 cmH2O−1. This increase was not significantly different from that without RHC80267. The time course of the increase in Lp was not significantly different between the two conditions either (see Fig. 7). Interestingly, perfusion of vessels with RHC80267 alone caused a transient increase in Lp. Lp measurements were made on three vessels during the first 3 min of perfusion with RHC80267. This resulted in a 5.7 (± 1.0)-fold transient increase in Lp from 1.1 (± 0.02) × 10−7 to 16.9 (± 5.25) × 10−7 cm s−1 cmH2O−1, which returned to control values within 3 min.
Figure 7. The effect of a DAG lipase inhibitor on the VEGF-mediated increase in Lp.
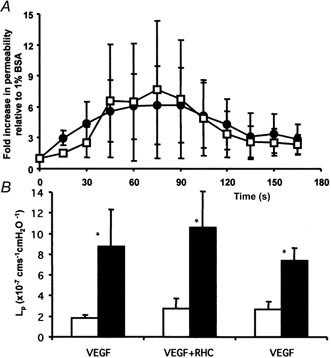
A, time-averaged Lp measurements on 10 vessels during 1 nm VEGF perfusion with (□) and without (•) 20 min pre-treatment with 50 μm RHC80267. Values are means ±s.e.m. B, median baseline (□) and peak (▪) Lp before, during and after perfusion with 50 μm RHC80267. * Significantly increased compared to baseline (P < 0.01). The peak increases are not significantly different from each other.
DISCUSSION
The signal transduction pathways of VEGF-mediated increases in Lp
Both vascular VEGF receptors (flk-1/KDR and flt-1) are receptor tyrosine kinases that have been shown to stimulate PLC by tyrosine phosphorylation in endothelial cells of a variety of origins in culture (Guo et al. 1995; Seymour et al. 1996; Sawano et al. 1997) and in vivo (Mukhopadhyay et al. 1998). PLC is known to generate IP3 and DAG in response to VEGF in cultured endothelial cells (Xia et al. 1996). IP3 in turn stimulates the release of calcium from endoplasmic reticulum stores. However, we have previously shown that VEGF increases endothelial [Ca2+]i and microvascular permeability in vivo through a calcium store-independent mechanism (Pocock et al. 2000). Since there is some confusion in the literature over the role of PLC in the VEGF-mediated increase in permeability (Waltenberger et al. 1994), and the only studies so far published on intact vessels (on isolated coronary venules ex vivo; Wu et al. 1999) are inconclusive since neither tachyphylaxis nor inactive enantiomer effects were investigated, we determined to investigate whether PLC inhibition would attenuate the increase in permeability. Our data show that the Lp increase induced by VEGF was attenuated in the presence of PLC inhibition, suggesting that VEGF increases microvascular permeability via a PLC-dependent mechanism. The fact that U73343 did not affect VEGF-mediated increases in Lp suggests that this is not a non-specific inhibition. Furthermore, ATP-induced increases in Lp, which have been shown to be dependent on calcium store release (Pocock et al. 2000), were also attenuated by PLC inhibition, by a similar magnitude to the attenuation of VEGF responses. These data agree with previous findings in endothelial cells grown in culture, and in isolated coronary venules ex vivo. However, this is the first study to show that the VEGF-mediated permeability response in vivo is reduced by PLC inhibitors.
Does VEGF signal through PKC to increase microvascular permeability?
We have shown, for the first time, that PKC inhibition with BIM did not affect the acute regulation of vascular permeability by VEGF in vivo. Although there is substantial evidence for the involvement of PKC and MAP kinase in VEGF-mediated angiogenesis (Yoshiji et al. 1999) there is less conclusive evidence to indicate the importance of this pathway in mediating the acute permeability changes described here. PKC inhibition has been shown to attenuate VEGF-induced increases in Lp in cultured HUVECs (Chang et al. 2000), but a different study (Kevil et al. 1998) showed that the VEGF-mediated increase in permeability across HUVEC monolayers was not PKC dependent. These studies investigated more chronic changes in permeability, occurring minutes to hours after VEGF perfusion. It may be that the chronic increase in permeability that we have previously described in vivo would be blocked by perfusion with PKC inhibitors (Bates & Curry, 1996; Bates, 1998). PKC inhibitors have also been shown to block increases in retinal transcapillary solute flux in vivo (Aiello et al. 1997), but, as far as we are aware, this is the first time that inhibitors such as BIM have been used in this frog model at room temperature. It is possible that BIM did not affect PKC activation in this model. However, our data from frog lung show that the previously described MEK activation (Bates et al. 2001) is inhibited by incubation with tissue for the same amount of time and at the same dose as used in the single vessel preparation. Therefore it seems likely that PKC is not involved in the acute permeability increase. The study that investigated the effect of PLC inhibitors on albumin permeability ex vivo also provided evidence that inhibition of PKC with BIM did attenuate the acute increase in diffusive permeability to albumin brought about by VEGF (Wu et al. 1999). However, the authors did not manage to demonstrate whether VEGF induced a second increase in permeability in the absence of BIM (or U73122). We have previously shown that VEGF does not elicit an equivalent second increase in Lp in the frog mesentery until 20 min after it has been washed out (Bates & Curry, 1996), whereas the experiments carried out by Wu et al. (1999) were only 10 min apart. This could be the reason that they failed to elicit a second response to VEGF. In contrast to some of these previous studies, but in agreement with Kevil et al. (1998), the experiments performed in the current study show that inhibiting PKC, using the inhibitor BIM, has no significant effect on the acute VEGF-induced Lp changes. These results therefore suggest that PKC activation is not important in mediating increases in microvascular permeability in vivo. This finding leads to the suggestion that PKC activation of MEK is unlikely to be involved in the permeability response to VEGF. The lack of an effect of PKC inhibitors on permeability does not rule out the possibility that VEGF may signal through the ras-raf-MEK-ERK pathway to induce permeability changes.
