

Abstract
We characterized the mechanisms in vascular smooth muscle cells (VSMCs) that produce asynchronous, wave-like Ca2+ oscillations in response to phenylephrine (PE). Confocal imaging was used to observe [Ca2+]i in individual VSMCs of intact inferior vena cava (IVC) from rabbits.
It was found that the Ca2+ waves were initiated by Ca2+ release from the sarcoplasmic reticulum (SR) via inositol 1,4,5-trisphosphate-sensitive SR Ca2+ release channels (IP3R channels) and that refilling of the SR Ca2+ store through the sarcoplasmic-endoplasmic reticulum Ca2+-ATPase (SERCA) was required for maintained generation of the repetitive Ca2+ waves.
Blockade of L-type voltage-gated Ca2+ channels (L-type VGCCs) with nifedipine reduced the frequency of PE-stimulated [Ca2+]i oscillations, while additional blockade of receptor-operated channels/store-operated channels (ROCs/SOCs) with SKF96365 abolished the remaining oscillations. Parallel force measurements showed that nifedipine inhibited PE-induced tonic contraction by 27% while SKF96365 abolished it. This indicates that stimulated Ca2+ entry refills the SR to support the recurrent waves of SR Ca2+ release and that both L-type VGCCs and ROCs/SOCs contribute to this process.
Application of the Na+-Ca2+ exchanger (NCX) inhibitors 2′,4′-dichlorobenzamil (forward- and reverse-mode inhibitor) and KB-R7943 (reverse-mode inhibitor) completely abolished the nifedipine-resistant component of [Ca2+]i oscillations and markedly reduced PE-induced tone.
Thus, we conclude that each Ca2+ wave depends on initial SR Ca2+ release via IP3R channels followed by SR Ca2+ refilling through SERCA. Na+ entry through ROCs/SOCs facilitates Ca2+ entry through the NCX operating in the reverse mode, which refills the SR and maintains PE-induced [Ca2+]i oscillations. In addition some Ca2+ entry through L-type VGCCs and ROCs/SOCs serves to modulate the frequency of the oscillations and the magnitude of force development.
An increase in [Ca2+]i from 100 nm or less to values up to 1 μm initiates smooth muscle contraction. Conduit arteries and capacitance veins when challenged with a maintained dose of the neurotransmitter noradrenaline or other pharmacological agonists respond with a biphasic tonic contraction. These same agonists initiate a whole-tissue Ca2+ signal, which has a similar profile to the contraction, albeit with a relatively faster onset and lower plateau value. In addition, removal of external Ca2+ abolishes the plateau, but not the initial transient. These observations led to the generally accepted theory that the initial phase is initiated by Ca2+ release from the sarcoplasmic reticulum (SR) and the tonic phase is supported by sustained Ca2+ influx through L-type voltage-gated Ca2+ channels (L-type VGCCs) and/or receptor-operated channels (ROCs). This view was challenged by Iino and collaborators (Iino et al. 1994) who first reported that noradrenaline elicits asynchronous oscillatory Ca2+ waves in vascular smooth muscle cells (VSMCs) within the intact wall of the rat tail artery. They postulated that agonist-induced vascular tone is maintained by asynchronous repetitive SR Ca2+ release rather than by sustained Ca2+ influx. Several subsequent reports have confirmed the presence of asynchronous Ca2+ waves in vascular smooth muscle fibres in isolated, intact blood vessels (Miriel et al. 1999; Asada et al. 1999; Ruehlmann et al. 2000). In addition, we have related these individual-cell Ca2+ signals quantitatively to the contractile force generated by the whole blood vessel wall (Ruehlmann et al. 2000). Increasing concentrations of phenylephrine (PE) applied to the rabbit inferior vena cava (IVC) resulted in the graded recruitment of responding cells, as well as an increase in the frequency of [Ca2+]i oscillations. These parameters of single cell Ca2+ signalling were thus shown to underlie the PE dose-related tonic constriction of the IVC. During the maintained [Ca2+]i oscillations, a significant amount of cytoplasmic Ca2+ will be extruded to the extracellular space via the plasma membrane Ca2+-ATPase (PMCA) or the plasma membrane Na+-Ca2+ exchanger (NCX) (Nazer & van Breemen, 1999). Therefore, stimulated Ca2+ entry is required to compensate for the loss of Ca2+ from the smooth muscle cells in order to sustain the [Ca2+]i oscillations. Several modes of Ca2+ entry have been documented in VSMCs, including L-type VGCCs, ROCs, store-operated channels (SOCs) and the NCX operating in the reverse mode. In addition there is a significant, though poorly defined, basal Ca2+ leak (Khalil et al. 1987). The relative importance of these pathways varies with the type of blood vessel. L-type VGCCs are the principle route of Ca2+ entry for initiating myogenic tone in resistance arteries (Davis & Hill, 1999), while aortic smooth muscle is relatively insensitive to membrane potential and relies mainly on ROCs to maintain its tone (Cauvin et al. 1985; Karaki et al. 1997). Recently, Blaustein and collaborators (Arnon et al. 2000) made the intriguing proposal that the NCX operating in the reverse mode plays an important role in agonist-induced [Ca2+]i elevation in vascular smooth muscle.
In our current report, we investigated the mechanism of the asynchronous [Ca2+]i oscillations in the rabbit IVC, focusing on the mode(s) of Ca2+ entry involved in sustaining the PE-induced cyclical release of Ca2+ from the SR.
METHODS
Solutions and chemicals
Normal physiological salt solution (PSS), containing (mm): NaCl 140, KCl 5, CaCl2 1.5, MgCl2 1, glucose 10 and Hepes 5 (pH 7.4 at 37 °C), was used for all the studies. High K+ (80 mm [K+]o) PSS was identical in composition to normal PSS with the exception of (mm): NaCl 65 and KCl 80. All the reagents were purchased from Sigma and were of the highest analytical grade. Fluo-3 AM, Pluronic F-127 and 2′,4′-dichlorobenzamil were purchased from Molecular Probes and were dissolved in dimethyl sulfoxide (DMSO). PE (Sigma), caffeine (Sigma), thapsigargin (Sigma), phentolamine (Sigma) and SKF96365 (Calbiochem) were prepared in normal PSS. Stocks of KB-R7943 (Tocris) and cyclopiazonic acid (CPA, Calbiochem) were prepared in DMSO while stocks of nifedipine (Sigma) and diphenylboric acid 2-aminoethyl ester (2-APB, Sigma) were prepared in ethanol and methanol, respectively. All experiments were performed at 37 °C.
