

Abstract
Whole-cell patch clamp recordings were made from rat rostral ventromedial medulla (RVM) neurons in vitro to investigate the cellular actions of the opioid-like receptor ORL1 (NOP), ligand nociceptin/orphanin FQ and other putative prepronociceptin products.
Primary and secondary RVM neurons were identified as responding to the κ-opioid receptor agonist U-69593 (300 nm to 1 μm) and the μ- and δ-opioid receptor agonist met-enkephalin (10 μm), respectively. Both primary and secondary RVM neurons responded to nociceptin (3 nm to 1 μm) with an outward current that reversed polarity at –115 mV in brain slices and with inhibition of Ca2+ channel currents in acutely isolated cells.
The putative ORL1 antagonist J-113397 (1 μm) produced no change in membrane current and abolished the outward current produced by nociceptin (100 nm). In contrast, Phe1ψ(CH2-NH)Gly2]-nociceptin-(1-13)NH2 (300 nm to 1 μm) alone produced an outward current and partially reduced the outward current produced by nociceptin (300 nm) when co-applied.
In brain slices nociceptin (300 nm) reduced the amplitude of evoked GABAA receptor-mediated inhibitory postsynaptic currents (IPSCs) but not non-NMDA receptor-mediated excitatory postsynaptic currents (EPSCs).
Met-enkephalin (10 μm), but not nociceptin (300 nm), reduced the rate of spontaneous miniature IPSCs in normal external potassium solution (K+ 2.5 mm). In high external potassium (K+ 17.5 mm), nociceptin reduced the rate of miniature IPSCs in the presence (Ca2+ 2.4 mm, Mg2+ 1.2 mm) but not in the absence of external calcium (Ca2+ 0 mm, Mg2+ 10 mm, Cd2+ 10 μm). Nociceptin and met-enkephalin had no effect on the amplitude of miniature IPSCs.
The putative nociceptin precursor products nocistatin (rat prepronociceptin125–132) and rat prepronociceptin154–181 had no effect on membrane currents, evoked IPSCs and evoked EPSCs.
These results indicate that nociceptin acts via the ORL1 receptor to directly inhibit both primary and secondary RVM neurons by activating a potassium conductance and by inhibiting calcium conductances. In addition, nociceptin inhibits GABA release within the RVM via a presynaptic Ca2+-dependent mechanism. Thus, nociceptin has the potential to exert both disinhibitory and inhibitory effects on neuronal action potential firing within the RVM.
The rostral ventromedial medulla (RVM) forms part of a descending inhibitory network that modulates nociceptive neurotransmission within the dorsal horn of the spinal cord and is a major site of the antinociceptive actions of μ-opioids (Fields et al. 1991). μ-Opioid receptor agonists reduce GABAergic influences on primary RVM neurons, which are thought to represent serotonergic neurons that project to the spinal dorsal horn (Pan et al. 1990, 1997). In addition, μ-opioid receptor agonists directly hyperpolarise secondary RVM neurons by increasing a K+ conductance. These observations have led to the hypothesis that μ-opioids produce analgesia within the RVM by disinhibiting primary neurons via (1) inhibition of secondary GABAergic interneurons and (2) presynaptic inhibition of transmitter release from GABAergic nerve terminals.
The heptadecapeptide nociceptin/orphanin FQ is an endogenous ligand for the opioid-like receptor ORL1 (NOP), which is highly homologous to the cloned μ- (MOP), δ- (DOP) and κ-opioid receptors (KOP) (Mollereau et al. 1994; Lachowicz et al. 1995; Meunier et al. 1995; Reinscheid et al. 1995). While, like opioids, nociceptin modulates ion channels, synaptic transmission and second messenger cascades, it also has distinct and complex effects on analgesia (Calo et al. 2000; Grisel & Mogil, 2000). Nociceptin is one of a number of putative peptide products of the prepronociceptin gene that have been shown to modulate analgesia (Houtani et al. 1996; Okuda-Ashitaka et al. 1998; Rossi et al. 1998).
In situ hybridization, autoradiographic and immunohistochemical studies have demonstrated that the rat RVM contains high levels of nociceptin, prepronociceptin (Neal et al. 1999) and the ORL1 receptor (Lachowicz et al. 1995; Anton et al. 1996). Microinjection of nociceptin into the RVM has no nociceptive/ antinociceptive effect alone but reduces the antinociception produced by μ-opioids (Heinricher et al. 1997; Pan et al. 2000). A recent in vitro study has demonstrated that nociceptin activates a potassium conductance in all RVM neurons (Pan et al. 2000). The present study examined the mechanism of action of nociceptin and other putative prepronociceptin products on potassium and calcium conductances and synaptic transmission in RVM neurons in vitro.