Does VEGF signal through the MEK-ERK pathway to increase microvascular permeability?
VEGF-induced activation of ERK has been demonstrated in vitro (Wheeler-Jones et al. 1997) and in vivo (Hillman et al. 2001). This pathway does not involve ras activation (Doanes et al. 1999). In this study, we have confirmed that VEGF indeed stimulates ERK in vivo, and that it is attenuated b. PKC inhibition. However, we have also shown that inhibiting ERK activation using PD98059 had no significant effect on the permeability increase induced by VEGF, indicating that, despite the fact that ERK was actually being activated, this pathway is not responsible for the increase in permeability in vivo. We have recently shown that MEK inhibition does not block the chronic increase in Lp brought about by VEGF perfusion in vivo either (Bates et al. 2001). It does, however, block the increase in compliance and diameter brought about by VEGF, showing both that PD98059 is effective in this model, and that ERK activation is part of the VEGF signalling cascade. This is in contrast to studies of endothelial cells in vitro, where ERK1/2 has been implicated in the regulation of permeability. This again may be due to the difference between the chronic change in permeability seen in vitro and the acute change seen in vivo. One likely explanation for this would be the more significant contribution of the extracellular matrix to the in vitro hydraulic conductivity, since the Lp values of monolayers reported in vitro are one to two orders of magnitude higher than those measured here (Albelda et al. 1988). This is therefore the first demonstration that the ERK1/2 pathway is not involved in the acute regulation of permeability by VEGF in capillaries in vivo.
Does VEGF increase microvascular permeability by DAG metabolism?
The finding that VEGF-mediated acute increases in Lp appear to be dependent on activation of PLC, but not PKC or MEK, together with previous findings that they are not dependent on release of calcium from stores, suggested that the products of DAG breakdown might be responsible. Since the acute increase in Lp is not dependent on the release of calcium from intracellular stores (Pocock et al. 2000), presumably IP3 activation of inositol phosphate receptors is not required. Therefore we turned our attention to the effects of DAG other than activation of PKC. DAG is broken down by DAG lipase to form arachidonic acid. This provides a substrate for prostaglandin production, which is known to mediate the VEGF-induced increase in vascular permeability in endothelial monolayers (Wheeler-Jones et al. 1997). However, surprisingly, there was no effect of DAG lipase inhibition by RHC80267 on the VEGF-mediated increase in Lp. On the contrary, there may have been a small (but not significant) increase in the VEGF-mediated increase in Lp. Furthermore, perfusion of vessels with RHC80267 on its own resulted in a transient increase in permeability. Since RHC80267 perfusion would be expected to increase DAG concentrations if it was effective in blocking DAG lipase, this is additional evidence for the hypothesis that DAG may directly increase permeability. Although it is difficult to draw any solid conclusions from these data, they are at least consistent with the hypothesis that VEGF increases vascular permeability by DAG activation of second messenger-operated calcium channels. DAG-mediated calcium channels have been described, and are members of the transient receptor potential channel family, for example TrpC6 (Hofmann et al. 1999).
In conclusion, we have demonstrated that VEGF signals through a pathway that involves activation of PLC to increase microvascular permeability in vivo and is not dependent on the activation of PKC or MAP kinase, despite the fact that these pathways are stimulated by VEGF. In addition a DAG lipase inhibitor did not reduce the increase in permeability. Our findings are therefore consistent with the hypothesis that VEGF increases vascular permeability through DAG production. One route for this activation would be through DAG-mediated second messenger-operated calcium channels.
