

Abstract
Studies of the effect of vagus nerve stimulation on ventricular myocardial function in mammals are limited, particularly in the human.
The present study was designed to determine the effect of direct electrical stimulation of the left vagus nerve on left ventricular contractile state in hearts paced at 10 % above the natural rate, in anaesthetised pigs and anaesthetised human subjects undergoing open chest surgery for coronary artery bypass grafting.
Contractility of the left ventricle was determined from a series of pressure-volume loops obtained from a combined pressure and conductance (volume) catheter placed in the left ventricle. From the measurements a regression slope of the end-systolic pressure-volume relationship was determined to give end-systolic elastance (Ees), a load-independent measure of contractility.
In six anaesthetised open chest pigs, stimulation of the peripheral cut end of the left cervical vagus nerve induced a significant decrease in Ees of 26 ± 14 %.
In nine patients electrical stimulation of the left thoracic vagus nerve close to its cardiac branch resulted in a significant drop in Ees of 38 ± 16 %.
The effects of vagal stimulation were blocked by the muscarinic antagonist glycopyrronium (5 mg kg−1).
Administration of the β-adrenoreceptor antagonist esmolol (1 mg kg−1) also attenuated the effect of vagal stimulation, indicating a degree of interaction of vagal and sympathetic influences on contractility.
These studies show that in the human and pig heart the left vagus nerve can profoundly decrease the inotropic state of the left ventricular myocardium independent of its bradycardic effect.
The mammalian heart is innervated by both parasympathetic (vagus) and sympathetic nerves. The latter have been subject to detailed study probably because they are principally responsible for the increase in cardiac output that enables animals to respond to threatening stimuli. Recently, there has been a revival of interest in the influence of vagal nerve fibres on the heart (Xenopoulos & Applegate, 1994). Clinical trials have shown that reduced heart rate variability and baroreflex sensitivity, both markers of cardiac vagal control, are powerful and independent indicators of adverse prognosis in patients with cardiac failure or myocardial infarction (Huikuri et al. 1999). This suggests preservation of the action of cholinergic nerves may protect the compromised cardiac ventricle (Schwartz, 1998). This effect could be related to a reduction in the energy requirements of myocardial contraction.
In several species acetylcholine has been shown to have a powerful antiadrenergic effect on the ventricular myocardium (Loffelholz & Pappano, 1985). Furthermore, in the human several studies have shown that there is a significant cholinergic innervation of the ventricle (Kent et al. 1974; Loffelholz & Pappano, 1985; Du et al. 1995) as well as a dense distribution of muscarinic M2 receptors (Fields et al. 1978; Goyal, 1989; Deighton et al. 1990). However, a load-independent assessment of the influences of vagal nerve stimulation on contractility, in mammals other than the dog, is lacking. The introduction of a catheter that can be placed within the ventricle to measure pressure and volume enables a direct measurement of ventricular contractility and we have taken advantage of this to study the influence of the vagus nerve in the pig and human heart in vivo.
METHODS
The contractility of cardiac muscle is an index of its ability to generate force independent of muscle fibre length (‘load’). Traditionally left ventricle (LV) contractility has been measured in the beating heart by registering the change in LV pressure with time, dP/dt. However, in an intact animal dP/dt is load and heart rate dependent (Mason, 1969), both of which are altered by vagal nerve stimulation. It is now possible to quantify LV contractility by measuring the end-systolic pressure-volume relationship, an index of contractility that is relatively independent of loading conditions. The development of a conductance catheter with a pressure sensor has enabled the end-systolic pressure-volume relationship to be examined in the human heart in vivo. (Baan et al. 1984; Kass et al. 1986). We have recently validated this technique for a broad range of conditions experienced during cardiac surgery (Al-Khalidi et al. 1998). The conductance method of determining left ventricular volume is based upon the measurement of blood conductivity within the left ventricle. We used a 12-electrode Millar conductance catheter (Millar instruments, TX, USA). Signals were processed with a customised software package (Leycom Sigma-5DF, Cardiodynamics BV, Zoetermeer, The Netherlands). We calculated the regression slope of the end-systolic pressure-volume relationship, the end-systolic elastance (Ees) obtained from a family of pressure-volume curves during transient inferior vena cava occlusion (Fig. 1 and Fig. 3). The Ees is a widely accepted assessment of load-independent contractility (Sagawa et al. 1988).