METHODS
The experimental protocol was approved by the University of Sydney Animal Care Ethics Committee. Male and female Sprague-Dawley rats (15-26 days old) were anaesthetised with halothane, decapitated and coronal brain slices were cut (250 μm) at the level of the facial nerve in ice-cold artificial cerebrospinal fluid (ACSF). The slices were maintained at 34 °C in a submerged chamber containing ACSF equilibrated with 95 % O2 and 5 % CO2. For experiments on synaptic currents and postsynaptic K+ currents, the slices were then transferred to a chamber and superfused continuously (2 ml min−1) with ACSF (32 °C) of composition (mm): NaCl 126, KCl 2.5, NaH2PO4 1.4, MgCl2 1.2, CaCl2 2.4, glucose 11 and NaHCO3 25, as previously described (Vaughan et al. 1999). RVM neurons were visualised in the triangular midline region dorsal to the pyramidal tracts using infra-red Nomarski optics on an upright microscope (Olympus BX50).
For experiments on postsynaptic K+ currents, a potassium gluconate-based internal solution was used that contained (mm): potassium gluconate 95, KCl 30, NaCl 15, MgCl2 2, Hepes 10, EGTA 11, MgATP 2 and NaGTP 0.25. For experiments on synaptic currents, a CsCl-based internal solution was used that contained (mm): CsCl 140, EGTA 10, Hepes 5, CaCl2 2 and MgATP 2 (pH 7.3, osmolarity 270-290 mosmol l−1). Voltage clamp recordings (holding potential -60 mV) were made in the whole-cell configuration using an Axopatch 200B (Axon Instruments, Foster City, CA, USA) with patch clamp electrodes (2-5 MΩ). Series resistance (< 20 MΩ) was compensated by 80 % and continuously monitored during experiments. Liquid junction potentials of -12 mV for potassium gluconate-based and -4 mV for CsCl-based internal solutions were corrected. Postsynaptic K+ current recordings were filtered (100 Hz low-pass filter) and sampled (200 Hz) for off-line analysis (Axograph 4, Axon Instruments). Inhibitory and excitatory postsynaptic currents (IPSCs and EPSCs) were filtered (1 and 2 kHz low-pass filter) and sampled (5 and 10 kHz) for on-line and later off-line analysis (Axograph 4). Electrically evoked IPSCs and EPSCs were elicited in neurons via bipolar tungsten stimulating electrodes placed 100-500 μm from the recording electrode (0.05-0.1 Hz stimuli: 5-70 V, 20-400 μs). Spontaneous miniature IPSCs and EPSCs (mIPSCs and mEPSCs) were obtained in neurons in the presence of TTX (0.3 μm). mIPSCs and mEPSCs above a preset threshold (4-6 standard deviations above baseline noise) were automatically detected by a sliding template algorithm (Axograph 4), then manually checked off-line. mIPSCs and mEPSCs were counted in 12 s epochs every 15 s to construct plots of event rate versus time.
For experiments on postsynaptic Ca2+ currents, cells were dissociated as previously described (Connor & Christie, 1998). Slices were transferred to a dissociation buffer of composition (mm): Na2SO4 82, K2SO4 30, Hepes 10, MgCl2 5 and glucose 10, containing 20 units ml−1 papain, pH 7.3 and incubated for 2-3 min at 35 °C. The slices were then placed in fresh dissociation buffer containing 1 mg ml−1 bovine serum albumin (BSA) and 1 mg ml−1 trypsin inhibitor, and the RVM was subdissected out by cutting from just lateral to each pyramid to the midline half-way up the slice with a fine tungsten wire. Cells were dissociated from the slices by gentle trituration, plated onto plastic culture dishes and kept at room temperature in dissociation buffer. Whole-cell patch clamp recordings of currents through Ca2+ channels were made at room temperature (22-24 °C). Immediately prior to recording, dishes of cells were superfused with a buffer of composition (mm): NaCl 140, KCl 2.5, CaCl2 2.5, MgCl2 1.5, Hepes 10 and glucose 10, pH 7.3, in order to wash off the dissociation buffer. For calcium channel current recordings (IBa), cells were perfused in solution containing (mm): tetraethylammonium chloride 140, BaCl2 2, MgCl2 1, CsCl 2.5, Hepes 10 and glucose 10, pH 7.3. Whole-cell patch clamp recordings were made with an intracellular solution containing (mm): CsCl 130, MgATP 5, Na2GTP 0.2, EGTA 10, CaCl2 2, NaCl 5 and Hepes 10, pH 7.3. Series resistance (≈3 MΩ) was compensated by 80 % and continuously monitored during experiments. Leak current was subtracted on line using a P/8 protocol; typically the leak conductance was of the order of 100 pS. IBa evoked by stepping the membrane potential from a holding potential of -90 mV was filtered (2 kHz low-pass filter) and sampled (5-10 kHz) for later analysis (pCLAMP, Axograph 3, Axon Instruments). Cells were exposed to drugs via a series of flow pipes positioned above the cells. The inhibition by drugs was quantified by measuring the current amplitude isochronically with the peak of the control IBa.