Tissue preparation
All experiments and procedures were carried out in accordance with the guidelines of the University of British Columbia Animal Care Committee (protocol no. A990290). Female New Zealand White rabbits (1.5-2.5 kg, obtained from Animal Care, University of British Columbia), were killed by a rising concentration of CO2 and then exsanguinated. The IVC was removed, cleaned of surrounding connective tissues and then inverted. The endothelium was removed by gently wiping it with filter paper and the inverted vessel was cut into multiple ring segments that were 4-5 mm in width.
Whole-tissue contraction study and confocal [Ca2+]i imaging
Detailed methods have previously been described for this preparation (Ruehlmann et al. 2000). Briefly, to study contraction the inverted rings were attached to isometric force transducers and the resting tension was set at 0.4 g. The bath solution was changed by rapidly draining and refilling the tissue bath. Data were acquired and analysed using Chart version 3.4.5 (ABI instruments). For confocal imaging experiments, inverted rings were loaded with Fluo-3 AM (5 μm, with 5 μm Pluronic F-127, 90 min, 25 °C) followed by a 30 min equilibration period in normal PSS. The vessel rings were then mounted isometrically on a custom-made microscope stage. [Ca2+]i imaging was accomplished with the use of a Noran Oz laser-scanning confocal microscope with a 100 μm slit through an air ×20 lens (numerical aperture, 0.45) on an inverted Nikon microscope. The tissue was illuminated by the 488 nm line of an argon-krypton laser and a high-gain photomultiplier tube collected the emission after it had passed through a 525/25 bandpass filter. Such a filter will only allow light from a narrow band of 525 ± 25 nm to pass through (525 nm is the emission wavelength of Fluo-3). The scanned regions correspond to a 232 μm × 217 μm area on the tissue and yield a 512 pixel × 479 pixel image. The image acquisition was set at 133 ms per frame. All data analysis was performed in ImagePro Plus using customized routines. The representative experimental fluorescence traces shown reflect the averaged fluorescence signals from a 3 pixel × 3 pixel region (1.36 μm2) in a single cell. Changes in this regional fluorescence level (F525) directly reflect changes in the Ca2+ concentration in this region of the cell. Prior to stimulation, the basal fluorescence allows for delineation of the outline of the ribbon-shaped VSMC. The 3 pixel × 3 pixel region was positioned towards the midline of the ribbon-shaped smooth muscle cell. Given that the focus of this study is on cytoplasmic Ca2+ signals, the region was positioned away from the highly fluorescent nuclear region of the cell, as Fluo-3 is known to accumulate in this region of the cell (Perez-Terzic et al. 1997). All the numerical data were analysed in Excel and SigmaPlot. One-sample non-parametric tests (Wilcoxon signed-rank test) and two-sample non-parametric tests (Mann-Whitney U test) were used to assess statistical significance. All the fluorescence traces shown represent findings from a minimum of 60 cells in four different tissues and all the contraction traces shown represent findings from a minimum of eight different tissues. Analysis of the wave velocity and the frequency of oscillations has been described previously (Ruehlmann et al. 2000).
RESULTS
SR Ca2+ release and SERCA Ca2+ re-uptake
We have previously described caffeine- and PE-stimulated Ca2+ waves in VSMCs within the intact vessel wall of the rabbit IVC (Ruehlmann et al. 2000). Figure 1A shows a comparison of this wave-like pattern generated by caffeine and PE with the non-wave-like pattern of [Ca2+]i elevation initiated by high K+ depolarization. After addition of 25 mm caffeine to the bathing solution, Ca2+ waves originate from distinct intracellular loci and travel along the longitudinal axis of the long ribbon-shaped vascular smooth muscle cells, probably as a consequence of regenerative SR Ca2+ release. This wave-like pattern is very different from the non-wave-like rise in [Ca2+]i along the length of the cells when the L-type VGCCs are activated by elevating [K+]o to 80 mm. The difference between these spatiotemporal patterns of the increments in [Ca2+]i strongly suggest that when the IVC is stimulated by PE, the initial event is intracellular Ca2+ release, rather than Ca2+ influx from the extracellular space.
Figure 1. PE-mediated [Ca2+]i oscillations result from repetitive SR Ca2+ release through the IP3R channel followed by influx-dependent SR store refilling via SERCA.