Figure 1. The effect of left cervical vagal nerve stimulation on the end-systolic pressure-volume relationship in the pig.
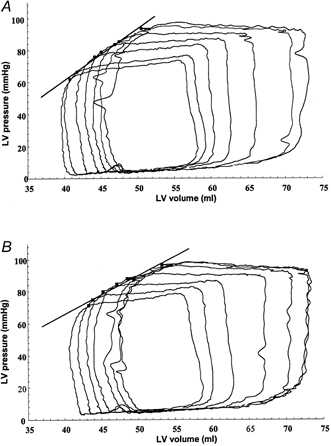
Pig 6, atrioventricular pacing at a rate of 130 bpm. Inferior vena cava occlusion, providing a family of pressure-volume loops (7) without (a) and with (B) vagal stimulation. A, end-systolic elastance (Ees) = 3.23 mmHg ml−1, r2= 0.99; B, Ees = 2.44 mmHg ml−1, r2= 0.97 (regression line drawn between the end-systolic pressure-volume points of each loop).
Figure 3. The effect of left thoracic vagal nerve stimulation on the end-systolic pressure-volume relationship in one human.
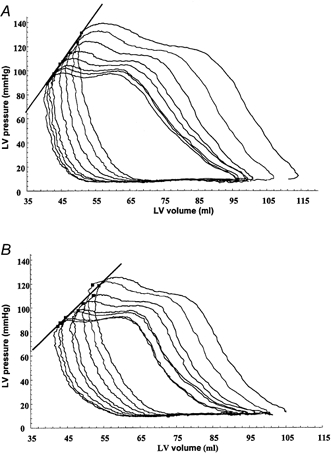
Patient 4, atrioventricular pacing at a rate of 100 bpm. Inferior vena cava occlusion, providing a family of pressure-volume loops (8) without (a) and with (B) vagal stimulation. A, Ees = 4.04 mmHg ml−1, r2= 0.99; B, Ees = 3.01 mmHg ml−1, r2= 0.96 (regression line drawn between the end-systolic pressure-volume points of each loop).
Pig experiment
Six pigs were anaesthetised with 10 mg kg−1 of ketamine intramuscularly, followed by the inhalation of isoflurane in oxygen-enriched air at a FI,O2 (inspired O2 fraction) of 0.6. The animals were intubated with a cuffed endotracheal tube and placed on a positive pressure volume ventilator. Intravenous access was established by a suitable ear vein.
Subsequent total intravenous anaesthesia was commenced using bolus doses of midazolam (0.5 mg kg−1), fentanyl citrate (100 μg kg−1) and fluanisone (3 mg kg−1). Anaesthesia was maintained using continuous infusions of midazolam at 1 mg kg−1 h−1, fentanyl at 200 μg kg−1 h−1 and fluanisone at 6 mg kg−1 h−1. Venous and arterial lines were placed in the right external jugular vein and right carotid artery for infusion of drugs and monitoring arterial blood pressure, respectively. Limb leads were placed for electrocardiographic recording. The tidal volume of the ventilator was adjusted to maintain arterial blood oxygen tension around 100 mmHg and arterial pH between 7.35 and 7.4. A median sternotomy was performed to expose the heart. The conductance-pressure catheter was introduced into the left ventricle via an incision and purse string suture in the apex. The position was confirmed by observing appropriate pressure-volume signals on a visual display unit. Atrial pacing wires were attached to the right atrium and ventricle. A tape snare was placed around the inferior vena cava. The left cervical vagus nerve was exposed and cut and temporary pacing wires attached to the peripheral end for electrically stimulating the nerve. Square wave pulses (6 s train of square wave negative pulses at 10 Hz, 20 V and 0.1 ms duration) were provided via an isolation unit and constant voltage stimulator (Digitimer). This stimulus was sufficient to produce a 20 ± 1 beats per minute (bpm) decrease in heart rate.