Stock solutions of all drugs were diluted to working concentrations using ACSF immediately before use and applied by superfusion. Nociceptin (Phe-Gly-Gly-Phe-Thr-Gly-Ala-Arg-Lys-Ser-Ala-Arg-Lys-Leu-Ala-Asn-Gln) and nocistatin (rat prepronociceptin125-132, mPNP-3-8P, Glu-Val-Glu-Gln-Lys-Gln-Leu-Gln) were obtained from Auspep (Parkville, Victoria, Australia). Rat prepronociceptin154-181 (mouse prepronociceptin160-187, Phe-Ser-Glu-Phe-Met-Arg-Gln-Tyr-Leu-Val-Leu-Ser-Met-Glu-Ser-Ser-Glu-Arg-Arg-Arg-Thr-Leu-His-Gln-Asn-Gly-Asn-Val) was made by solid phase synthesis by Research Genetics (Huntsville, AL, USA). Met-enkephalin (methionine-enkephalin), bicuculline methiodide, strychnine hydrochloride, BSA and trypsin inhibitor (Type II-O) were obtained from Sigma (Sydney, Australia). CTAP (d-Phe-Cys-Tyr-d-Trp-Arg-Pen-Thr-NH2) was from Phoenix Pharmaceuticals (Mountain View, CA, USA). CNQX (6-cyano-7-nitroquinoxaline-2,3-dione disodium salt) was from Tocris Cookson (Bristol, UK). Naloxone hydrochloride, nor-BNI (nor-binaltorphimine dihydrochloride) and U-69593 were from Research Biochemicals Inc. (Natick, MA, USA). Tetrodotoxin (TTX) was from Alomone Laboratories (Jerusalem, Israel). Papain was from Worthington Biochemical Corporation (Freehold, NJ, USA). Phe-ψ-nociceptin (Phe1ψ(CH2-NH)Gly2]-nociceptin-(1-13)NH2) was provided by Dr R. Guerrini (University of Ferrara, Italy). J-113397 (1-[(3R,4R)-1-cyclo-octylmethyl-3-hydroxymethyl-4-piperidyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one) was provided by Banyu Pharmaceutical Co. Ltd (Tokyo, Japan). CGP-55845A was from Novartis. All pooled data are expressed as means ±s.e.m. Unless otherwise stated, all statistical comparisons were made using Student's paired t test.
RESULTS
Nociceptin acts via the ORL1 receptor to increase a potassium current in all RVM neurons
In brain slices, RVM neurons were classified as being either primary or secondary cells on the basis of their postsynaptic responses to μ-opioid and/or κ-opioid receptor activation (Pan et al. 2000). In primary neurons, superfusion of the selective κ-opioid receptor agonist U-69593 (300 nm to 1 μm) produced an outward current (48 ± 6 pA, n = 13) that was abolished by addition of the κ-opioid receptor antagonist norBNI (300 nm, n = 8, Fig. 1A). The selective μ-opioid receptor agonist DAMGO (1-3 μm) and the μ- and δ-opioid receptor agonist met-enkephalin (10 μm, Fig. 1A) produced no change in membrane current in primary neurons (1 ± 1 pA, n = 13). In secondary neurons, met-enkephalin (10 μm) produced an outward current (72 ± 8 pA, n = 31, Fig. 1B) that was abolished by naloxone (1 μm, n = 11) or the μ-opioid receptor antagonist CTAP (1 μm, n = 5). U-69593 (300 nm to 1 μm) had no effect on membrane currents in the secondary neurons tested (0 ± 1 pA, n = 28, Fig. 1B). Nociceptin (300 nm) produced an outward current in all primary (102 ± 13 pA, n = 10, Fig. 1A) and secondary neurons tested (100 ± 10 pA, n = 30, Fig. 1B).
Figure 1. Nociceptin produces an outward current in both primary and secondary RVM neurons.