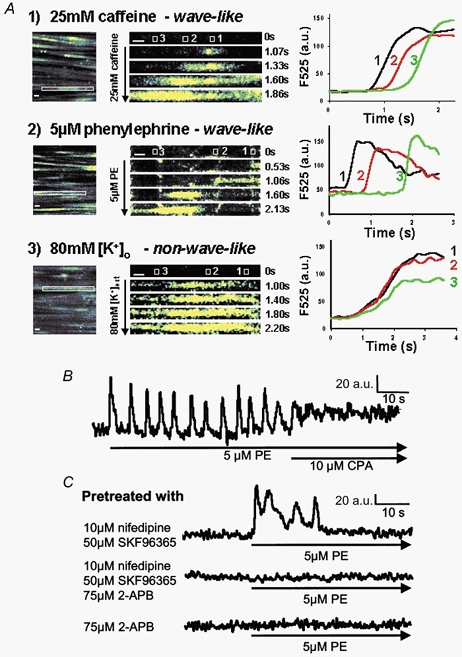
A, sets of time-series images (1-3) are displayed to identify the spatiotemporal patterns of [Ca2+]i rise elicited by different stimuli. Still images of a field of smooth muscle cells are shown to illustrate the rectangular regions (outlined in white in the leftmost images) from which each set of time-series images was derived. The rectangular regions contain a segment of one representative ribbon-shaped SMC and are enlarged and contrast-enhanced to facilitate the visualization of the spatiotemporal patterns of [Ca2+]i rise in these time-series. Variations in fluorescence intensity (F525) directly reflect changes in [Ca2+]i. In addition, the changes in F525 over time from selected areas (area 1, 2 and 3 outlined in white) spaced out across the longitudinal axis of each depicted SMC are illustrated on the right in the F525-time traces (a.u., arbitrary units). SR Ca2+ release with 25 mm caffeine caused an initial [Ca2+]i elevation that was initiated at a distinctive intracellular locus (time 1.07 s) and subsequently propagated along the longitudinal axis of the SMC in a regenerative, wave-like fashion. The F525-time traces (set 1) from the three intracellular areas indicate a sequential rise in [Ca2+]i over time as the Ca2+ signal was initiated at area 1 and subsequently propagated in a wave-like fashion through area 2 and finally to area 3. Stimulation by 5 μm PE also elicited a wave-like Ca2+ signal as the F525-time traces (set 2) demonstrated a sequential rise of [Ca2+]i over time in the three intracellular areas. In contrast, Ca2+ influx across the plasma membrane (PM) stimulated with 80 mm extracellular K+ PSS (80 mm [K+]o), caused an initial Ca2+ elevation that appears to be non-wave-like in nature. Such a non-wave-like pattern is clearly demonstrated by the F525-time traces (set 3), which indicate a synchronized rise in [Ca2+]i from the three intracellular areas. Thus, the wave-like pattern of the [Ca2+]i rise within a series of PE-induced [Ca2+]i oscillations resembles that seen in response to caffeine. This suggests that individual Ca2+ spikes elicited by PE are the result of regenerative SR Ca2+ release. The scale bars shown represent 5 μm. B, a representative Ca2+ trace is shown that displays the temporal changes in [Ca2+]i determined in a 1.36 μm2 cytoplasmic region of a single SMC from the rabbit IVC. SERCA blockade with 10 μm CPA resulted in both the abolishment of PE-induced [Ca2+]i oscillations and a sustained elevation in [Ca2+]i. C, PE-elicited transient [Ca2+]i oscillations in tissues pretreated with 10 μm nifedipine (L-type VGCC blocker) and 50 μm SKF96365 (ROC/SOC blocker); additional pretreatment with 75 μm 2-APB (IP3R channel blocker) abolished this transient Ca2+ signal. Pretreatment of the tissues with 75 μm 2-APB alone also abolished PE-mediated Ca2+ signals completely.
Figure 1B explores the source of the PE-induced Ca2+ oscillations. SERCA blockade with 10 μm CPA or 2 μm thapsigargin (data not shown) practically abolished the waves while the [Ca2+]i remained elevated just below wave peak value, confirming the critical role of the SR in the generation of the repetitive Ca2+ waves. Pretreatment of the IVC with a combination of nifedipine to block the L-type VGCCs and SKF96365 to block ROCs/SOCs caused a delayed inhibition of the [Ca2+]i oscillations and maintained the [Ca2+]i near the basal value (Fig. 1C). This result indicates that stimulated Ca2+ entry is required to refill the SR in order to maintain the periodic Ca2+ release, which is responsible for each up-stroke of the PE-induced [Ca2+]i oscillations. Thus, in the absence of stimulated Ca2+ entry, PE-induced [Ca2+]i oscillations can only persist for a few cycles during which Ca2+ is lost from the SR to the extracellular space. To test the hypothesis that the Ca2+ waves originate from the opening of inositol 1,4,5-trisphosphate-sensitive Ca2+ release channels (IP3R channels), 2-APB (a selective IP3R channel inhibitor; Ascher-Landsberg et al. 1999; Ma et al. 2000) was added to the pre-incubation solution before stimulating with 5 μm PE. Figure 1C shows that blockade of the IP3R channel prevented all PE-induced Ca2+ waves. Control experiments performed on the IVC confirmed that 75 μm 2-APB did not abolish the caffeine-induced [Ca2+]i transient (data not shown). These results indicate that PE-induced [Ca2+]i oscillations are a direct consequence of Ca2+ release from the SR via IP3R channels and, in addition, rely on stimulated Ca2+ entry for refilling of the SR. Interestingly, 2-APB pretreatment alone also prevented any change in [Ca2+]i, induced by PE, indicating that activation of IP3R channels is the very first and requisite event for PE-mediated [Ca2+]i signalling in VSMCs from the rabbit IVC.
Relative contributions by the L-type VGCCs and ROCs/SOCs
Nifedipine (L-type VGCC blocker) and SKF96365 (ROC/SOC and L-type VGCC blocker) were employed to investigate the routes of Ca2+ entry. However, due to the non-selective actions of SKF96365, its effects on PE-induced [Ca2+]i oscillations and tonic contraction were only assessed in tissues pretreated with 10 μm nifedipine. As shown in Fig. 2A, 10 μm nifedipine abolished 80 mm [K+]o-induced tonic contraction (n = 8 rings) in rabbit IVC pretreated with 10 μm phentolamine (an α-adrenergic receptor antagonist used to inhibit the actions of any noradrenaline released from the nerve endings). However, as shown in Fig. 3, 10 μm nifedipine pretreatment failed to abolish the asynchronous oscillatory Ca2+ waves elicited by 5 μm PE. This is similar to what has been reported by Miriel et al. (1999) in the rat mesenteric artery. Furthermore, in a detailed analysis of the spatiotemporal characteristics of the oscillations before and after addition of nifedipine, we found that nifedipine pretreatment significantly reduced the frequency of the oscillations from 0.44 ± 0.02 Hz to 0.28 ± 0.02 Hz (mean ± s.e.m., n = 86 cells). In contrast, nifedipine had no significant effect on the amplitude of the oscillations and the velocity of the Ca2+ waves. These results suggest that there is a relatively large nifedipine-insensitive component of Ca2+ entry that is capable of refilling the SR and maintaining PE-induced [Ca2+]i oscillations. It is clear from Fig. 3 that blockade of ROCs/SOCs with SKF96365 (50 μm) abolished the nifedipine-insensitive component of the PE-induced Ca2+ signal.
Figure 2. Effects of L-type VGCC blockade and ROC/SOC blockade on PE-mediated tonic contraction.