Before commencing the experimental protocols, blood temperature, and resistivity were measured (Kass et al. 1986; Al-Khalidi et al. 1998). The heart was then paced using a dual chamber pacing box with atrial and ventricular leads to give a captured rate 10 % above the natural rate. Next, ventilation was briefly interrupted and, whilst partially occluding the vena cava to reduce systolic blood pressure by 25 mmHg over 6 s, a series of pressure-volume curves were obtained (Fig. 1). This procedure was repeated whilst stimulating the left vagus nerve (4 s of vagal stimulation). Further studies were undertaken in two pigs with and without vagal stimulation after treatment with glycopyrrolate (15 mg kg−1 bolus, a muscarinic antagonist) and in a further two pigs with and without vagal stimulation after treatment with esmolol (a short-acting β-adrenoreceptor antagonist, 1 mg kg−1 bolus).
This animal study was conducted under the Home Office (UK) rules for the use of experimental animals in research and approved under a specific Project Licence. Animals were humanely killed at the end of the experiment by an overdose of an intravenous infusion of phenobarbitone.
Human experiment
To determine whether human left ventricular myocardium was influenced by efferent vagal nerve fibres we conducted a similar series of tests in a group of 10 patients undergoing open chest surgery for coronary artery bypass grafting (CABG). Local ethics committee (South Birmingham) approval was obtained and studies were performed according to the Declaration of Helsinki. All patients gave informed written consent. All patients had good ventricular function (ejection fraction > 50 %), triple vessel coronary disease and no significant co-morbidity. Following induction of anaesthesia (fentanyl 15 μg kg−1, propofol 3 mg kg−1, etomidate 0.3 mg kg−1 and pancuronium 100 μg kg−1) the patient was intubated with a cuffed endotracheal tube, and ventilation commenced at 10 ml kg−1 tidal volume. FI,O2 and ventilation were adjusted to maintain arterial blood oxygen tension at around 100 mmHg and pH in the range 7.35–7.4. Anaesthesia was maintained using alfentanyl at 50 μg kg−1 h−1 and a target-controlled infusion of propofol. A central venous line and Swan Ganz catheter were inserted through the right internal jugular vein and arterial pressure was monitored via a right radial line. The chest was opened by median sternotomy, internal mammary artery harvest performed and preparations made to commence cardiopulmonary bypass. The left vagus nerve was visualised as it passed over the arch of the aorta (Matheny & Shaar, 1997) and temporary pacing wires were attached to it just proximal to the origin of the cardiac vagal branch. These pacing wires were used to stimulate the nerve with negative (cathode placed distally) square wave pulses in a 6 s train at 25 Hz, 20 V and 0.1 ms width. This stimulation resulted in an immediate reduction in heart rate causing asystole and has been described as a means of providing cardioplegia during CABG (Matheny & Shaar, 1997). Temporary right atrial and ventricular pacing wires were attached and the conductance-pressure catheter introduced via a purse string in the ascending aorta and advanced into the left ventricle. Its position was confirmed by monitoring the pressure and volume signals. The heart was then paced (via atrioventricular pacing wires) at 10 % greater than natural rate (mean paced rate, 114 ± 6 bpm). A series of baseline pressure-volume curves were obtained (Fig. 3) with ventilation of the lungs stopped and caval occlusion as performed in the pig experiments. Investigation proceeded as described for the pig pressure-volume measurements (muscarinic receptor antagonism was achieved with glycopyrrolate, 5 mg kg−1, 3 patients, and β-adrenoreceptor antagonism with esmolol, 1 mg kg−1 bolus, 3 patients). Following completion of the investigations, the CABG operation was performed according to standard management. All patients underwent uncomplicated recovery.
