

Abstract
The effects of activation of protein kinase C (PKC) on membrane currents gated by capsaicin, protons, heat and anandamide were investigated in primary sensory neurones from neonatal rat dorsal root ganglia (DRG) and in HEK293 cells (human embryonic kidney cell line) transiently or stably expressing the human vanilloid receptor hVR1.
Maximal activation of PKC by a brief application of phorbol 12-myristate 13-acetate (PMA) increased the mean membrane current activated by a low concentration of capsaicin by 1.65-fold in DRG neurones and 2.18-fold in stably transfected HEK293 cells. Bradykinin, which activates PKC, also enhanced the response to capsaicin in DRG neurones. The specific PKC inhibitor RO31-8220 prevented the enhancement caused by PMA.
Activation of PKC did not enhance the membrane current at high concentrations of capsaicin, showing that PKC activation increases the probability of channel opening rather than unmasking channels.
Application of PMA alone activated an inward current in HEK293 cells transiently transfected with VR1. The current was suppressed by the VR1 antagonist capsazepine. PMA did not, however, activate a current in the large majority of DRG neurones nor in HEK293 cells stably transfected with VR1.
Removing external Ca2+ enhanced the response to a low concentration of capsaicin 2.40-fold in DRG neurones and 3.42-fold in HEK293 cells. Activation of PKC in zero Ca2+ produced no further enhancement of the response to capsaicin in either DRG neurones or HEK293 cells stably transfected with VR1.
The effects of PKC activation on the membrane current gated by heat, anandamide and low pH were qualitatively similar to those on the capsaicin-gated current.
The absence of a current activated by PMA in most DRG neurones or in stably transfected HEK293 cells suggests that activation of PKC does not directly open VR1 channels, but instead increases the probability that they will be activated by capsaicin, heat, low pH or anandamide. Removal of calcium also potentiates activation, and PKC activation then has no further effect. The results are consistent with a model in which phosphorylation of VR1 by PKC increases the probability of channel gating by agonists, and in which dephosphorylation occurs by a calcium-dependent process.
Heat stimuli of above about 43 °C elicit a sensation of pain in humans, and initiate action potentials in nociceptive nerve terminals in humans and animals (Belmonte & Giraldez, 1981; Robinson et al. 1983). In experiments on isolated nociceptive neurones, heat was found to activate an inward current that has the properties expected from psychophysical and whole-animal experiments for the detector of painful levels of heat (Cesare & McNaughton, 1996; Reichling & Levine, 1997; Kirschstein et al. 1997; Nagy & Rang, 1999). Inflammation causes the threshold for the initiation both of a sensation of pain and of action potentials in nociceptive nerve fibres to fall to a lower temperature, a process known as sensitization (Treede et al. 1992). The heat-gated current also shows sensitization, in that the temperature threshold for activation of the current is lowered on exposure of the nociceptor to bradykinin, a nonapeptide released during inflammation (Cesare & McNaughton, 1996). Bradykinin sensitizes the heat-gated current by activating the ɛ isoform of protein kinase C (PKCɛ) (Cesare et al. 1999a).
An important molecular sensor for heat is the receptor for capsaicin, which was recently cloned and named vanilloid receptor 1, or VR1 (Caterina et al. 1997). Capsaicin, the active ingredient of chilli peppers, excites nociceptive neurones (Baumann et al. 1991; LaMotte et al. 1992) resulting in a burning pain sensation (Buck & Burks, 1986). VR1 is a non-selective cation channel that is activated by a broad spectrum of stimuli, including capsaicin, heat, low pH and the endocannabinoid anandamide (Caterina et al. 1997; Tominaga et al. 1998; Zygmunt et al. 1999; Smart et al. 2000). Targeted disruption of the VR1 gene in mice produces animals that do not respond adversely to even high doses of capsaicin (Caterina et al. 2000), showing that VR1 is the only receptor for capsaicin linked to pain perception. VR1 knockout animals still respond to noxious heat, showing that other heat-sensitive nociceptor mechanisms exist in addition to VR1. A major difference between wild-type and VR1 knockout mice, however, was that the heat hyperalgesia observed in inflamed tissue was greatly reduced in the knockout, suggesting that VR1 is the principal mechanism responsible for heat hyperalgesia (Davis et al. 2000; Caterina et al. 2000).
The experiments in the present study explore the role of PKC in the sensitization of VR1, using both nociceptive neurones and heterologously expressed VR1. In nociceptive neurones we found that the response to capsaicin is sensitized by PKC activation, in a similar manner to the response to heat, adding to the evidence that the same molecular mechanism is responsible for the response to the two stimuli. In HEK293 cells expressing VR1 (hVR1-HEK293 cells) the responses to heat, capsaicin, low pH and anandamide were found to be enhanced by PKC activation. The enhancement of current in response to capsaicin, in both nociceptive neurones and hVR1-HEK293 cells, was abolished in the absence of external Ca2+, consistent with the demonstration by other authors that desensitization of VR1 occurs by a calcium-sensitive process (Cholewinski et al. 1993; Docherty et al. 1996; Liu & Simon, 1996; Koplas et al. 1997). Finally, while heat, capsaicin, low pH and anandamide activate current through VR1, activation of PKC potentiates the effect of these stimuli but does not itself activate current, showing that PKC activation does not directly gate the VR1 channel. Preliminary reports of the data reported here have appeared elsewhere (Vellani et al. 1999).
METHODS
Preparation of rat dorsal root ganglion neurone cultures
Experiments were performed on cultured dorsal root ganglion (DRG) neurones taken from neonatal rats. Rat pups (Wistar, postnatal day (P)2-P7) were killed by cervical dislocation followed by decapitation. Culture methods and recording conditions were as previously described (Cesare & McNaughton, 1996). In brief, neurones were enzymatically isolated from DRG ganglia, plated on plastic Petri dishes (Nunc) and maintained in culture in Dulbecco's Modified Eagle's Medium (DMEM, Gibco) in 5 % CO2 and in the presence of 50 ng ml−1 nerve growth factor (NGF) for 3-4 days before being whole-cell patch clamped (see below). In some experiments neurones from adult mice (killed by cervical dislocation followed by decapitation) were used for comparison with those of neonatal rat (noted where appropriate). Isolation and culture methods were identical to those for rat neurones.