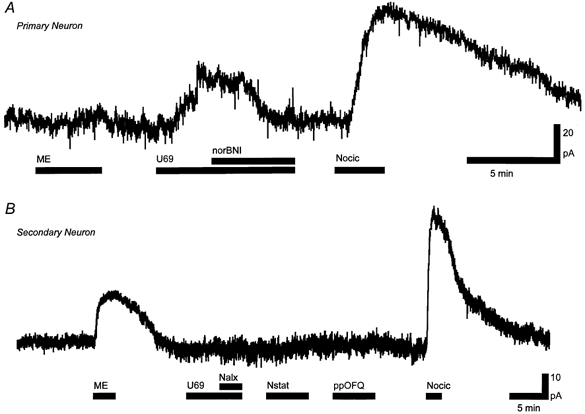
A, U-69593 (U69, 300 nm) produced a nor-BNI (300 nm)-sensitive outward current and met-enkephalin (ME, 10 μm) produced no currents in a primary neuron. B, met-enkephalin but not U-69593 produced an outward current in a secondary neuron. Nociceptin (Nocic, 300 nm) produced an outward current in both primary (A) and secondary (B) neurons. Nocistatin (Nstat, 1 μm) and prepronociceptin154-181 (ppOFQ, 300 nm) produced no currents in the secondary neuron (B). Nalx, naloxone (1 μm).
The outward current produced by nociceptin was concentration dependent with a pEC50 of 7.8 ± 0.1 (Fig. 2) and a Hill slope for the curve of 1.2 ± 0.2. At concentrations of 300 nm and above the outward current produced by nociceptin partly desensitised following the peak outward current. The outward current produced by nociceptin (100 nm, 51 ± 6 pA) was abolished by the addition of the putative ORL1 antagonist J-113397 (1 μm, 1 ± 2 pA, n = 9, Fig. 3A). The outward current produced by met-enkephalin (10 μm) was similar in the absence (28 ± 5 pA) and presence (32 ± 7 pA) of J-113397 (1 μm, n = 5, Fig. 3A). J-113397 (1 μm) alone produced no change in membrane current (0 ± 2 pA, n = 4). Another putative ORL1 ligand Phe-ψ-nociceptin (300 nm) produced an outward current of 71 ± 24 pA (n = 5, Fig. 3B). Application of Phe-ψ-nociceptin (1 μm) in the continued presence of nociceptin (300 nm) reversibly reduced the outward current caused by nociceptin to 46 ± 4 % of the value before application of Phe-ψ-nociceptin (n = 4, Fig. 3B). The outward current produced by nociceptin (300 nm) was unaffected by naloxone (1 μm, n = 15; not shown).
Figure 2. Nociceptin-induced outward currents and inhibition of Ca2+ channel currents are dose dependent.
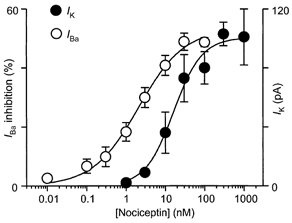
Concentration-response relationships of nociceptin-induced outward currents (IK in brain slices, •) and inhibition of Ca2+ channel currents (IBa in isolated cells, ○) in RVM neurons. Each point shows the mean (±s.e.m.) of responses of several different neurons (n = 5-29). A logistic function was fitted to determine the pEC50.
Figure 3. J-113397 behaves as a pure antagonist and Phe-ψ-nociceptin behaves as a partial agonist at the ORL1 receptor in RVM neurons.
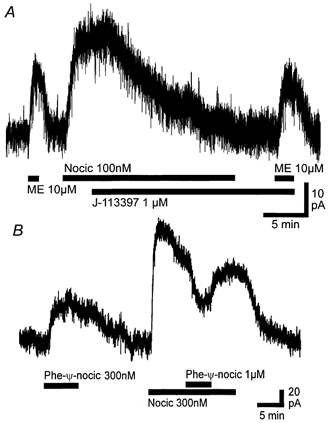
A, application of nociceptin (Nocic, 100 nm) produced an outward current that was abolished after addition of J-113397 (1 μm). Met-enkephalin (ME, 10 μm) produced a similar current before and after addition of J-113397. B, application of Phe-ψ-nociceptin (Phe-ψ-nocic, 300 nm) produced an outward current. Subsequent application of nociceptin (300 nm) produced an outward current that was partially antagonised by the addition of Phe-ψ-nociceptin (1 μm). A and B are from different neurons.