Data acquisition and analysis
All data were gathered at 500 Hz via an analog-digital conversion board (DAS1600 series, Keithley, Metrabyte, MA, USA) and processed and stored on a Compaq portable 480C computer (Compaq Corp., USA). In an attempt to avoid effects of vagal stimulation caused by activating afferent fibres, which might cause reflex changes in sympathetic drive to the heart, we restricted analysis to the pressure-volume loops of the first 10 cardiac cycles (6 s). Statistical analysis was performed on an IBM compatible computer using Arcus software (Research Solutions, Adison Wesley Longman, Cambridge, UK). Data were first analysed for non-normality and subsequently subjected to Student's paired two-way t test. Significance was set at differences of P < 0.05. Results are expressed as means ±s.d.
RESULTS
Pig experiments
The results showed a significant reduction in Ees (Fig. 1) during vagal stimulation in all six experiments (Fig. 2) (P < 0.002) from a value of 2.64 ± 1.36 mmHg ml−1 to 1.95 ± 1.03 mmHg ml−1, a change of 0.69 mmHg ml−1 or 26 ± 14 %. As the vagus nerve is reported to exert an inhibitory effect upon sympathetic neurotransmission, this ‘indirect’ action (Levy, 1998) was assessed by repeating the two series of tests following β-1 adrenoreceptor antagonism with intravenous esmolol. Esmolol induced a decrease of 36.2 ± 16.5 % in baseline Ees indicating significant cardiac sympathetic antagonism. Vagal stimulation during β-adrenoreceptor antagonism produced a further small (mean 7 %) but non-significant reduction in Ees. Stimulation of efferent vagal nerve fibres releases acetylcholine, which acts on cardiac muscarinic M2 receptors. We therefore determined the effect of a muscarinic antagonist on the vagally induced decrease in contractility. In two pigs in which vagal stimulation decreased Ees by 40 % in one and 45 % in the other, glycopyrronium reduced this to 3 and 22 %, respectively.
Figure 2. Negative inotropic effect of vagal stimulation in pigs.
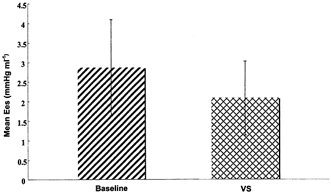
Bar graph of mean value of Ees, indicating contractility, for all pigs with (VS) and without (baseline) vagal stimulation (P < 0.002).
Human experiments
For the 10 patients Ees baseline was 4.8 ± 2.4 mmHg ml−1. In 9 of the 10 patients vagal stimulation produced a significant (P < 0.006) reduction in Ees (Fig. 4) to 3.2 ± 1.9 mmHg ml−1 (ranging from 10 to 60 %, mean 38 ± 16 %). In one patient an apparent small increase in Ees occurred. We believe this may have been due to stimulating electrode displacement.
Figure 4. Change in inotropic state of left ventricle during vagal stimulation in humans.
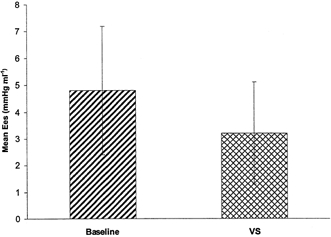
Graph shows change in contractility (vertical axis) from baseline Ees with vagal stimulation (VS) in humans (P < 0.006).
To establish whether these effects were due to efferent vagal stimulation, tests were repeated in three patients following partial muscarinic receptor antagonism with a dose of glycopyronnium. The negative inotropic actions of vagus nerve stimulation were reduced from 1.93 ± 1.83 to 0.3 ± 0.12 mmHg ml−1 (85 % reduction in effect).