Cell culture of HEK293 cells and expression of VR1
Full-length human VR1 (hVR1) cDNA was amplified from brain cDNA, inserted into the expression vector pcDNA3.1 and double-strand sequenced (Hayes et al. 2000). A stable clone of human embryonic kidney (HEK293) cells expressing hVR1 (hVR1-HEK293) was obtained by transfection using Lipofectamine Plus (Life Technologies) followed by selection in 400 μg ml−1 geneticin (Life Technologies) and colony cloning. HEK293 cells transiently transfected with VR1 typically gave larger and more reproducible currents in response to capsaicin application than the stably transfected cells, and for this reason transiently transfected cells were used in experiments with anandamide, where the response was smaller than with capsaicin (Smart et al. 2000). HEK293 cells were co-transfected with hVR1 and green fluorescent protein (GFP, 10:1; plasmid pGreen lantern; Life Technologies) using the calcium phosphate precipitation method of transfection (Clontech).
Wild-type HEK293 and hVR1-HEK293 cells were routinely grown as monolayers and were maintained in DMEM supplemented with 10 % fetal bovine serum, 2 mml-glutamine and 90 μg ml−1 streptomycin-penicillin, in 5 % CO2 at 37 °C. Cells were passaged every 3-4 days and were not used above passage number 30. Wild-type HEK293 and hVR1-HEK293 cells were plated on poly-l-lysine-coated 35 mm diameter plastic culture dishes (Nunc) at a density of 125 000 cells per dish. Isolated hVR1-HEK293 cells or transiently transfected HEK293 cells (identified by expresssion of GFP) were selected for patch-clamp recording.
Electrophysiological recordings
All recordings were made from the somata of small-diameter DRG neurones (diameter, 15-25 μm) or from HEK293 cells, at ambient room temperature (20-24 °C), with the whole-cell patch-clamp technique using a List EPC-7 amplifier and pCLAMP software (Axon Instruments). Patch pipettes of resistance 1.5-4 MΩ were prepared from borosilicate glass using a Sutter Instruments P-87 (Novato, CA, USA) horizontal micropipette puller. Pipette capacitance was compensated electronically before breaking into the whole-cell mode, and the series resistance after establishing the whole-cell mode was calculated from the time constant of current decay in response to a voltage step. Series resistance (from 4 to 7 MΩ) was routinely checked to ensure that it did not increase in the course of an experiment, but was not compensated. Only one recording was performed on each culture dish, in order to ensure that data were not obtained from cells that had been inadvertently exposed to capsaicin, heat or other test treatments. All experiments were performed at room temperature (20-24 °C).
Measuring [Ca2+]i
The Ca2+-dependent fluorescence in cultured DRG neurones was imaged using an MRC-600 confocal microscope (Bio-Rad) as previously described (Nadal et al. 1996). Cells were loaded with the Ca2+-sensitive dye Fluo-4 (10 μm for 10 min at room temperature). At the end of the experiment the Ca2+-dependent fluorescence was calibrated by first elevating [Ca2+]i to a saturating level, by permeabilizing the neurone with ionomycin (10 μm) in the presence of 30 mm[Ca2+]o, followed by lysis in digitonin (20-50 μm) in order to measure background fluorescence from loaded organelles and from instrumental sources. All experiments were performed at room temperature (20-24 °C).
Immunocytochemistry and Western blots
The presence in HEK293 cells of the δ and ɛ isoforms of PKC was confirmed using both immunocytochemistry and Western blotting, according to the methods described in Cesare et al. (1999a).
Solutions
Cells were normally superfused with a solution of the following composition (mm): NaCl, 140; CaCl2, 1.8; MgCl2, 1; KCl, 4; Hepes, 10; and glucose, 4; neutralized to pH 7.4 with NaOH. Ultrapure water (MilliQ) was used in the preparation of all solutions. To produce nominally Ca2+-free external solutions 5 mm EGTA was added to the external solution and Mg2+ was increased to 1.6 mm, in order to obtain the same free Mg2+ concentration. Mg2+ was retained in the zero Ca2+ solution to suppress the large inward Na+ current passing through Ca2+ channels, which is observed on total removal of divalent ions (Carbone et al. 1997). For anandamide experiments, the extracellular solution was supplemented with 0.2 % lipid-free bovine serum albumin, to prevent non-specific adsorption of the lipophilic anandamide to the plastic tubing. To investigate the effect of pH, in solutions of pH 6.7 and 5.4 the extracellular Hepes was replaced with 10 mm Mes. The patch-clamp pipette solution was (mm): KCl, 135; MgCl2, 1.6; EGTA, 2; MgATP, 2.5; Li2GTP, 0.2; and Hepes, 10; neutralized to pH 7.3 with NaOH. In some experiments calcium was buffered with BAPTA (potassium salt, 10 mm) or EGTA was lowered to 0.2 mm (specified in the figure legends). Chemicals were prepared as concentrated stock solutions in either distilled water or ethanol as appropriate, then diluted to the final concentration using superfusate solution or pipette solution as appropriate. All chemicals were supplied by Sigma, except for NGF (Promega) and Fluo-4 (Molecular Probes).
A rapid extracellular solution change (< 50 ms) was achieved using a BioLogic RSC-100 multibarrel perfusion pipette, moved in front of the recorded cell by a computer-controlled stepping motor. The temperature of one barrel could be controlled using a Peltier device as previously described (Cesare & McNaughton, 1996), with the temperature monitored by a miniature thermocouple close to the solution exit from the tube.
Statistics
Means are given ± standard error of the mean (s.e.m.), with the value of n referring to the number of separate cells on which the experiment was performed. Significance was established with Student's two-tailed t test (paired or unpaired depending on whether the data were obtained from the same or different cells) and the values of the level of significance, P, obtained are given.
RESULTS
Protein kinase C activation enhances capsaicin-gated membrane current
Capsaicin is a lipophilic compound that readily penetrates the cell membrane, and activates cation-permeable ion channels by an action at a vanilloid receptor located at the internal surface of the membrane (Jung et al. 1999). With long exposure the current amplitude declines, an effect that has been shown to be due to calcium influx through capsaicin-gated channels, followed by activation of calcineurin, a calcium-sensitive phosphatase (Cholewinski et al. 1993; Docherty et al. 1996; Liu & Simon, 1996; Koplas et al. 1997). To minimize the calcium influx and the consequent current decline we used brief pulses of capsaicin for experiments in this study. Figure 1 shows typical inward currents activated in a small-diameter neurone from the DRG by pulses of capsaicin of increasing duration (Fig. 1A) or of increasing concentration (Fig. 1B). The effects of PKC activation on capsaicin-gated currents were apparent only at subsaturating concentrations of capsaicin (see below), so for the majority of experiments on DRG neurones an approximately half-saturating concentration of 0.5 μm was chosen. An application time of 1 s was chosen as it gave reproducible current responses and produced only modest desensitization with repeated applications. In hVR1-HEK293 cells a lower test concentration of 0.1 μm capsaicin was routinely used.