The resting membrane conductance showed inward rectification with slope conductances of 7.1 ± 1.0 and 15.4 ± 2.5 nS when measured between -60 and -90 mV and between -110 and -130 mV, respectively (n = 12). Nociceptin increased the slope conductances to 8.7 ± 1.2 and 17.5 ± 2.8 nS, when measured over the same potentials (n = 12). The nociceptin-induced current reversed polarity at -115 ± 3 mV (n = 12).
Nociceptin inhibits calcium currents in all RVM neurons
When acutely isolated RVM neurons were stepped from a holding potential of -90 mV inward currents in most cells began to activate at about -40 mV and were invariably greatest at membrane potentials between -10 and 0 mV. The peak inward current was 148 ± 7 pA pF−1(n = 42). The inward current could be abolished by Cd2+ (30 μm, n = 8, data not shown), suggesting that it was being carried by Ba2+ through calcium channels (IBa). In most RVM neurons tested, superfusion of high concentrations of the μ-opioid receptor agonist DAMGO and/or the κ-opioid receptor agonist U-69593 inhibited the peak IBa evoked by stepping the membrane potential from -90 to 0 mV every 30 s (Fig. 4A). An inhibition of greater than 10 % of the peak current, which recovered after washout, was considered significant. U-69593 (1 μm) inhibited IBa in 9 of 26 neurons tested, by an average of 22 ± 4 %. DAMGO (3 μm) inhibited IBa in 12 of 25 neurons tested, by an average of 34 ± 4 %. In 1 of 25 neurons, both DAMGO (3 μm) and U-69593 (1 μm) significantly inhibited IBa, while in 4 of 25 neurons neither DAMGO nor U-69593 significantly inhibited IBa. Nociceptin produced an inhibition of the peak inward IBa in all RVM neurons examined (n = 36, Fig. 4). The maximum inhibition of the peak IBa by nociceptin was about 50 %. Nociceptin inhibition of IBa was dose dependent with a pEC50 of 8.7 ± 0.2 (Fig. 2) and a Hill slope of 0.8 ± 0.1. The inhibition of IBa by nociceptin was apparent when the current was evoked at range of membrane potentials (n = 6, data not shown).
Figure 4. Nociceptin inhibits IBa in RVM neurons.
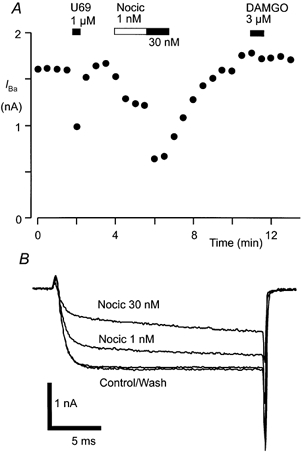
A, time plot of the peak amplitude of IBa during the application of U-69593 (U69, 1 μm), nociceptin (Nocic, 1 or 30 nm) and DAMGO (3 μm). B, example raw current traces of IBa. IBa was elicited by repetitively stepping the membrane potential of an RVM neuron from -90 to 0 mV every 30 s. A and B are taken from one neuron.
The inhibition of IBa by nociceptin was characterised by a significant slowing of the activation of IBa and could be attenuated by a strong positive depolarising step shortly before the test step (Table 1, Fig. 5A). In these experiments cells were stepped twice to 0 mV, with an 80 ms depolarising step to +60 mV between the test steps. In control conditions, amplitudes of the first (T1) and second (T2) test steps were not different (Fig. 5A and B). In the presence of nociceptin (100 nm), T1 was inhibited by 53 ± 3 % while T2 was inhibited by 21 ± 3 %, which is significantly less than the inhibition of T1 (P < 0.0002, unpaired t test, n = 6). Superfusion of nociceptin caused a 4-fold increase in the 0-95 % risetime of T1 but only a small change in the 0-95 % risetime of T2 (Table 1). If there was no step to +60 mV between T1 and T2, there was no difference in the amount by which nociceptin inhibited T1 and T2 (56 ± 2 vs. 55 ± 2 %, P = 0.65, unpaired t test, n = 6), nor was there any difference in the nociceptin-induced slowing of the risetime between T1 and T2 (Table 1, Fig. 5B).
Table 1.
Effects of a depolarising conditioning step on nociceptin modulation of IBa in RVM neurons
| Control | Nociceptin | |
|---|---|---|
| With conditioning step | ||
| Ratio T2:T1 amplitude | 1.00 ± 0.03 | 1.74 ± 0.13** |
| 0–95% risetime of T1 (ms) | 1.5 ± 0.1 | 6.5 ± 0.9** |
| 0–95% risetime of T2 (ms) | 1.4 ± 0.1 | 1.6 ± 0.1*† |
| Without conditioning step | ||
| Ratio T2:T1 amplitude | 0.98 ± 0.01 | 1.0 ± 0.01 |
| 0–95% risetime of T1 (ms) | 1.7 ± 0.2 | 7.1 ± 0.3** |
| 0–95% risetime of T2 (ms) | 1.7 ± 0.1 | 6.9 ± 0.4** |
RVM neurons were stepped twice from −90 to 0 mV, with or without an 80 ms conditioning step to +60 mV preceding the second step (see Fig. 5). T1 is the first test step, T2 the second test step. Cells were exposed to 100 nM nociceptin.