β-1 adrenoreceptor antagonism reduced baseline Ees by a mean of 33 ± 11 % in three patients, indicating a strong dependence of cardiac contractility on sympathetic tone under these conditions of anaesthesia and open chest surgery. β-1 adrenoreceptor antagonism consistently reduced the negative inotropic action of vagal stimulation in the three patients (by 17, 53 and 55 %, respectively).
DISCUSSION
This series of experiments clearly establishes that efferent vagal activity to the pig and human LV induces a significant negative inotropic effect and shows that the dog heart (Henning & Levy, 1991; Xenopoulos & Applegate, 1994) is not alone amongst mammals in possessing this important action of the vagus nerve. Although textbooks have long played down the importance of the vagal action on ventricular muscle, histochemical evidence has increasingly questioned this viewpoint. Studies of the regional distribution of muscarinic cholinergic receptors in the hearts of a number of species, including humans, demonstrate that they are widespread in the ventricles and conducting tissue (Fields et al. 1978; Wei & Sulakhe, 1978; Yamada et al. 1980; Deighton et al. 1990); indeed they are far more abundant than A1-adenosine or β-adrenoreceptors (Bohm et al. 1990). Details of the precise cellular location of the receptors and nerve endings are currently lacking.
Our results provide a firm experimental basis for the interpretation of results obtained using pulsed-wave aortic doppler ultrasound in humans suggesting a vagally mediated baroreflex control of myocardial contractility (Casadei et al. 1992). The data also suggest that part of this effect is due to an interaction of the vagus nerve with sympathetic nerve influences. This action, known as ‘accentuated antagonism’, appears to involve both pre- and post-junctional mechanisms. In both locations the nitric oxide cyclic GMP pathway may play an important role (Paterson, 2001), although this remains controversial (Vandecasteele et al. 1999; Belevych & Harvey, 2000). Whether there is some further direct independent action of the vagus nerve on Ca2+ delivery in the cardiac muscle cell merits further study in view of our finding that vagal effects were still present after β-1 adrenoreceptor antagonism – an effect that has also been observed in the dog (Xenopoulos & Applegate, 1994).
The use of a human model prevented us from testing the effect of complete block of vagal activity in each patient. Therefore a criticism of this work could be that we are unable to distinguish efferent from afferent vagus effects. We attempted to overcome this problem in two ways. Firstly we took only the first six heart beats following inferior vena cava occlusion. Secondly, we used glycopyrrolate in three humans to block muscarinic effects. This abolished the effect of vagal stimulation on both heart rate and contractility. Unfortunately, due to constraints of patient safety, we were not able to give glycopyrrolate to all our patients (we had preset a study time limit of 30 min). We did attempt the administration of lignocaine to the proximal portion of the vagus nerve to overcome afferent effects (Carlsten et al. 1957). We attempted this in two patients not included in this study, but found it impossible thereafter to stimulate the vagus nerve, presumably because the local anaesthetic provided an extended block to conduction along the nerve sheath.
Since these patients were suffering from coronary artery disease we felt it was not acceptable to test the level of β-1 adrenoreceptor antagonism, with an adrenaline bolus, in those patients treated with esmolol. It is therefore possible that these patients were not fully β-blocked. However treatment with esmolol infusion did result in a mean reduction in heart rate of 21 ± 6 %, suggesting a high level of antagonism. However, the number of patients treated with esmolol was small and therefore it is impossible to determine whether the effects of vagal stimulation depend completely or only partly upon adrenergic prestimulation of the heart.
Autonomic control of the diseased human heart has great clinical significance. Suppression of sympathetic tone by chronic β-adrenoreceptor antagonism is a well-established mode of treatment that reduces mortality in heart failure (Anonymous, 1999) and after myocardial infarction (Hjalmarson, 1997). Vagal activity appears to exert similar actions to β-adrenoreceptor antagonism; reducing heart rate and contractility. Our results, taken together with the known powerful influences of vagal activity on the susceptibility of ischaemic myocardium to ventricular arrhythmia (Schwartz, 1998), suggest that the vagus nerve might be a novel target for treatment. Future research might usefully be directed at further examining the cellular and molecular physiology of the actions of the cardiac vagal stimulation in animal models and at methods of increasing cardiac parasympathetic activity in patients with heart disease.