Figure 1. Membrane current in a nociceptive neurone in response to application of capsaicin.

Membrane current recorded from a DRG neurone in response to brief pulses of capsaicin of increasing duration (A, concentration 1 μm) and of increasing concentration (B, duration of exposure 1 s).
Figure 2A shows the effect of PKC activation on capsaicin-gated membrane currents in a small-diameter DRG neurone. The mean peak current of 0.91 ± 0.07 nA in response to 0.5 μm capsaicin immediately before application of PMA, a specific PKC activator, increased to 1.50 ± 0.09 nA within 10-15 s of application of PMA (1 μm, n = 9). The increase was highly significant (P = 2 × 10−4; paired t test), and the ratio between the mean current before and after PMA was 1.65. The enhancement was maximal within 15 s (Fig. 2B), and further 10 s applications or a more prolonged exposure caused no further enhancement (see below, Fig. 5C). The rapid and irreversible effect of PMA is in agreement with a previous study in which the heat-activated current in DRG neurones was found to be permanently enhanced by a single brief exposure to PMA (Cesare & McNaughton, 1996).
Figure 2. PKC activation enhances the capsaicin-activated current in nociceptive neurones.

A, membrane current activated by a 1 s application of 0.5 μm capsaicin before and 15 s after maximal activation of PKC with PMA (1 μm, 10 s application), in a DRG neurone in 1.8 mm Ca2+ (left panel) or in zero Ca2+, 5 mm EGTA (right panel). The inset in each case shows that application of PMA itself did not activate detectable membrane current. B, mean current in DRG neurones in response to application of 0.5 μm capsaicin, and the effect of 1 μm PMA, in 1.8 mm Ca2+ (left panel) and zero Ca2+ (right panel). Error bars show ±s.e.m. (n = 5 in 1.8 mm Ca2+ and n = 14 in zero Ca2+, where n gives the number of cells on which the experiment was repeated, *** P < 0.0005). Protocol as shown in A. Mean current in this figure is from cells in which stable recordings were obtained for the complete duration of the time course shown; other cells gave stable recordings during and after application of PMA, but not for the entire time course, and these cells were used in addition to those shown here for the data quoted in the text and shown in Fig. 8.
Figure 5. Direct activation of VR1 by PMA.

A, 1 μm PMA repeatedly activated an inward current, which was blocked by the VR1 antagonist capsazapine (CPZ, 10 μm), in HEK cells transiently transfected with hVR1. Mean current amplitude in responding cells was 2.60 ± 0.38 nA (n = 7). This cell responded to 0.5 μm capsaicin with 5.1 nA membrane current. B, lack of effect of 10 μm PMA in 11/13 capsaicin-sensitive DRG neurones (example in left-hand panel) and a small inward current seen in 2/13 DRG neurons (largest example shown in right-hand panel). Mean current amplitude in the 2 PMA-responsive neurones was 38 ± 8 pA. The amplitude of the current gated by 0.5 μm capsaicin in the cells shown was 0.78 nA (left) and 0.27 nA (right). C, a single exposure to 1 μm PMA caused maximal sensitization of the capsaicin-gated current in DRG neurones, as further exposures caused no additional sensitization. Similar results were seen in 4 other experiments. Longer exposures (30 s) or higher concentrations (10 μm) were also ineffective after an initial exposure to PMA.
PKC activation caused a qualitatively similar enhancement of the capsaicin-gated current in hVR1-HEK293 cells (Fig. 3A). The peak current of 0.22 ± 0.06 nA in response to 0.1 μm capsaicin (n = 14) increased to 0.48 ± 0.08 nA on application of PMA (1 μm). The increase was highly significant (P = 2 × 10−5; paired t test), and the ratio between the mean current before and after PMA was 2.18. As in DRG neurones, the effect was rapid, though the maximum current increase typically occurred ≈45 s after PMA application (see Fig. 3B).
Figure 3. PKC activation enhances the capsaicin-activated current in hVR1-HEK293 cells.

A, membrane current activated by a 1 s application of 0.1 μm capsaicin before and 15 s after activation of PKC with PMA (1 μm, 15 s application), in a stably transfected hVR1-HEK293 cell in 1.8 mm Ca2+ (left panel) or in zero Ca2+, 5 mm EGTA (right panel). The inset shows that application of PMA itself did not activate any detectable membrane current in HEK293 cells stably transfected with hVR1. B, mean current in response to application of 0.1 μm capsaicin, and the effect of 1 μm PMA, in 1.8 mm Ca2+ (left panel) and zero Ca2+ (right panel). Error bars show ±s.e.m. (n = 14 in 1.8 mm Ca2+ and n = 12 in zero Ca2+, *** P < 0.0005). Protocol as shown in A.
The dose-response relation for activation of membrane current by capsaicin in DRG neurones is shown in Fig. 4A. With the brief 1 s exposures employed in the present study the current was half-activated at 0.57 μm, and the dose-response curve was well fitted by a Hill equation with a Hill coefficient of 1.8. Exposure to PMA (1 μm) shifted the K1/2 to 0.3 μm. There was no change in the maximal current activated by a saturating dose of capsaicin (see Fig. 4B).
Figure 4. Effect of PKC activation on the relation between membrane current and capsaicin concentration in DRG neurones.

A, mean values of membrane current vs. capsaicin concentration in nociceptive neurones immediately before (○) and 15 s after (•) activation of PKC with 1 μm PMA. Error bars give ±s.e.m. (n at least 4 for each point). Points were fitted with a Hill equation with nH= 1.8 and the value of K1/2 shown. B, traces showing that PKC activation enhances capsaicin-activated current in a nociceptive neurone at a sub-saturating capsaicin concentration (0.5 μm) but not at a saturating concentration (20 μm).
Membrane current directly activated by PMA
A recent report has claimed that PKC activation directly gates VR1 (Premkumar & Ahern, 2000), and we therefore investigated in detail a possible activation of VR1 by PMA. In agreement with Prekumar & Ahern (2000) we found that PMA activates significant membrane current in HEK293 cells transiently transfected with VR1 (Fig. 5A). This current was not observed in untransfected HEK293 cells (not shown), and moreover was suppressed by the VR1-specific antagonist capsazepine (see Fig. 5A), showing that it is carried by VR1. The current was, however, not observed in the stably transfected hVR1-HEK293 cell line (Fig. 3A, inset), in contrast to the large current observed in the majority of transiently transfected HEK293 cells (Fig. 5A). Moreover, no current (< 10 pA) was activated in any DRG neurone on exposure to 1 μm PMA (Fig. 2A, inset), although the large currents activated by capsaicin application (typically 3-8 nA in response to a saturating concentration of capsaicin) show that VR1 was abundantly expressed in these cells. With a higher concentration of 10 μm PMA a small inward current was observed in 2 out of 13 capsaicin-sensitive DRG neurones (see Fig. 5B). The presence of this PMA-activated current did not correlate with the amplitude of the capsaicin-gated current (see legend to Fig. 5B).