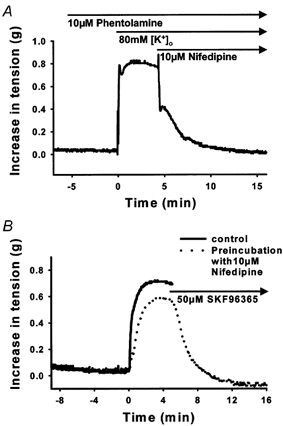
A, a representative tension trace showing that 10 μm nifedipine completely abolished an 80 mm [K+]o-mediated tonic contraction in rings of rabbit IVC pretreated with 10 μm phentolamine. B, L-type VGCC blockade with 10 μm nifedipine partially inhibited a PE-mediated tonic contraction (P < 0.05, Wilcoxon signed-rank test), while additional ROC/SOC blockade with 50 μm SKF96365 completely abolished the remaining tonic contraction.
Figure 3. Effects of L-type VGCC blockade and ROC/SOC blockade on PE-mediated [Ca2+]i oscillations.
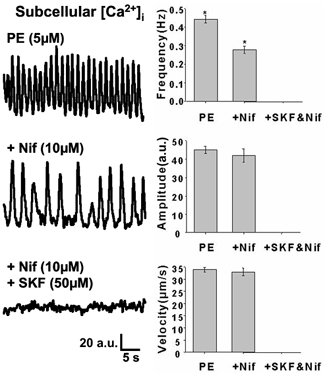
As shown in the representative traces, which indicate temporal changes in [Ca2+]i from a 1.36 μm2 sub-cellular region, L-type VGCC blockade with 10 μm nifedipine (Nif) did not abolish PE-induced [Ca2+]i oscillations. It reduced the frequency of PE-induced [Ca2+]i oscillations, but did not affect the amplitude or the apparent velocity of the oscillatory Ca2+ waves. ROC/SOC blockade with 50 μm SKF96365 (SKF) in addition to L-type VGCC blockade abolished PE-induced [Ca2+]i oscillations completely. The frequency of the [Ca2+]i oscillations was derived by counting the number of Ca2+ waves generated in a single SMC over a time period of 30-60 s. For estimation of the apparent velocity of the Ca2+ wave, two subcellular 1.36 μm2 regions separated by distance x (Δx) were selected and the time lag (Δt) in the onset of Ca2+ rise between the two regions was determined. The fraction of Δx over Δt yields the apparent velocity of the Ca2+ wave. (* Statistical significance with Mann-Whitney U test.)
In a parallel contraction study using identical protocols, we found that nifedipine pretreatment significantly (P < 0.05) reduced PE-induced tonic contraction by 27 ± 6 % (mean ± s.e.m., n = 15 rings; Fig. 2B) while addition of SKF96365 completely abolished the remaining contraction. This observation again supports the existence of a nifedipine-insensitive, SKF96365-sensitive component of Ca2+ entry, which is responsible for 73 ± 6 % (mean ± s.e.m., n = 15 rings) of tonic contraction.
Calcium entry through reverse-mode NCX
Arnon et al. (2000) have recently shown that in the mesenteric artery agonists stimulate Ca2+ entry via reverse-mode NCX as a consequence of Na+ entry through SOCs (see Discussion). We therefore tested the possibility that at least part of the SKF96365-sensitive component of stimulated Ca2+ entry in the IVC is mediated by the NCX. Two distinct inhibitors of NCX, 2′,4′-dichlorobenzamil (2,4-DCB, forward- and reverse-mode inhibitor; Blaustein & Lederer, 1999) and KB-R7943 (selective reverse-mode inhibitor; Ladilov et al. 1999) were employed in parallel [Ca2+]i and contraction studies. For this series of studies all tissues were pretreated with 10 μm nifedipine to eliminate Ca2+ entry through L-type VGCCs.
In a series of contraction studies, it was found that 2,4-DCB at both 10 μm (data not shown) and 100 μm (Fig. 4A) significantly (P < 0.05) reduced PE-induced tonic contraction in tissues pretreated with nifedipine by 81 ± 6 % (n = 8 rings) and 80 ± 2 % (n = 16 rings), respectively. The remaining 20 % of contraction was completely inhibited with 50 μm SKF96365. In a parallel [Ca2+]i study, it was found that 2,4-DCB (100 μm) abolished PE-induced [Ca2+]i oscillations in tissues pretreated with nifedipine (Fig. 4B), but did not reduce the steady-state [Ca2+]i to the baseline level. However, subsequent addition of SKF96365 did reduce the level of non-oscillating [Ca2+]i to the baseline level. These results indicate that the [Ca2+]i oscillations are, in large part, dependent on Ca2+ entry via the NCX.
Figure 4. Effects of NCX inhibitor 2′,4′-dichlorobenzamil on the nifedipine-resistant component of PE-mediated [Ca2+]i oscillations and tonic contraction.
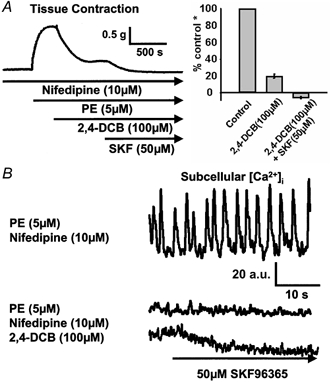
Blockade of Ca2+ entry through NCX with 100 μm 2′,4′-dichlorobenzamil (2,4-DCB) led to a large inhibition of PE-induced tonic contraction (A) and a complete inhibition of PE-induced [Ca2+]i oscillations (B) in rings pretreated with 10 μm nifedipine. The remaining contraction was abolished by application of 50 μm SKF96365 (SKF) while the non-oscillating [Ca2+]i was correspondingly reduced. (* Control refers to the amplitude of PE-mediated tonic contraction in rings pretreated with 10 μm nifedipine.)