Acknowledgments
We thank the British Heart Foundation and the National Heart Research Fund for their generous support. We would also like to thank Karen Parkes and the anaesthetic and theatre staff of the Cardiothoracic Surgical Unit of the Queen Elizabeth Hospital, Birmingham.
References
- Al-Khalidi AH, Townend JN, Bonser RS, Coote JH. Validation of the conductance catheter method for measurement of ventricular volumes under varying conditions relevant to cardiac surgery. American Journal of Cardiology. 1998;82:1248–1252. doi: 10.1016/s0002-9149(98)00613-4. [DOI] [PubMed] [Google Scholar]
- Anonymous. The cardiac insufficiency bisoprolol study II (CIBIS-II): a randomised trial. Lancet. 1999;353:9–13. [PubMed] [Google Scholar]
- Baan J, van der Velde ET, de Bruin HG, Smeenk GJ, Koops J, van Dijk AD, Temmerman D, Senden J, Buis B. Continuous measurement of left ventricular volume in animals and humans by conductance catheter. Circulation. 1984;70:812–823. doi: 10.1161/01.cir.70.5.812. [DOI] [PubMed] [Google Scholar]
- Belevych AE, Harvey RD. Muscarinic inhibitory and stimulatory regulation of the L-type Ca2+ current is not altered in cardiac ventricular myocytes from mice lacking endothelial nitric oxide synthase. Journal of Physiology. 2000;528:279–289. doi: 10.1111/j.1469-7793.2000.00279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm M, Gierschik P, Jakobs KH, Pieske B, Schnabel P, Ungerer M, Erdmann E. Increase of Gi alpha in human hearts with dilated but not ischemic cardiomyopathy. Circulation. 1990;82:1249–1265. doi: 10.1161/01.cir.82.4.1249. [DOI] [PubMed] [Google Scholar]
- Carlsten A, Folkow B, Hamberger C-A. Cardiovascular effects of direct vagal stimulation in man. Acta Physiologica Scandanavia. 1957;41:68–76. doi: 10.1111/j.1748-1716.1957.tb01510.x. [DOI] [PubMed] [Google Scholar]
- Casadei B, Meyer TE, Coats AJ, Conway J, Sleight P. Baroreflex control of stroke volume in man: an effect mediated by the vagus. Journal of Physiology. 1992;448:539–550. doi: 10.1113/jphysiol.1992.sp019056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deighton NM, Motomura S, Borquez D, Zerkowski HR, Doetsch N, Brodde OE. Muscarinic cholinoceptors in the human heart: demonstration, subclassification, and distribution. Naunyn-Schmiedeberg's Archives of Pharmacology. 1990;341:14–21. doi: 10.1007/BF00195052. [DOI] [PubMed] [Google Scholar]
- Du XY, Schoemaker RG, Bos E, Saxena PR. Characterization of the positive and negative inotropic effects of acetylcholine in the human myocardium. European Journal of Pharmacology. 1995;284:119–127. doi: 10.1016/0014-2999(95)00384-w. [DOI] [PubMed] [Google Scholar]
- Fields JZ, Roeske WR, Morkin E, Yamamura HI. Cardiac muscarinic cholinergic receptors. Biochemical identification and characterization. Journal of Biological Chemistry. 1978;253:3251–3258. [PubMed] [Google Scholar]
- Goyal RK. Muscarinic receptor subtypes. Physiology and clinical implications. New England Journal of Medicine. 1989;321:1022–1029. doi: 10.1056/NEJM198910123211506. [DOI] [PubMed] [Google Scholar]