In order to investigate more fully the possible presence of a PMA-gated current in DRG neurones we used calcium imaging, which allows a larger number of cells to be studied. Figure 6 shows that only a small minority of capsaicin-sensitive neurones (3.5 % in rat and 3.8 % in mouse) showed an increase in [Ca2+]i in response to 10 μm PMA, and that there was no correlation between the presence or absence of a response to PMA and the amplitude of the response to capsaicin. In a large sample of stably transfected hVR1-HEK293 cells PMA did not induce an increase in [Ca2+]i in any cell, although a large increase was observed in response to capsaicin (Fig. 6C).
Figure 6. Increases in [Ca2+]i caused by PKC activation and capsaicin.

A, PMA application (10 μm) caused no change in [Ca2+]i in the majority of capsaicin-sensitive rat DRG neurones. Capsaicin concentration, 10 μm. B, in 6 out of 174 capsaicin-responsive DRG neurones a small increase in [Ca2+]i was observed (largest example shown here). Mean amplitude (F/Fmax) was 0.12 ± 0.3. In similar experiments on DRG neurones from adult mice 2 out of 52 capsaicin-sensitive neurones responded to 1 μm PMA with an increase in [Ca2+]i. C, no significant increase in [Ca2+]i was seen in response to 10 μm PMA in HEK293 cells stably transfected with the hVR1 gene (10 coverslips, 216 cells). Capsaicin concentration was 1 μm. Untransfected HEK293 cells also did not respond to PMA (5 coverslips, 285 cells).
We conclude that direct activation of VR1 by PMA is variable and cell specific, as a large-amplitude current was regularly observed in HEK293 cells transiently transfected with VR1, but activation of VR1 was infrequently observed, and was of small amplitude when present, in stably transfected HEK293 cells or in DRG neurones from either rat or mouse. In addition, there was no correlation between the amplitude of a current evoked by PMA and the amplitude of the capsaicin-gated current.
A second difference between the action of PMA in enhancing the capsaicin-gated current and in activating current directly was the repeatability of the effect. The capsaicin-gated current was maximally enhanced by a single 10 s application of 1 μm PMA, and further applications were ineffective (Fig. 5C). The heat-gated current is similarly activated irreversibly by a single application of PMA (Cesare & McNaughton, 1996). By contrast, in transiently transfected HEK293 cells PMA was able to repeatedly activate an inward current (Fig. 5A).
Calcium dependence of PKC action
Repeated exposure to even brief pulses of capsaicin in the presence of 1.8 mm extracellular calcium caused a steady decline in the membrane current elicited by each exposure, in both DRG neurones (Fig. 2B) and hVR1-HEK293 cells (Fig. 3B). This decline has been attributed to calcium entry through the calcium-permeable VR1 channel, which in turn activates calcineurin, a calcium-dependent phosphatase (Cholewinski et al. 1993; Docherty et al. 1996; Liu & Simon, 1996; Koplas et al. 1997). In agreement with this proposal, removal of external calcium caused a substantial increase in the current activated by a subsaturating pulse of capsaicin in both DRG neurones and hVR1-HEK293 cells (Fig. 2B and Fig. 3B, right-hand panels). In DRG neurones the mean current elicited by the first 0.5 μm capsaicin application was 1.14 ± 0.18 nA in 1.8 mm Ca2+ and 2.74 ± 0.39 nA in zero Ca2+, an increase of 2.40 times (significant, P = 2 × 10−7, unpaired t test), and in hVR1-HEK293 cells the current elicited by the first application of 0.1 μm capsaicin was 0.24 ± 0.06 nA in 1.8 mm Ca2+ and 0.82 ± 0.13 nA in zero Ca2+, an increase of 3.42 times (significant, P = 3 × 10−4, unpaired t test). A plausible explanation for the increase in capsaicin-gated current in zero Ca2+ is that calcium withdrawal leads to deactivation of calcineurin, and that in the absence of phosphatase action VR1 becomes fully phosphorylated by background kinase activity. In agreement with this idea, activation of PKC in zero Ca2+ produced no further enhancement in current in response to capsaicin in either DRG neurones or hVR1-HEK293 cells, as expected if VR1 is fully phosphorylated in zero Ca2+ (Fig. 2B and Fig. 3B). In hVR1-HEK293 cells, application of PMA in zero Ca2+ in fact caused a small and transient reduction in current amplitude in some cells (see Fig. 3B, right-hand panel). This reduction was also observed when the inactive phorbol ester 4α-phorbol 12,13-didecanoate (4αPDD) was used or when phosphorylation was inhibited by the specific PKC inhibitor RO31-8220 (not shown) suggesting that the reduction is a non-specific effect.
An alternative explanation for the increase in capsaicin-gated current in zero Ca2+ is that Ca2+ may interact with and partially block ion flow through the channel, as is observed in the case of calcium channels (Carbone et al. 1997). A direct action at a site accessible from the external medium would be expected to be rapid. In Fig. 7A, however, elevation of [Ca2+]o caused only a gradual decline in the amplitude of the capsaicin-gated current, and the current recovered slowly on removal of Ca2+. In Fig. 7B a brief elevation of [Ca2+]o from 0 to 1.8 mm, in the presence of strong intracellular Ca2+ buffering with 5 mm BAPTA, caused no significant decline in the capsaicin-gated current. These experiments argue against a direct interaction of calcium ions with the VR1 channel.
Figure 7. External calcium does not rapidly block the capsaicin-gated current in DRG neurons.

A, elevating [Ca2+]o from zero (5 mm EGTA) to 1.8 mm did not rapidly reduce the amplitude of capsaicin-gated current in nociceptive neurones. Mean suppression in 1.8 mm Ca2+ was 5.05 (± 1.1)-fold (n = 9). Time constant of the recovery on return to zero Ca2+ was 122 ± 37 s. Intracellular solution contained 0.2 mm EGTA. B, rapid elevation of [Ca2+] from zero (5 mm EGTA) to 1.8 mm Ca2+ had no immediate effect on capsaicin-gated current (current decreased by 0.008 (± 0.012)-fold, n = 7). Internal calcium in this experiment was stabilized with 5 mm BAPTA added to the patch pipette.