To test whether the NCX operating in the reverse mode provides Ca2+ to sustain PE-mediated [Ca2+]i oscillations and tonic contraction in IVC pretreated with nifedipine, we used KB-R7943, which specifically blocks this mode of Na+-Ca2+ exchange. As shown in Fig. 5A, 10 μm KB-R7943 also significantly reduced PE-induced tonic contraction in vessels pretreated with nifedipine by 75 ± 5 % (mean ± s.e.m., n = 9 rings) while the remaining 25 % of contraction was completely inhibited by 50 μm SKF96365. In a parallel [Ca2+]i study (Fig. 5B) the same concentration of KB-R7943 also abolished PE-induced [Ca2+]i oscillations. Thus, the fact that two structurally unrelated inhibitors of NCX similarly abolished the PE-induced [Ca2+]i oscillations indicates that there is a large reverse-mode Na+-Ca2+ exchange component of Ca2+ entry, which is required for refilling the SR and allowing the [Ca2+]i oscillations to persist. Interestingly, there is a relatively smaller 2,4-DCB-insensitive and KB-R7943-insensitive component within the SKF96365-sensitive component, which may reflect Ca2+ entry through ROCs/SOCs.
Figure 5. Effects of the selective reverse-mode NCX inhibitor KB-R7943 on the nifedipine-resistant component of PE-mediated [Ca2+]i oscillations and tonic contraction.
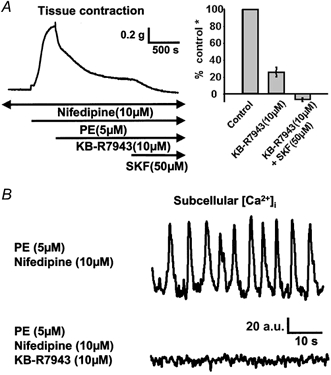
Blockade of the NCX operating in the reverse with 10 μm KB-R7943 led to a large inhibition of PE-induced tonic contraction (A) in rings pretreated with 10 μm nifedipine and complete inhibition of PE-induced [Ca2+]i oscillations (B). The remaining contraction was abolished with 50 μm SKF96365 (SKF). (* Control refers to the amplitude of PE-mediated tonic contraction in rings pretreated with 10 μm nifedipine.)
DISCUSSION
The concept that tonic vasoconstriction is based on asynchronous Ca2+ waves in individual smooth muscle cells has a profound impact on our views on the molecular mechanisms underlying Ca2+ signalling in smooth muscle. Iino and collaborators (Iino et al. 1994) proposed that agonist-induced Ca2+ waves are propagated by regenerative Ca2+ release from the SR network. This hypothesis is supported by our data on the inhibitory effect of 2-APB, which shows that the obligatory first step in generating an agonist-induced Ca2+ wave is the opening of IP3-sensitive channels. In smooth muscle, the release of Ca2+ from the SR is always accompanied by Ca2+ extrusion from the cells and in a Ca2+-free medium all Ca2+ released from the SR is irreversibly lost to the extracellular space (Leijten & van Breemen, 1986). Thus, it follows that refilling of smooth muscle SR involves Ca2+ entry across the cell membrane. In this paper we present evidence to support the view that agonist-induced [Ca2+]i oscillations are generated by cyclical movements of Ca2+. This Ca2+ cycle, depicted in Fig. 6, starts with Ca2+ release from the SR through IP3R channels into the myoplasm. A large fraction of the elevated myoplasmic Ca2+ is then extruded via the PMCA to the extracellular space, while the remaining fraction of the Ca2+ is taken up by the SERCA. Ca2+ entry via the NCX operating in the reverse mode and Ca2+ channels subsequently supplies Ca2+ to a sub-plasmalemmal, cytoplasmic microdomain from where it is pumped back into the SR via the SERCA. The mechanisms contributing to the individual components of this cyclical movement of Ca2+ are discussed below.
Figure 6. Model for Ca2+ movements during PE-induced [Ca2+]i oscillations in VSMCs from rabbit IVC.
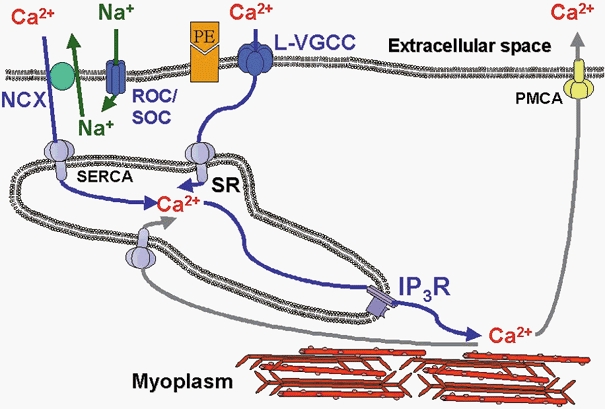
IP3 generated as a consequence of α-adrenergic activation releases Ca2+ towards the myoplasm to activate the myofilaments. Subsequent Ca2+ removal occurs partially through the PMCA, while the remainder is taken up into the SR by the SERCA. ROCs/SOCs, activated by receptor activation and SR depletion, allow entry of mainly Na+ and some Ca2+. This Na+ entry raises [Na+]i in a restricted space between the plasma membrane and the SR membrane and drives NCX in the reverse mode to supply Ca2+ to SERCA to refill the SR lumen, thereby completing the cycle. PE, phenylephrine; NCX, sodium/calcium exchanger; ROC/SOC, receptor-operated channel/store-operated channel; SERCA, sarcoplasmic- endoplasmic reticulum Ca2+-ATPase; L-type VGCC, L-type voltage-gated Ca2+ channel; PMCA, plasma membrane Ca2+-ATPase; IP3R, IP3-sensitive SR Ca2+ release channel; SR, sarcoplasmic reticulum.
Several investigators initially suggested that the SR could be directly filled by ROCs (Fasolato et al. 1994), capacitative Ca2+ entry (Casteels & Droogmans, 1981; Putney, 1986) or VGCCs (Gagov et al. 1994). However, the advent of SERCA blockers, like thapsigargin and CPA, provided evidence that refilling requires SERCA activity (Deng & Kwan, 1991). It was found that refilling of smooth muscle SR is not accompanied by the expected rise in global [Ca2+]i and myofilament activation (Aaronson & van Breemen, 1981; Casteels & Droogmans, 1981). The results reported herein indicate that each upstroke of the [Ca2+]i oscillations is associated with release through IP3R channels rather than with the influx of Ca2+, again suggesting that refilling does not proceed via the bulk of the cytoplasm. This and other functional evidence has led to the hypothesis that refilling of smooth muscle SR takes place in part across a restricted cytoplasmic space, or micro-domain, located between the cell membrane and the peripheral SR (for reviews see van Breemen et al. 1995 and Karaki et al. 1997). Although much of the evidence for cytoplasmic micro-domains is indirect and deduced from functional data, narrow spaces of approximately 15 nm in width between the plasma membrane (PM) and the SR have been visualized by electron microscopy (Somlyo, 1985). In addition, ion transport proteins residing in the PM have been co-localized with the superficial SR using deconvolution and confocal microscopy of fluorescent antibodies (Moore, 1993; Juhaszova et al. 1994, 1997).