Acknowledgments
The authors would like to thank Becky Foster, Cathy Powell and Cheryl Whittles for their technical assistance and support. Both D.O.B. and T.M.P. are supported by the British Heart Foundation, and this work was supported by the British Heart Foundation (PG97198, PG200030, FS200057) and the Wellcome Trust (no. 50742).
References
- Aiello LP, Bursell SE, Clermont A, Duh E, Ishii H, Takagi C, Mori F, Ciulla TA, Ways K, Jirousek M, Smith LE, King GL. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes. 1997;46:1473–1480. doi: 10.2337/diab.46.9.1473. [DOI] [PubMed] [Google Scholar]
- Albelda SM, Sampson PM, Haselton FR, McNiff JM, Mueller SN, Williams SK, Fishman AP, Levine EM. Permeability characteristics of cultured endothelial cell monolayers. Journal of Applied Physiology: Respiratory Environmental and Exercise Physiology. 1988;64:308–322. doi: 10.1152/jappl.1988.64.1.308. [DOI] [PubMed] [Google Scholar]
- Bates DO. The chronic effect of vascular endothelial growth factor on individually perfused frog mesenteric microvessels. Journal of Physiology. 1998;513:225–233. doi: 10.1111/j.1469-7793.1998.225by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates DO, Curry FE. Vascular endothelial growth factor increases hydraulic conductivity of isolated perfused microvessels. American Journal of Physiology. 1996;271:H2520–2528. doi: 10.1152/ajpheart.1996.271.6.H2520. [DOI] [PubMed] [Google Scholar]
- Bates DO, Curry FE. Vascular endothelial growth factor increases microvascular permeability via a Ca2+-dependent pathway. American Journal of Physiology. 1997;273:H687–694. doi: 10.1152/ajpheart.1997.273.2.H687. [DOI] [PubMed] [Google Scholar]
- Bates DO, Heald RI, Curry FE, Williams B. Vascular endothelial growth factor induces chronically increased vascular permeability and compliance through different signalling pathways. Journal of Physiology. 2001;533:263–272. doi: 10.1111/j.1469-7793.2001.0263b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates DO, Lodwick D, Williams B. Vascular endothelial growth factor and microvascular permeability. Microcirculation. 1999;6:83–96. [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Chang YS, Munn LL, Hillsley MV, Dull RO, Yuan J, Lakshminarayanan S, Gardner TW, Jain RK, Tarbell JM. Effect of vascular endothelial growth factor on cultured endothelial cell monolayer transport properties. Microvascular Research. 2000;59:265–277. doi: 10.1006/mvre.1999.2225. [DOI] [PubMed] [Google Scholar]
- Curry FE, Huxley VH, Sarelius IH. Cardiovascular Physiology. Techniques in the Life Sciences. New York: Elsevier; 1983. Techniques in microcirculation: measurement of permeability, pressure and flow; pp. 1–34. [Google Scholar]
- Doanes AM, Hegland DD, Sethi R, Kovesdi I, Bruder JT, Finkel T. VEGF stimulates MAPK through a pathway that is unique for receptor tyrosine kinases. Biochemical and Biophysical Research Communications. 1999;255:545–548. doi: 10.1006/bbrc.1999.0227. [DOI] [PubMed] [Google Scholar]
- Dusting GJ, Moncada S, Vane JR. Vascular actions of arachidonic acid and its metabolites in perfused mesenteric and femoral beds of the dog. European Journal of Pharmacology. 1978;49:65–72. doi: 10.1016/0014-2999(78)90222-4. [DOI] [PubMed] [Google Scholar]
- Guo D, Jia Q, Song HY, Warren RS, Donner DB. Vascular endothelial cell growth factor promotes tyrosine phosphorylation of mediators of signal transduction that contain SH2 domains. Association with endothelial cell proliferation. Journal of Biological Chemistry. 1995;270:6729–6733. doi: 10.1074/jbc.270.12.6729. [DOI] [PubMed] [Google Scholar]
- Hillman NJ, Whittles CE, Pocock TM, Williams B, Bates DO. Differential effects on microvascular hydraulic conductivity (Lp) of vascular endothelial growth factor C (VEGF-C) and placental growth factor-1 (PlGF-1) Journal of Vascular Research. 2001;38:176–186. doi: 10.1159/000051044. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- Kevil CG, Payne DK, Mire E, Alexander JS. Vascular permeability factor/vascular endothelial cell growth factor-mediated permeability occurs through disorganization of endothelial junctional proteins. Journal of Biological Chemistry. 1998;273:15099–15103. doi: 10.1074/jbc.273.24.15099. [DOI] [PubMed] [Google Scholar]