Figure 8A summarizes data from a series of experiments on the PKC and calcium dependence of the capsaicin-gated current in DRG neurones. Bradykinin, which activates PKCɛ in DRG neurones (Cesare et al. 1999a), caused an increase in the current activated by a 1 s pulse of 0.5 μm capsaicin from 0.95 ± 0.07 to 1.38 ± 0.16 nA, or an increase of 1.45-fold (significant, P = 1.2 × 10−4, n = 5). The more potent PKC activation caused by PMA application enhanced the current from 0.91 ± 0.07 nA to 1.5 ± 0.095 pA, or an increase of 1.65-fold (significant, P = 1 × 10−4, n = 9). The enhancement caused by PMA was completely blocked by the specific PKC inhibitor RO31-8220. A larger enhancement, to 2.46 ± 0.36 nA (significant, P = 2 × 10−7, n = 4), was observed when PMA was pre-applied (fifth bar). Note, however, that in these experiments capsaicin was applied for the first time after a long exposure to PMA, while in those shown in the third bar of Fig. 8A PMA was applied during a train of capsaicin exposures. The larger enhancement shown in the fifth bar should not be taken to mean that a more prolonged exposure to PMA causes a greater increase in capsaicin-gated current, as with further exposures to capsaicin after PMA preapplication the enhancement declined to the value shown in the third bar. This experiment can, however, be validly compared with experiments in zero Ca2+ (sixth and seventh bars), in which the first capsaicin response was measured after 5 min in zero Ca2+. In these experiments zero Ca2+ enhanced the current amplitude to 2.74 ± 0.39 nA, a value similar to that obtained with preapplied PMA. In the presence of zero Ca2+, application of PMA then caused no further enhancement of the capsaicin response (last bar in Fig. 8A). These experiments show that lowering Ca2+ has effects that are similar to those of maximal PKC activation (see Discussion).
Figure 8. Enhancement by PKC activation of capsaicin-gated current in DRG neurons (A) and hVR1-HEK293 cells (B).

A, DRG neurones. Bars give mean peak current (error bars show ±s.e.m.) in nociceptive neurones in response to (from left): a control 1 s application of 0.5 μm capsaicin in 1.8 mm Ca2+; 15 s after application of bradykinin (BK, 1 μm for 10 s, significantly different from control, ***P = 1.2 × 10−4); 15 s after application of PMA (1 μm for 10 s, significantly different from control, ***P = 1 × 10−4); 15 s after application of PMA (1 μm, 10 s) with the specific PKC inhibitor RO31-8220 (1 μm, preapplied in the external solution for 5 min; no significant difference from control); and with PMA preapplied 5 min before (significantly different from control, ***P = 2 × 10−7). Last two bars show capsaicin-gated current in zero Ca2+ (+ 5 mm EGTA), and in zero Ca2+ 15 s after application of PMA (1 μm, 10 s). B, stably transfected HEK293 cells. Bars give mean current relative to current in 1.8 mm Ca2+ (error bars show ±s.e.m.) in hVR1-HEK293 cells in response to (from left): application of 100 nm capsaicin in 1.8 mm Ca2+; 15 s after application of the phorbol ester 4αPDD, which does not activate PKC (1 μm, 15 s; no significant difference from control); 15 s after application of PMA (1 μm, 15 s; significantly different from control, ***P = 2 × 10−5); and 15 s after application of PMA (1 μm, 15 s) with the specific PKC inhibitor RO31-8220 (2 μm in the internal solution; no significant difference from control). Last two bars show the fifth response to 100 nm capsaicin in zero Ca2+ (5 mm EGTA), and in zero Ca2+ 15 s after application of PMA (1 μm, 15 s), as already shown in the time course experiment of Fig. 3B).
Comparable results were obtained in HEK293 cells stably transfected with human VR1 (see Fig. 8B). The results in Fig. 8B are normalized to the current observed in response to capsaicin in 1.8 mm Ca2+ (first bar) because although the relative increases in current with (for example) PMA were reproducible in all cells, there was a much larger variation in current amplitude amongst different cells than for DRG neurones. The increase was completely blocked by RO31-8220. The inactive phorbol ester 4αPDD caused no enhancement, showing that a non-specific effect of PMA, unrelated to PKC activation, is not responsible. The enhancement caused by zero Ca2+ was qualitatively similar to that observed in DRG neurones. As noted above (see Fig. 3B), a small transient suppression in capsaicin-gated current was observed in hVR1-HEK293 cells on application of PMA (final bar in Fig. 8B), but the effect was non-specific in origin as a similar suppression was also seen with the inactive phorbol ester 4αPDD (not shown).
VR1 activation by heat, protons and anandamide is potentiated by PKC activation
Acidification causes an aching pain in vivo, an effect that can be at least partly attributed to a direct activation of VR1 by protons binding at an external site (Bevan & Geppetti, 1994; Tominaga et al. 1998). Figure 9A shows that the membrane current induced by acidification in hVR1-HEK293 cells was enhanced by PKC activation, in a similar manner to the capsaicin-gated current. Wild-type HEK293 cells express an endogenous proton-gated ion channel that activates transiently but then inactivates following acidification to pH 6.7 (Cesare et al. 1999b), while VR1 is not significantly activated by acidification to pH 6.7, but is strongly activated at pH 5.4 (Tominaga et al. 1998). In these experiments current generated by VR1 was therefore separated from the endogenous proton-gated current by applying pH 5.4 from a bath solution of pH 6.7. In wild-type HEK293 cells a small inward current (53 ± 7 pA, n = 16) was activated by acidification from pH 6.7 to 5.4, and PKC activation had no significant effect on the amplitude of this current (mean increase 1.20 (± 0.12)-fold, not significant, n = 24; see Fig. 9A, upper panel). In hVR1-HEK293 cells the inward current activated by acidification from pH 6.7 to 5.4, which mainly reflects gating of VR1 by protons, was much larger (311 ± 47 pA) and was significantly enhanced by PMA (mean increase 3.50 (± 0.58)-fold, significant, P = 7 × 10−5, n = 20; see Fig. 9A, lower panel).
Figure 9. Enhancement by PKC activation of currents gated by protons, anandamide and heat in hVR1-HEK293 cells.

A, pH-gated current. Acidification from pH 6.7 to 5.4 activated a sustained inward current that was larger in transfected than in untransfected HEK293 cells (compare upper and lower sets of traces). PMA (1 μm for 15 s, applied 60 s before pH 5.4 application) enhanced the current in transfected cells but had little effect in untransfected cells. Increase of current activated by pH 5.4 after PMA in transfected cells was 3.50 (± 0.58)-fold (mean ±s.e.m., n = 20). B, anandamide-gated current. For these experiments, HEK293 cells transiently transfected with hVR1 were used. Application of 10 μm anandamide activated an inward current that was enhanced by PMA (1 μm for 15 s, applied 30 s before trace shown). Increase of current activated by anandamide after PMA was 8.15 (± 3.47)-fold, (n = 12, P = 0.008). No current was activated by anandamide in untransfected cells. C, heat-gated current. The inward current activated by heat (50 °C) was enhanced by PMA (1 μm for 15 s, applied 30 s before trace shown). Increase of current activated by heat after PMA was 3.49 (± 0.39)-fold (n = 14, P = 4 × 10−5). A small inward current was activated by heat in untransfected cells (amplitude, 54 ± 13 pA; n = 10) but was not changed by PMA (not shown).