A major finding in this report is that blocking the ROCs/SOCs with SKF96365 prevented the maintenance of repetitive Ca2+ waves. Unfortunately, the identity of the ROCs/SOCs has not been resolved at this time. α-Adrenergic receptor activation has been reported to open ROCs, which are cation channels permeable to both Ca2+ and Na+ (Fasolato et al. 1994). Since the α-adrenergic receptor is in itself not a channel, or channel activator, intermediate signalling possibly involving a second messenger (Gα, IP3, inositol tetrakisphosphate (IP4) or Ca2+) is required (Fasolato et al. 1994; Barritt, 1999). Another hypothetical mechanism, which links emptying of the SR store to the opening of SOCs, has been described for smooth muscle and non-excitable cells (Casteels & Droogmans, 1981; Cauvin et al. 1984; Putney, 1986). However, the molecular mechanism, which may involve either a calcium-influx factor (Trepakova et al. 2000) or direct mechano-coupling with the IP3R channel (Putney, 1999; Ma et al. 2000), remains to be determined. It has also been reported that different modes of activation function in parallel to open a common channel identified as either ROC or SOC (Wang & van Breemen, 1997). In any case the influx of Na+ and Ca2+ through these non-selective cation channels depolarizes the plasma membrane potential sufficiently to open L-type VGCCs. This was confirmed by the inhibitory effect of nifedipine on the PE-induced contraction.
The effect of nifedipine was interesting in that it did not affect either the amplitude or velocity of the repetitive Ca2+ waves, but did reduce their frequency. There are two plausible mechanisms through which the frequency could be decreased. Firstly, the absence of stimulated Ca2+ influx through L-type VGCCs may reduce the rate of refilling of the SR Ca2+ store. Given that the SR luminal [Ca2+] can regulate IP3R channel opening probability (Meldolesi & Pozzan, 1998), a reduced rate of SR Ca2+ refilling can result in a decreased frequency of SR Ca2+ release at the wave initiation site. Secondly, Ca2+ influx through L-type Ca2+ channels can elevate [Ca2+]i in regions adjacent to IP3Rs, which facilitates calcium-induced calcium release (CICR) mediated by both types of SR Ca2+ release channel. Thus, removal of Ca2+ entry through L-type VGCCs can result in a delay of regenerative CICR and thereby reduce the frequency of the [Ca2+]i oscillations.
The nifedipine-insensitive component was not only blocked by SKF96365, but was also greatly inhibited by blockers of the NCX. This implicates reverse-mode NCX as a physiological route of Ca2+ entry in the VSMC. Since the NCX-mediated Ca2+ entry is also sensitive to SKF96365, it follows that the reverse-mode NCX Ca2+ entry is dependent on the stimulation of ROCs/SOCs. While evidence indicates that at a resting [Na+]i of ≈10 mm the NCX typically operates in the forward Ca2+ extrusion mode, when [Na+]i is elevated above 20 mm, the energetics favour the reverse-mode activity. In order to significantly raise [Na+]i, Na+ must enter the cell, putatively, via the non-selective cation-permeable ROCs/SOCs into a restricted space, which is considerably smaller than the bulk cytoplasm. In contrast to L-type VGCCs (permeability ratio of Ca2+ to Na+, PCa/PNa > 1000; Bean, 1989), ROCs/SOCs are believed to be much more permeable to Na+ with a PCa/PNa < 10 (Philipp et al. 1996). With 140 mm extracellular Na+ as compared to 1.5 mm extracellular Ca2+, opening of ROCs/SOCs should result in a large Na+ influx in addition to the Ca2+ influx. It has previously been proposed that Na+ entering the cardiac myocyte accumulates in a restricted sub-plasmalemmal space (fuzzy space) formed by the close apposition between the superficial SR and the PM (Philipson & Nicoll, 2000), similar to the one discussed above for smooth muscle. Thus, conditions may exist to temporarily reverse the balance between the Na+ and Ca2+ gradients to promote Ca2+ entry via the NCX. In fact our findings support a mechanism, similar to that previously proposed by Blaustein and colleagues (Arnon et al. 2000), whereby Ca2+ entry through reverse-mode NCX is serially coupled to Na+ entry through the activated ROCs/SOCs. Therefore, either blockade of the ROCs/SOCs or blockade of the NCX (reverse mode) abolishes the [Ca2+]i oscillations. This model is further supported by the structural findings that the plasmalemmal NCX and the high ouabain affinity isoform (α3) of the Na+-K+-ATPase appear to colocalize with the superficial SR and SERCA in smooth muscles (Moore et al. 1993; Juhaszova et al. 1994; Juhaszova et al. 1997). This close spatial arrangement would allow Ca2+ entry through reverse-mode NCX to refill the superficial SR via its SERCA pumps. Since the SR in SMCs is believed to be an interconnecting tubular network, refilling of the deeper SR would occur through SR Ca2+ redistribution from the superficial SR. We have previously shown that at rest the close association between the superficial SR and NCX functions in the forward mode to unload the SR (Nazer & van Breemen, 1998). These studies also provided evidence in support of Ca2+ redistribution within the SR. The mechanisms described above may apply to other cell types, as Graier and coworkers recently reported that reverse-mode NCX-dependent endoplasmic reticulum Ca2+ refilling supports agonist-mediated [Ca2+]i oscillations in endothelial cell lines (Paltauf-Doburzynska et al. 2000).
Ca2+ entry through ROC/SOC-coupled reverse-mode NCX, L-type VGCCs and ROCs/SOCs all contribute to force generation. However, Ca2+ entry through the ROCs/SOCs alone is incapable of supporting the recurrent Ca2+ waves. This may be due to the relatively slow rate of Ca2+ entry through these non-selective cation channels, which is not able to refill the SR Ca2+ store to a sufficiently high level to maintain the repetitive firing of Ca2+ waves. However, the slow Ca2+ entry through the ROCs/SOCs does elevate steady-state [Ca2+]i to support sub-maximal force development as shown in Fig. 4.