- Henning RJ, Levy MN. Effects of autonomic nerve stimulation, asynchrony, and load on dP/dtmax and on dP/dtmin. American Journal of Physiology. 1991;260:H1290–1298. doi: 10.1152/ajpheart.1991.260.4.H1290. [DOI] [PubMed] [Google Scholar]
- Hjalmarson A. Effects of beta blockade on sudden cardiac death during acute myocardial infarction and the postinfarction period. American Journal of Cardiology. 1997;80:35–39J. doi: 10.1016/s0002-9149(97)00837-0. [DOI] [PubMed] [Google Scholar]
- Huikuri HV, Makikallio T, Airaksinen KE, Mitrani R, Castellanos A, Myerburg RJ. Measurement of heart rate variability: a clinical tool or a research toy? Journal of the American College of Cardiology. 1999;34:1878–1883. doi: 10.1016/s0735-1097(99)00468-4. [DOI] [PubMed] [Google Scholar]
- Kent KM, Epstein SE, Cooper T, Jacobowitz DM. Cholinergic innervation of the canine and human ventricular conducting system. Anatomic and electrophysiologic correlations. Circulation. 1974;50:948–955. doi: 10.1161/01.cir.50.5.948. [DOI] [PubMed] [Google Scholar]
- Levy MN. Neurocardiology. Vol. 275. Mt Kisco, NY, USA: Futura Publishing Co.; 1998. Sympathetic-parasympathetic interactions in the heart; pp. 85–98. [Google Scholar]
- Loffelholz K, Pappano AJ. The parasympathetic neuroeffector junction of the heart. Pharmacological Reviews. 1985;37:1–24. [PubMed] [Google Scholar]
- Mason DT. Usefulness and limitations of the rate of rise of intraventricular pressure (dp-dt) in the evaluation of myocardial contractility in man. American Journal of Cardiology. 1969;23:516–527. doi: 10.1016/0002-9149(69)90005-8. [DOI] [PubMed] [Google Scholar]
- Matheny RG, Shaar CJ. Vagus nerve stimulation as a method to temporarily slow or arrest the heart. Annals of Thoracic Surgery. 1997;63(suppl.):S28–29. doi: 10.1016/s0003-4975(97)00423-2. [DOI] [PubMed] [Google Scholar]
- Paterson DJ. Nitric oxide and the autonomic regulation of cardiac excitability. Experimental Physiology. 2001;86:1–12. doi: 10.1113/eph8602169. [DOI] [PubMed] [Google Scholar]
- Sagawa K, Maughan L, Suga H, Sunagawa K. Cardiac Contraction and the Pressure-Volume Relationship. New York: Oxford University Press; 1988. [Google Scholar]
- Schwartz PJ. The autonomic nervous system and sudden death. European Heart Journal. 1998;19:F72–80. [PubMed] [Google Scholar]
- Vandecasteele G, Eschenhagen T, Scholz H, Stein B, Verde I, Fischmeister R. Muscarinic and beta-adrenergic regulation of heart rate, force of contraction and calcium current is preserved in mice lacking endothelial nitric oxide synthase. Nature Medicine. 1999;5:331–334. doi: 10.1038/6553. [DOI] [PubMed] [Google Scholar]
- Wei JW, Sulakhe PV. Regional and subcellular distribution of myocardial muscarinic cholinergic receptors. European Journal of Pharmacology. 1978;52:235–238. doi: 10.1016/0014-2999(78)90212-1. [DOI] [PubMed] [Google Scholar]
- Xenopoulos NP, Applegate RJ. The effect of vagal stimulation on left ventricular systolic and diastolic performance. American Journal of Physiology. 1994;266:H2167–2173. doi: 10.1152/ajpheart.1994.266.6.H2167. [DOI] [PubMed] [Google Scholar]
- Yamada S, Yamamura HI, Roeske WR. Alterations in cardiac autonomic receptors following 6-hydroxydopamine treatment in rats. Molecular Pharmacology. 1980;18:185–192. [PubMed] [Google Scholar]