Anandamide, an endogenous cannabinoid, has recently been shown to activate VR1, an effect that may underlie the vasodilatory action of cannabinoids (Zygmunt et al. 1999). Figure 9B shows that the current activated by 10 μm anandamide in HEK293 cells transiently transfected with hVR1 is, like the capsaicin-gated current, enhanced by PKC activation (increase of 8.15 (± 3.47)-fold, significant, P = 0.008, n = 12). No anandamide-gated current was observed in untransfected HEK293 cells.
Heat-activated currents in DRG neurones have been shown to be enhanced by PKC activation (Cesare & McNaughton, 1996), and Fig. 9C shows that a similar enhancement is observed with heterologously expressed VR1. The current activated by a 50 °C pulse in HEK293 cells stably expressing hVR1 was enhanced by a factor of 3.49 ± 0.39 (significant, P = 4 × 10−5, n = 14). In parallel experiments carried out on untransfected HEK293 cells, heat activated a much smaller inward current (54 ± 9 pA, n = 10) which was unaffected by PKC activation (ratio 1.41 ± 0.28, not significant, n = 5). The current vs. temperature relationship in hVR1-HEK293 cells shifted to lower temperatures after PKC activation (Fig. 10), in a similar manner to the shift observed in DRG neurones (Cesare & McNaughton, 1996).
Figure 10. Dependence of heat-activated current on temperature in hVR1-HEK293 cells.

Temperature steps of duration 400 ms were applied to hVR1-HEK293 cells before (○) and after (•) application of PMA (cf. Fig. 9C). Current (ordinate) is expressed as a percentage of current evoked by a 50 °C step before PMA. Error bars show ±s.e.m. (5 measurements per point).
Enhancement by PKC activation of the heat-gated current in DRG neurones
Figure 11 summarizes experiments in which the effects of PKC activation on the membrane currents elicited by a brief pulse of heat in DRG neurones were investigated. The experiments were carried out in parallel with those shown in Fig. 8A, in which enhancement of the capsaicin-gated current was studied, in order to allow a direct comparison between the two sets of data. PKC activation by bradykinin enhanced the heat-gated current, from 0.29 ± 0.07 to 0.60 ± 0.18 nA, or a factor of 2.05 (significant, P = 0.045, n = 6) but PMA caused a more pronounced enhancement, from 0.30 ± 0.03 to 1.16 ± 0.13 nA, a factor of 3.80 (significant, P = 3 × 10−6, n = 20), presumably because PMA achieves a more complete and long-lasting activation of PKC than does bradykinin. The enhancement caused by PMA was completely blocked by the specific PKC inhibitor RO31-8220. The results are qualitatively similar to those obtained with capsaicin (Fig. 8A), but the enhancement of the heat-gated current caused by both bradykinin and PMA was proportionally greater than the enhancement of the capsaicin-gated current.
Figure 11. Enhancement by PKC activation of the current elicited by heat in nociceptors.

Bars give mean current (error bars show ±s.e.m.) in nociceptive neurones in response to (from left): a control 400 ms step to 50 °C in 1.8 mm Ca2+; the same step 30 s after application of bradykinin (1 μm for 10 s, significantly different from control, *P = 0.045); 30 s after application of PMA (1 μm, 10 s; significantly different from control, ***P = 3 × 10−6); and 30 s after application of PMA (1 μm) with the specific PKC inhibitor RO31-8220 (1 μm, 10 s; no significant difference from control). Last two bars show heat-gated current in zero Ca2+ (5 mm EGTA) and in zero Ca2+ 30 s after application of PMA (1 μm, difference is significant, *P = 0.01).
Removal of external calcium enhanced the magnitude of the heat-gated current in a qualitatively similar manner to the enhancement of the capsaicin-gated current (cf. Fig. 8A). Activation of PKC caused a small but significant increase (from 1.01 ± 0.25 to 1.20 ± 0.28 nA, a factor of 1.19, difference significant, P = 0.010) in the heat-gated current in zero Ca2+, in contrast to the capsaicin-gated current where PKC activation caused no increase in zero Ca2+.
DISCUSSION
Enhancement of the capsaicin-gated current by PKC activation
The heat-sensitive current in nociceptive neurones is enhanced, or sensitized, by activation of PKC (Cesare & McNaughton, 1996; Cesare et al. 1999a), and one of the aims of the present study was to determine whether the capsaicin-gated current is similarly sensitized. Figure 2 and Figure 3 show that the current gated by capsaicin was enhanced by PKC activation in a similar way to the current gated by heat. At a sub-maximal concentration of capsaicin the response amplitude was enhanced approximately 2-fold in both sensory neurones and hVR1-HEK293 cells. Two possible explanations for the enhancement of capsaicin-gated current after PKC activation are that phosphorylation could increase the probability that channels will open in response to agonist application, or that it may unmask latent channels, without changing the characteristics of gating of individual channels by capsaicin. In the first model the current activated by a saturating concentration of capsaicin would not be changed by phosphorylation, while in the second the current at a high capsaicin concentration would increase in proportion to the increase observed at a lower concentration. Figure 4 supports the first possibility, in that PMA application produced a substantial enhancement in the membrane current activated by low capsaicin concentrations, but did not affect the current activated by high capsaicin concentrations (Fig. 4B). The effect of PKC activation is therefore to enhance the apparent potency of capsaicin by a factor of about two, with no significant effect on either the form of the dose-response relation or its maximum amplitude (Fig. 4A). Phosphorylation does not seem, therefore, to unmask latent channels, but instead increases the probability that channels will open in response to capsaicin. In a similar way, a shift of the temperature vs. current relation to lower temperatures observed after PKC activation in both DRG neurones (Cesare & McNaughton, 1996) and hVR1-HEK293 cells (Fig. 10 of the present study), suggests that PKC activation also increases the probability of channel opening in response to an elevation in temperature, although limitations of cell viability mean that a temperature sufficient to saturate channel opening cannot be applied.