Although the mechanisms underlying the PE-mediated [Ca2+]i oscillations are now being resolved, the physiological advantage of repetitive waves of SR Ca2+ release over steady-state Ca2+ elevation is rather poorly understood. In the temporal domain, oscillations of varying frequencies can regulate frequency-sensitive enzyme(s) that serve to amplify downstream signals (De Koninck & Schulman, 1998). It is also possible that recurrent Ca2+ waves, given their transient nature, may result in organellar Ca2+ concentrations (e.g. nuclear [Ca2+]) that are different from those that would result if [Ca2+]i were held at a steady-state level. Thus, Ca2+ oscillations may provide a means to selectively activate the contractile machinery in the myoplasm without disrupting Ca2+-sensitive processes that occur in the nuclear compartment.
In summary, our data show that the tonic contraction of a large vein in response to α-adrenergic stimulation is maintained by repetitive, asynchronous Ca2+ waves, which are initiated by the opening of IP3-sensitive SR channels. This oscillating SR Ca2+ release is maintained by stimulated Ca2+ entry through ROC/SOC-coupled reverse-mode NCX, L-type VGCCs and ROCs/SOCs, which in the IVC are responsible for 59, 27 and 14 % of the tonic contraction, respectively.
Acknowledgments
C.-H.L. is a recipient of CIHR MD/PhD studentship; D.O.R. was supported by a grant from the BC/Yukon Heart & Stroke Foundation. The research was supported by an operating grant from the BC/Yukon Heart & Stroke Foundation. We would like to thank the St Paul's Hospital Foundation for their generous support.
References
- Aaronson P, van Breemen C. Effects of sodium gradient manipulation upon cellular calcium, 45Ca fluxes and cellular sodium in the guinea-pig taenia coli. Journal of Physiology. 1981;319:443–461. doi: 10.1113/jphysiol.1981.sp013920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon A, Hamlyn JM, Blaustein MP. Na+ entry via store-operated channels modulates Ca2+ signaling in arterial myocytes. American Journal of Physiology. 2000;278:C163–173. doi: 10.1152/ajpcell.2000.278.1.C163. [DOI] [PubMed] [Google Scholar]
- Asada Y, Yamazawa T, Hirose K, Takasaka T, Iino M. Dynamic Ca2+ signalling in rat arterial smooth muscle cells under the control of local renin-angiotensin system. Journal of Physiology. 1999;521:497–505. doi: 10.1111/j.1469-7793.1999.00497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascher-Landsberg J, Saunders T, Elovitz M, Phillipe M. The effects of 2-aminoethoxydiphenyl borate, a novel inositol 1,4,5-triphosphate receptor modulator on myometrial contractions. Biochemical and Biophysical Research Communications. 1999;264:979–982. doi: 10.1006/bbrc.1999.1602. [DOI] [PubMed] [Google Scholar]
- Barritt GJ. Receptor-activated Ca2+ inflow in animal cells: a variety of pathways tailored to meet different intracellular Ca2+ signalling requirements. Biochemical Journal. 1999;337:153–169. [PMC free article] [PubMed] [Google Scholar]
- Bean BP. Classes of calcium channels in vertebrate cells. Annual Review of Physiology. 1989;51:367–384. doi: 10.1146/annurev.ph.51.030189.002055. [DOI] [PubMed] [Google Scholar]
- Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiological Reviews. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- Casteels R, Droogmans G. Exchange characteristics of the noradrenaline-sensitive calcium store in vascular smooth muscle cells or rabbit ear artery. Journal of Physiology. 1981;317:263–279. doi: 10.1113/jphysiol.1981.sp013824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauvin C, Lukeman S, Cameron S, Hwang O, Meisheri K, Yamamoto H, van Breemen C. Theoretical bases for vascular selectivity of Ca2+ antagonists. Journal of Cardiovascular Pharmacology. 1984;6:S630–638. doi: 10.1097/00005344-198406004-00009. [DOI] [PubMed] [Google Scholar]
- Cauvin C, Lukeman S, Cameron J, Hwang O, van Breemen C. Differences in norepinephrine activation and diltiazem inhibition of calcium channels in isolated rabbit aorta and mesenteric resistance vessels. Circulation Research. 1985;56:822–828. doi: 10.1161/01.res.56.6.822. [DOI] [PubMed] [Google Scholar]
- Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiological Reviews. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- De Koninck P, Schulman H. Sensitivity of Ca2+/calmodulin-dependent protein kinase II to the frequency of Ca2+ oscillations. Science. 1998;279:227–230. doi: 10.1126/science.279.5348.227. [DOI] [PubMed] [Google Scholar]
- Deng HW, Kwan CY. Cyclopiazonic acid is a sarcoplasmic reticulum Ca2+-pump inhibitor of rat aortic muscle. Chung Kuo Yao Li Hsueh Pao. 1991;12:53–58. [PubMed] [Google Scholar]
- Fasolato C, Innocenti B, Pozzan T. Receptor-activated Ca2+ influx: how many mechanisms for how many channels. Trends in Pharmacological Sciences. 1994;15:77–83. doi: 10.1016/0165-6147(94)90282-8. [DOI] [PubMed] [Google Scholar]
- Gagov HS, Duridanova DB, Boev KK, Daniel EE. L-type calcium channels may fill directly the IP3-sensitive calcium stores. General Physiology and Biophysics. 1994;13:75–84. [PubMed] [Google Scholar]