P < 0.05 between control and nociceptin conditions (paired t test)
P≥0.005 between control and nociceptin conditions (paired t test)
P < 0.0005 between T1 and T2 in the same condition (unpaired t test).
Figure 5. Nociceptin inhibition of IBa in RVM neurons is relieved by a positive prepulse.
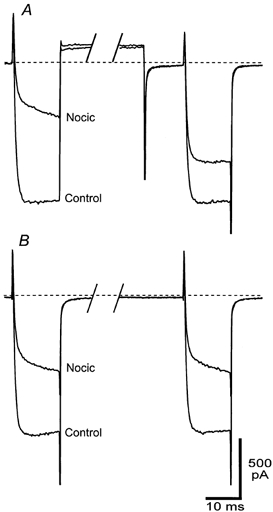
RVM neurons were voltage clamped at -90 mV and stepped twice to a test potential of -10 mV, with 90 ms between the test pulses. In A an 80 ms positive step to +60 mV was applied to the cell immediately after the first test pulse and the cell was returned to -90 mV 10 ms before the second test pulse. In B the cell was held at -90 mV for the 90 ms between test pulses. The resulting raw current traces for steps in the absence and presence of nociceptin (Nocic, 100 nm) are shown. The dashed line represents the zero current line because the complex step protocol leak subtraction was not used. A section of the trace between test pulses has been removed for clarity. A and B are taken from different neurons.
Nociceptin inhibits GABAergic but not glutamatergic synaptic transmission
Electrical stimulation in the presence of CNQX (3 μm) and strychnine (3 μm) evoked IPSCs in RVM neurons that were abolished by the GABAA antagonist bicuculline (30 μm) or picrotoxin (100 μm). The neurons could not be readily classified as being either primary or secondary cells because experiments were carried out using Cs+-filled, GTP-free pipettes to block agonist activation of postsynaptic K+ currents. Superfusion of nociceptin (300 nm) had a variable effect on the amplitude of evoked IPSCs. The mean amplitude of evoked IPSCs was reduced by 37 ± 5 % in the presence of nociceptin (range 6-62 %, P = 0.003, n = 11) and slowly recovered after washout (85 ± 4 % of control, P = 0.007 for nociceptin vs. wash, n = 9, Fig. 6A and B). In these neurons, met-enkephalin (10 μm) reduced the amplitude of evoked IPSCs by an average of 62 ± 7 % (range 38-79 %) and this was abolished by naloxone (1 μm, 86 ± 3 % of control, n = 5). There was no correlation between the percentage inhibition of evoked IPSCs produced by nociceptin and met-enkephalin (P = 0.7, n = 5, Spearman's rank correlation coefficient).
Figure 6. Nociceptin inhibits evoked but not miniature GABAergic synaptic currents in RVM neurons.
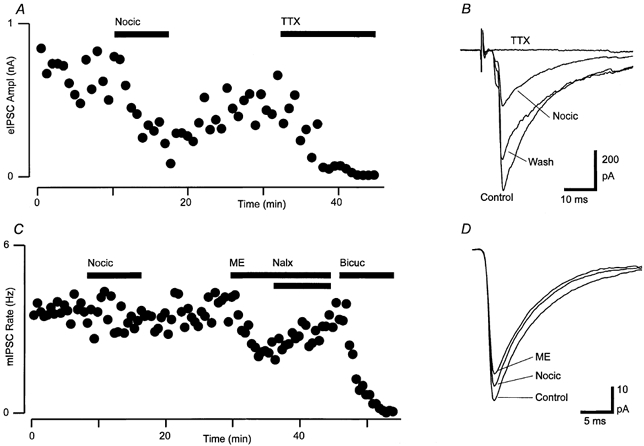
A, time course of the amplitude of evoked IPSCs (eIPSC Ampl) during application of nociceptin (Nocic, 300 nm), then TTX (300 nm). Each point is the mean of 3 consecutive evoked IPSCs. B, averaged raw traces of evoked IPSCs from before (Control), during (Nocic) and after (Wash) application of nociceptin, then after addition of TTX (average of 10-20 IPSCs). C, time course of mIPSC rate in the presence of TTX (300 nm) during superfusion of nociceptin (300 nm), then met-enkephalin (ME, 10 μm) and addition of naloxone (Nalx, 1 μm), then bicuculline (Bicuc, 30 μm). D, averaged raw traces of mIPSCs before (Control) and during superfusion of nociceptin and met-enkephalin. A-D are taken from one neuron in the presence of CNQX (3 μm) and strychnine (3 μm).