Heat, capsaicin, low pH and the endocannabinoid anandamide all activate VR1, and the fact that the activation can be observed in several heterologous expression systems implies that the activation is direct (Caterina et al. 1997; Tominaga et al. 1998; Zygmunt et al. 1999). Capsaicin acts at an internal site (Jung et al. 1999) and protons act externally (Jordt et al. 2000), while the binding site of anandamide, and the location of the conformational change caused by heat, have yet to be determined. Despite these different binding sites, the final pathway for channel activation by these agonists may be the same, because the current activated by each is enhanced in a similar way by PKC activation (Fig. 9).
Effect of calcium on the capsaicin- and heat-gated current
Lowering the calcium concentration enhances the amplitude of both the capsaicin-gated and the heat-gated current. Relief of a direct blocking action of external calcium on capsaicin-gated channels is not a major contributor to this enhancement, because rapid elevations of calcium cause little immediate change in current (Fig. 7). The most straightforward alternative explanation is that a reduction in internal calcium concentration produces an effect similar to that caused by PKC activation. Increased PKC activity caused by a reduction in internal [Ca2+] is unlikely to be responsible for increasing the phosphorylation of VR1, because a reduction in [Ca2+] inhibits rather than enhances the activity of calcium-sensitive PKC isoforms (Tanaka & Nishizuka, 1994), and because the ɛ isoform of PKC, which mediates the sensitization of the heat-sensitive current in nociceptors (Cesare et al. 1999a), is not calcium sensitive at normal cellular levels of Ca2+ (Tanaka & Nishizuka, 1994). A more likely explanation is provided by the demonstration that the capsaicin-sensitive channel is deactivated by the calcium-sensitive phosphatase calcineurin (Cholewinski et al. 1993; Docherty et al. 1996; Liu & Simon, 1996; Koplas et al. 1997). If background kinase activity in the resting cell causes phosphorylation of VR1, then inhibition of the phosphatase activity of calcineurin, caused by a reduction in [Ca2+]i, will lead to an increase in phosphorylation of VR1, causing in turn an enhanced membrane current gated by capsaicin and heat. In zero Ca2+ activation of PKC by application of PMA produced no further increase in the amplitude of the current gated by capsaicin (Fig. 2 and Fig. 3), suggesting that background kinase activity is sufficient to fully phosphorylate VR1 when calcineurin activity has been inhibited by low [Ca2+], and that activation of PKC then has no further effect.
Direct activation of VR1 by PKC
PKC activation can cause current flow through VR1 in some circumstances. In transiently transfected HEK293 cells a large membrane current could be repeatedly evoked by PKC activation, and the suppression of this current by the VR1 antagonist capsazepine shows that the current flowed through VR1 ion channels (Fig. 5). Premkumar & Ahern (2000) have made similar observations, and have proposed that PKC activation directly gates VR1. PKC-activated currents were not observed, however, in HEK293 cells stably expressing VR1 (Fig. 3B), and in DRG neurones few cells exhibited a direct response to PKC activation. Figure 5B shows that only a small current (< 40pA) was activated by PMA in DRG neurones, and even then only at high levels of PMA and in a small minority of neurones. The presence or absence of this PMA-activated current did not correlate with the amplitude of the capsaicin-gated current. In a larger sample of neurones examined by calcium imaging (Fig. 6A and B) less than 4 % of capsaicin-sensitive neurones responded to PMA with an increase in [Ca2+]i, and the amplitude of this reponse, as in the patch-clamp experiments, did not correlate with the amplitude of the response to capsaicin. In similar experiments on stably transfected hVR1-HEK293 cells no increase in [Ca2+]i was observed in reponse to PMA in any experiment (Fig. 6C). Direct activation of VR1 by PKC is therefore strongly dependent on the cell system, and on the individual cell, in contrast to gating of VR1 by capsaicin, heat, anandamide or low pH, and enhancement of these currents by PKC activation, which are observed in all VR1-expressing cells. This suggests that current activation by PKC depends on a mechanism other than direct phosphorylation of VR1. One possibility is that PKC activation may induce the release of an agonist able to activate VR1 from the cell itself or from adjacent cells, and that the release of this agonist depends on the individual cell and in particular on whether it has been subjected to the physiological stress of transient transfection.
PKC activation reproducibly enhanced the capsaicin-gated current in HEK293 cells and in neurones. This effect was maximal after a single exposure to PMA, and was not further enhanced by subsequent exposures to PMA (Fig. 5C). In this respect the enhancement of capsaicin-gated current differs from the direct activation of VR1 by PMA, which could be repeatedly observed in transiently transfected HEK293 cells (Fig. 5A). The different behaviour of the enhancement of capsaicin-gated current and the direct activation of current by PMA suggests that the two phenomena have different origins.
A possible model for PKC action
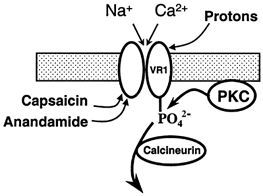
The above considerations suggest that PKC activation acts differently from capsaicin, heat, protons and anandamide, all of which directly activate current flow through VR1. The main action of PKC activation is to enhance the potency of these ‘agonists’, without in general activating current, showing that PKC activation must act at a different stage in the process of channel opening. The simplest explanation for the enhancement of channel gating caused by PKC activation is that the channel itself is directly phosphorylated by PKC. This model is summarized in Fig. 12. Capsaicin has been shown to gate channels by binding at an internal site (Jung et al. 1999), and anandamide, which has structural similarities to capsaicin (Zygmunt et al. 1999a) is therefore also shown acting at an internal site in Fig. 12, though there is no direct evidence for this. Channels can also be gated by protonation of an external site (Jordt et al. 2000). The site of gating by heat has not been determined. Channel gating is promoted by phosphorylation by PKC, and is antagonized when the channel is dephosphorylated by activation of the calcium-dependent phosphatase calcineurin. An important objective for future work will be to determine whether the channel is in fact directly phosphorylated by PKC, and if so, which phosphorylation sites are important in modulating channel gating.
Figure 12. Model for gating and phosphorylation of VR1.
Acknowledgments
We are grateful for helpful advice from Drs L. Dekker, D. B. Dixon, P. Cesare, M. J. Gunthorpe and A. D. Randall. We thank also Mr Harold Ayetey (supported by The Nuffield Foundation) for help with DRG experiments. We are grateful to Helen Bye, Andrea Margutti and Guiseppe Dia for excellent technical assistance. This work was supported by grants from the MRC and BBSRC to P.McN., a Marie Curie fellowship from the EC to V.V. and a studentship from King's College London to S.M.