- Iino M, Kasai H, Yamazawa T. Visualization of neural control of intracellular Ca2+ concentration in single vascular smooth muscle cells in situ. EMBO Journal. 1994;13:5026–5031. doi: 10.1002/j.1460-2075.1994.tb06831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhaszova M, Ambesi A, Lindenmayer GE, Bloch RJ, Blaustein MP. Na+-Ca2+ exchanger in arteries: identification by immunoblotting and immunofluorescence microscopy. American Journal of Physiology. 1994;266:C234–242. doi: 10.1152/ajpcell.1994.266.1.C234. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Blaustein MP. Na+ pump low and high ouabain affinity α subunit isoforms are differently distributed in cells. Proceedings of the National Academy of Sciences of the USA. 1997;94:1800–1805. doi: 10.1073/pnas.94.5.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaki H, Ozaki H, Hori M, Mitsui-Saito M, Amano K, Miyamoto S, Nakazawa H, Won KJ, Sato K. Calcium movements, distribution and functions in smooth muscle. Pharmacological Reviews. 1997;49:157–230. [PubMed] [Google Scholar]
- Khalil R, Lodge N, Saida K, van Breemen C. Mechanism of calcium activation in vascular smooth muscle. Journal of Hypertension. 1987;5:S5–15. doi: 10.1097/00004872-198712004-00003. [DOI] [PubMed] [Google Scholar]
- Ladilov Y, Haffner S, Balser-Schafer C, Maxeiner H, Piper HM. Cardioprotective effects of KB-R7943: a novel inhibitor of the reverse mode of Na+/Ca2+ exchanger. American Journal of Physiology. 1999;276:H1868–1876. doi: 10.1152/ajpheart.1999.276.6.H1868. [DOI] [PubMed] [Google Scholar]
- Leijten PA, van Breemen C. The relationship between noradrenaline-induced contraction and 45Ca2+ efflux stimulation in rabbit mesenteric artery. British Journal of Pharmacology. 1986;89:739–747. doi: 10.1111/j.1476-5381.1986.tb11178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H-T, Patterson RL, van Rossum DB, Birnbaumer L, Mikoshiba K, Gill DL. Requirement of the inositol triphosphate receptor for activation of store-operated Ca2+ channels. Science. 2000;287:1647–1651. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- Meldolesi J, Pozzan T. The endoplasmic reticulum Ca2+ stores: a view from the lumen. Trends in Biochemical Sciences. 1998;23:10–14. doi: 10.1016/s0968-0004(97)01143-2. [DOI] [PubMed] [Google Scholar]
- Miriel VA, Mauban JRH, Blaustein MP, Wier WG. Local and cellular Ca2+ transients in smooth muscle of pressurized rat resistance arteries during myogenic and agonist stimulation. Journal of Physiology. 1999;518:815–824. doi: 10.1111/j.1469-7793.1999.0815p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore ED, Etter EF, Philipson KD, Carrington WA, Fogarty KE, Lifshitz LM, Fay FS. Coupling of the Na+/Ca2+ exchanger, Na+/K+ pump and sarcoplasmic reticulum in smooth muscle. Nature. 1993;365:657–660. doi: 10.1038/365657a0. [DOI] [PubMed] [Google Scholar]
- Nazer MA, van Breemen C. Functional linkage of Na+-Ca2+ exchange and sarcoplasmic reticulum Ca2+ release mediates Ca2+ cycling in vascular smooth muscle. Cell Calcium. 1998;24:275–283. doi: 10.1016/s0143-4160(98)90051-3. [DOI] [PubMed] [Google Scholar]
- Nazer MA, van Breemen C. A role for the sarcoplasmic reticulum in Ca2+ extrusion from rabbit inferior vena cava smooth muscle. American Journal of Physiology. 1999;274:H123–131. doi: 10.1152/ajpheart.1998.274.1.H123. [DOI] [PubMed] [Google Scholar]
- Paltauf-Doburzynska J, Frieden M, Spitaler M, Graier WF. Histamine-induced Ca2+ oscillations in a human endothelial cell line depend on transmembrane ion flux, ryanodine receptors and endoplasmic reticulum Ca2+-ATPase. Journal of Physiology. 2000;524:701–713. doi: 10.1111/j.1469-7793.2000.00701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Terzic C, Stehno-Bittel L, Clapham DE. Nucleoplasmic and cytoplasmic differences in the fluorescence properties of the calcium indicator Fluo-3. Cell Calcium. 1997;21:275–282. doi: 10.1016/s0143-4160(97)90115-9. [DOI] [PubMed] [Google Scholar]
- Philipp S, Cavalie A, Freichel M, Wissenbach U, Zimmer S, Trost C, Marquart A, Murakami M, Flockerzi V. A mammalian capacitative calcium entry channel homologous to Drosophila TRP and TRPL. EMBO Journal. 1996;15:6166–6171. [PMC free article] [PubMed] [Google Scholar]
- Philipson KD, Nicoll DA. Sodium-calcium exchange: A molecular perspective. Annual Review of Physiology. 2000;62:111–133. doi: 10.1146/annurev.physiol.62.1.111. [DOI] [PubMed] [Google Scholar]
- Putney JW. A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Putney JW. TRP, inositol 1,4,5-triphosphate receptors, and capacitative calcium entry. Proceedings of the National Academy of Sciences of the USA. 1999;96:14669–14671. doi: 10.1073/pnas.96.26.14669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruehlmann DO, Lee CH, Poburko D, van Breemen C. Asynchronous Ca2+ waves in intact venous smooth muscle. Circulation Research. 2000;86:e72–79. doi: 10.1161/01.res.86.4.e72. [DOI] [PubMed] [Google Scholar]
- Somlyo AP. Excitation-contraction coupling and the ultrastructure of smooth muscle. Circulation Research. 1985;57:497–507. doi: 10.1161/01.res.57.4.497. [DOI] [PubMed] [Google Scholar]
- Trepakova ES, Csutora P, Hunton DL, Marchase RB, Cohen RA, Bolotina VM. Calcium influx factor directly activates store-operated cation channels in vascular smooth muscle cells. Journal of Biological Chemistry. 2000;275:26158–26163. doi: 10.1074/jbc.M004666200. [DOI] [PubMed] [Google Scholar]
- van Breemen C, Chen Q, Laher I. Superficial buffer barrier function of smooth muscle sarcoplasmic reticulum. Trends in Pharmacological Sciences. 1995;16:98–105. doi: 10.1016/s0165-6147(00)88990-7. [DOI] [PubMed] [Google Scholar]
- Wang X, van Breemen C. Multiple mechanisms of activating Ca2+ entry in freshly isolated rabbit aortic endothelial cells. Journal of Vascular Research. 1997;34:196–207. doi: 10.1159/000159223. [DOI] [PubMed] [Google Scholar]