Electrical stimulation in the presence of strychnine (3 μm) and bicuculline (30 μm) or picrotoxin (100 μm) evoked EPSCs in RVM neurons that were abolished by the non-NMDA glutamate receptor antagonist CNQX (3 μm). Superfusion of nociceptin (300 nm) had no significant effect on the amplitude of evoked EPSCs (Fig. 7). The mean amplitude of evoked EPSCs was reduced by 13 ± 2 % in the presence of nociceptin (range 3-17 %, P = 0.01, n = 10) but did not significantly recover after washout (94 ± 4 % of control, P = 0.2 for nociceptin vs. wash, n = 9). In these neurons, baclofen (10 μm) reduced the amplitude of evoked EPSCs by an average of 72 ± 7 % and this was abolished by the GABAB receptor antagonist CGP-55845A (1 μm, n = 5, Fig. 7).
Figure 7. Nociceptin has no effect on evoked glutamatergic synaptic currents in RVM neurons.
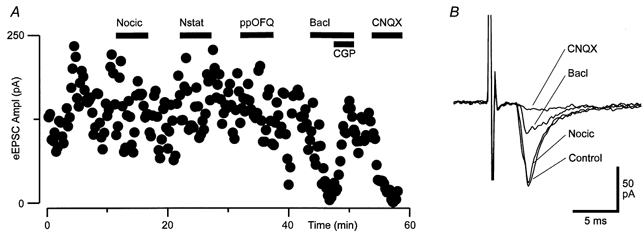
A, time course of the amplitude of evoked EPSCs during application of nociceptin (Nocic, 300 nm), nocistatin (Nstat, 1 μm), prepronociceptin154-181 (ppOFQ, 300 nm), baclofen (Bacl, 10 μm) and addition of CGP-55845A (CGP, 1 μm), then CNQX (3 μm). B, averaged raw traces of evoked EPSCs prior to (Control) and in the presence of nociceptin, baclofen and CNQX (average of 5-10 EPSCs). A and B are taken from one neuron in the presence of picrotoxin (100 μm) and strychnine (3 μm).
Nociceptin inhibits GABAergic synaptic transmission via a presynaptic Ca2+-dependent process
To determine whether nociceptin inhibits GABAA receptor-mediated synaptic transmission by a presynaptic reduction in the probability of GABA release or by a reduction in postsynaptic GABAA receptor sensitivity, we examined the effect of nociceptin on spontaneous mIPSCs. In the presence of CNQX (3 μm), strychnine (3 μm) and TTX (300 nm), mIPSCs were readily observed that were blocked by picrotoxin (100 μm) or bicuculline (30 μm, Fig. 6C). This concentration of TTX prevented Na+-dependent evoked IPSCs (Fig. 6A and B). Superfusion of nociceptin (300 nm) had no significant effect on the rate (5 ± 8 % reduction, P = 0.7), amplitude (0 ± 5 % reduction, P = 0.9) and kinetics of mIPSCs (n = 9, Fig. 6C and D). In two of these neurons, nociceptin produced a reduction in evoked IPSC amplitude prior to addition of TTX (Fig. 6A and B). Superfusion of met-enkephalin (10 μm) reduced the mean rate (50 ± 7 %, P = 0.001) but not the mean amplitude (7 ± 7 %, P = 0.07) of mIPSCs in these neurons (n = 5, Fig. 6C and D).
The inhibition of evoked IPSCs in RVM neurons may have been due to presynaptic inhibition of voltage-dependent calcium currents that were activated during action potential-evoked release but were inactive during Na+ channel-independent (TTX-resistant) spontaneous release. We therefore examined the effect of nociceptin (300 nm) on spontaneous mIPSCs during tonic activation of voltage-dependent calcium currents by increasing the external concentration of potassium (K+ increased from 2.5 to 17.5 mm). In high potassium normal calcium solutions (Ca2+ 2.4 mm, Mg2+ 1.2 mm), superfusion of nociceptin reduced the rate of mIPSCs by 52 ± 4 %(P = 0.005, n = 12, Fig. 8A). In contrast, nociceptin had no effect on the rate of mIPSCs (2 ± 9 % inhibition, P = 0.2, n = 6) in high potassium, calcium-free solutions (Ca2+ 0 mm, Mg2+ 10 mm, Cd2+ 10 μm, Fig. 8A). Nociceptin had no effect on the amplitude of mIPSCs in both normal calcium and calcium-free high potassium solutions (pooled 1 ± 3 % decrease, P = 0.8, n = 18, Fig. 8B). In high potassium solutions, the mIPSC rate in normal calcium solutions was 360 ± 90 % of that in calcium-free external solutions (n = 5, Fig. 8A).