References
- Baumann TK, Simone DA, Shain CN, LaMotte RH. Neurogenic hyperalgesia: The search for the primary cutaneous afferent fibers that contribute to capsaicin-induced pain and hyperalgesia. Journal of Neurophysiology. 1991;66:212–227. doi: 10.1152/jn.1991.66.1.212. [DOI] [PubMed] [Google Scholar]
- Belmonte C, Giraldez F. Responses of cat corneal sensory receptors to mechanical and thermal stimulation. Journal of Physiology. 1981;321:355–368. doi: 10.1113/jphysiol.1981.sp013989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan S, Geppetti P. Protons: Small stimulants of capsaicin-sensitive sensory nerves. Trends in Neurosciences. 1994;17:509–512. doi: 10.1016/0166-2236(94)90149-x. [DOI] [PubMed] [Google Scholar]
- Buck SH, Burks TF. The neuropharmacology of capsaicin: review of some recent observations. Pharmacological Reviews. 1986;38:179–226. [PubMed] [Google Scholar]
- Carbone E, Lux HD, Carabelli V, Aicardi G, Zucker H. Ca2+ and Na+ permeability of high-threshold Ca2+ channels and their voltage-dependent block by Mg2+ ions in chick sensory neurones. Journal of Physiology. 1997;504:1–15. doi: 10.1111/j.1469-7793.1997.001bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron. 1999a;23:617–624. doi: 10.1016/s0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- Cesare P, McNaughton PA. A novel heat-activated current in nociceptive neurons, and its sensitization by bradykinin. Proceedings of the National Academy of Sciences of the USA. 1996;93:15435–15439. doi: 10.1073/pnas.93.26.15435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare P, Young J, Wafford K, Clark S, England S, Delmas P, McNaughton PA, Wood JN. Endogenous proton-gated cation channels in cell lines and Xenopus oocytes. Journal of Physiology. 1999b;518.P:116P. [Google Scholar]
- Cholewinski A, Burgess GM, Bevan S. The role of calcium in capsaicin-induced desensitization in rat cultured dorsal root ganglion neurons. Neuroscience. 1993;55:1015–1023. doi: 10.1016/0306-4522(93)90315-7. [DOI] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- Docherty RJ, Yeats JC, Bevan S, Boddeke HWGM. Inhibition of calcineurin inhibits the desensitization of capsaicin-evoked currents in cultured dorsal root ganglion neurones from adult rats. Pflügers Archiv. 1996;431:828–837. doi: 10.1007/s004240050074. [DOI] [PubMed] [Google Scholar]
- Hayes P, Meadows HJ, Gunthorpe MJ, Harries MH, Duckworth DM, Cairns W, Harrison DC, Clarke CE, Ellington K, Prinjha RK, Barton AJL, Medhurst AD, Smith GD, Topp S, Murdock P, Sanger GJ, Terrett J, Jenkins O, Benham CD, Randall AD, Gloger IS, Davis JB. Cloning and functional expression of a human orthologue of rat vanilloid receptor 1. Pain. 2000;88:207–217. doi: 10.1016/S0304-3959(00)00353-5. [DOI] [PubMed] [Google Scholar]
- Jordt S-E, Tominaga M, Julius D. Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proceedings of the National Academy of Sciences of the USA. 2000;97:8134–8139. doi: 10.1073/pnas.100129497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Sun WH, Kwak J, Lee S-Y, Kang C-J, Won BK, Kim D, Oh U. Capsaicin binds to the intracellular domain of the capsaicin-activated ion channel. Journal of Neuroscience. 1999;19:529–538. doi: 10.1523/JNEUROSCI.19-02-00529.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschstein T, Busselberg D, Treede RD. Noxious heat activates an inward current in a subpopulation of dorsal root ganglion neurones. Pflügers Archiv. 1997;433:P187. [Google Scholar]
- Koplas PA, Rosenberg RL, Oxford GS. The role of calcium in the desensitization of capsaicin responses in rat dorsal root ganglion neurons. Journal of Neuroscience. 1997;17:3525–3537. doi: 10.1523/JNEUROSCI.17-10-03525.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMotte RH, Lundberg LER, Torebjork HE. Pain, hyperalgesia and activity in nociceptive C units in humans after intradermal injection of capsaicin. Journal of Physiology. 1992;448:749–764. doi: 10.1113/jphysiol.1992.sp019068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Simon SA. Capsaicin-induced currents with distinct desensitization and Ca2+ dependence in rat trigeminal ganglion cells. Journal of Neurophysiology. 1996;75:1503–1514. doi: 10.1152/jn.1996.75.4.1503. [DOI] [PubMed] [Google Scholar]
- Nadal A, Fuentes E, McNaughton PA. Albumin stimulates uptake of calcium into subcellular stores in rat cortical astrocytes. Journal of Physiology. 1996;492:737–750. doi: 10.1113/jphysiol.1996.sp021342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy I, Rang H. Noxious heat activates all capsaicin-sensitive and also a sub-population of capsaicin-insensitive dorsal root ganglion neurons. Neuroscience. 1999;88:995–997. doi: 10.1016/s0306-4522(98)00535-1. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Ahern GP. Induction of vanilloid receptor channel activity by protein kinase C. Nature. 2000;408:985–990. doi: 10.1038/35050121. [DOI] [PubMed] [Google Scholar]
- Reichling DB, Levine JD. Heat transduction in rat sensory neurons by calcium-dependent activation of a cation channel. Proceedings of the National Academy of Sciences of the USA. 1997;94:7006–7011. doi: 10.1073/pnas.94.13.7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson CJ, Torebjork HE, LaMotte RH. Psychophysical detection and pain ratings of incremental thermal stimuli: A comparison with nociceptor responses in humans. Brain Research. 1983;274:87–106. doi: 10.1016/0006-8993(83)90523-1. [DOI] [PubMed] [Google Scholar]
- Smart D, Gunthorpe MJ, Jerman JC, Nasir S, Gray J, Muir AI, Chambers JK, Randall AD, Davis JB. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1) British Journal of Pharmacology. 2000;129:227–230. doi: 10.1038/sj.bjp.0703050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka C, Nishizuka Y. The protein kinase C family for neuronal signaling. Annual Review of Neuroscience. 1994;17:551–567. doi: 10.1146/annurev.ne.17.030194.003003. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Treede RD, Meyer RA, Raja SN, Campbell JN. Peripheral and central mechanisms of cutaneous hyperalgesia. Progress in Neurobiology. 1992;38:397–421. doi: 10.1016/0301-0082(92)90027-c. [DOI] [PubMed] [Google Scholar]
- Vellani V, Moriondo A, McNaughton PA. The capsaicin-dependent current in isolated dorsal root ganglion neurones is enhanced by PKC activation. Journal of Physiology. 1999;518.P:157P. [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H-H, Sorgard M, Di Marzo V, Julius D, Hogestatt ED. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]