- Kroll J, Waltenberger J. The vascular endothelial growth factor receptor KDR activates multiple signal transduction pathways in porcine aortic endothelial cells. Journal of Biological Chemistry. 1997;272:32521–32527. doi: 10.1074/jbc.272.51.32521. [DOI] [PubMed] [Google Scholar]
- Michel CC, Mason JC, Curry FE, Tooke JE, Hunter PJ. A development of the Landis technique for measuring the filtration coefficient of individual capillaries in the frog mesentery. Quarterly Journal of Experimental Physiology and Cognitive Medical Science. 1974;59:283–309. doi: 10.1113/expphysiol.1974.sp002275. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D, Nagy JA, Manseau EJ, Dvorak HF. Vascular permeability factor/vascular endothelial growth factor-mediated signaling in mouse mesentery vascular endothelium. Cancer Research. 1998;58:1278–1284. [PubMed] [Google Scholar]
- Pocock TM, Williams B, Curry FE, Bates DO. VEGF and ATP act by different mechanisms to increase microvascular permeability and endothelial [Ca2+]i. American Journal of Physiology - Heart and Circulatory Physiology. 2000;279:H1625–1634. doi: 10.1152/ajpheart.2000.279.4.H1625. [DOI] [PubMed] [Google Scholar]
- Putney JW. Capacitative calcium entry revisited. Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- Sawano A, Takahashi T, Yamaguchi S, Shibuya M. The phosphorylated 1169-tyrosine containing region of flt-1 kinase (VEGFR-1) is a major binding site for PLCgamma. Biochemical and Biophysical Research Communications. 1997;238:487–491. doi: 10.1006/bbrc.1997.7327. [DOI] [PubMed] [Google Scholar]
- Seymour LW, Shoaibi MA, Martin A, Ahmed A, Elvin P, Kerr DJ, Wakelam MJ. Vascular endothelial growth factor stimulates protein kinase C-dependent phospholipase D activity in endothelial cells. Laboratory Investigation. 1996;75:427–437. [PubMed] [Google Scholar]
- Shen BQ, Lee DY, Zioncheck TF. Vascular endothelial growth factor governs endothelial nitric-oxide synthase expression via a KDR/Flk-1 receptor and a protein kinase C signaling pathway. Journal of Biological Chemistry. 1999;274:33057–33063. doi: 10.1074/jbc.274.46.33057. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Shibuya M. The 230 kDa mature form of KDR/Flk-1 (VEGF receptor-2) activates the PLC-gamma pathway and partially induces mitotic signals in NIH3T3 fibroblasts. Oncogene. 1997;14:2079–2089. doi: 10.1038/sj.onc.1201047. [DOI] [PubMed] [Google Scholar]
- Waltenberger J, Claessonwelsh L, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. Journal of Biological Chemistry. 1994;269:26988–26995. [PubMed] [Google Scholar]
- Wheeler-Jones C, Abu-Ghazaleh R, Cospedal R, Houliston RA, Martin J, Zachary I. Vascular endothelial growth factor stimulates prostacyclin production and activation of cytosolic phospholipase A2 in endothelial cells via p42/p44 mitogen-activated protein kinase. FEBS Letters. 1997;420:28–32. doi: 10.1016/s0014-5793(97)01481-6. [DOI] [PubMed] [Google Scholar]
- Williams TJ, Morley J. Prostaglandins as potentiators of increased vascular permeability in inflammation. Nature. 1973;246:215–217. doi: 10.1038/246215a0. [DOI] [PubMed] [Google Scholar]
- Wu HM, Yuan Y, Zawieja DC, Tinsley J, Granger HJ. Role of phospholipase C, protein kinase C, and calcium in VEGF-induced venular hyperpermeability. American Journal of Physiology. 1999;276:H535–542. doi: 10.1152/ajpheart.1999.276.2.H535. [DOI] [PubMed] [Google Scholar]
- Xia P, Aiello LP, Ishii H, Jiang ZY, Park DJ, Robinson GS, Takagi H, Newsome WP, Jirousek MR, King GL. Characterization of vascular endothelial growth factor's effect on the activation of protein kinase C, its isoforms, and endothelial cell growth. Journal of Clinical Investigation. 1996;98:2018–2026. doi: 10.1172/JCI119006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiji H, Kuriyama S, Ways DK, Yoshii J, Miyamoto Y, Kawata M, Ikenaka Y, Tsujinoue H, Nakatani T, Shibuya M, Fukui H. Protein kinase C lies on the signaling pathway for vascular endothelial growth factor-mediated tumor development and angiogenesis. Cancer Research. 1999;59:4413–4418. [PubMed] [Google Scholar]