Figure 8. Nociceptin inhibition of GABAergic synaptic currents is dependent on presynaptic calcium entry.
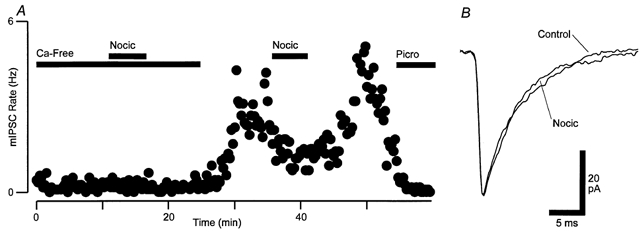
A, time course of mIPSC rate during superfusion of nociceptin (Nocic, 300 nm) in a high K+-containing solution. The external solution was initially Ca2+ free (0 mm Ca2+, 10 mm Mg2+, 10 μm Cd2+) and then switched to a normal Ca2+ solution (2.4 mm Ca2+, 1.2 mm Mg2+, 0 μm Cd2+). The mean mIPSC rate before/during nociceptin application was 0.24/0.23 Hz in Ca2+-free and 2.7/1.4 Hz in normal Ca2+ solutions. Picrotoxin (Picro, 100 μm) was added at the end of the experiment. B, averaged raw traces of mIPSCs before (Control) and during superfusion of nociceptin in the high K+, normal Ca2+ solution. A and B are taken from one neuron in the presence of high K+ (17.5 mm), CNQX (3 μm), strychnine (3 μm) and TTX (300 nm).
Other nociceptin precursors have no actions on RVM neurons
In brain slices, superfusion of nocistatin (1-3 μm) produced no change in membrane current in either primary (0 ± 1 pA, n = 6) or secondary neurons (0 ± 1 pA, n = 9, Fig. 1B). The outward current produced by nociceptin (300 nm) was similar in the absence (106 ± 13 pA, n = 16) and presence of nocistatin (1 μm, 110 ± 26 pA, n = 7). In addition, superfusion of prepronociceptin154-181 (1-3 μm) produced no change in membrane current in either primary (1 ± 1 pA, n = 6) or secondary neurons (0 ± 1 pA, n = 8, Fig. 1B). Superfusion of nocistatin (300 nm to 1 μm) also had no significant effect (P > 0.3, n = 6) on the amplitude of evoked IPSCs (6 ± 7 % reduction) or evoked EPSCs (1 ± 4 % reduction, Fig. 7). Similarly, superfusion of prepronociceptin154-181 (300 nm to 1 μm) had no significant effect (P > 0.05, n = 6) on the amplitude of evoked IPSCs (3 ± 5 % reduction) or evoked EPSCs (4 ± 4 % reduction, Fig. 7).
In acutely isolated RVM neurons nocistatin did not inhibit IBa in any RVM neuron tested, nor did it affect the inhibition of IBa by a subsequent co-superfusion of nociceptin. In the presence of nocistatin (10 μm) IBa was 100 ± 1 % of control (n = 8); subsequent co-application of nociceptin (3 nm) inhibited IBa by 28 ± 7 %, which is similar to the inhibition of IBa by nociceptin alone (30 ± 4 %, n = 6). In addition, prepronociceptin154-181 did not modulate IBa in any RVM neuron tested; IBa was 99 ± 1 % of control in the presence of prepronociceptin154-181 (1 μm, n = 7).
DISCUSSION
This study demonstrates that the endogenous ORL1 (NOP) receptor ligand nociceptin/orphanin FQ has both pre- and postsynaptic inhibitory actions on neurons of the rat RVM. Nociceptin increased a potassium conductance and inhibited calcium channel conductances in both primary and secondary RVM neurons. Nociceptin also inhibited GABAergic synaptic transmission within the RVM via a presynaptic calcium-dependent mechanism. The other putative peptide products of the prepronociceptin gene, nocistatin (rat prepronociceptin125-132) and rat prepronociceptin154-181, had no effect on RVM neurons.
Nociceptin activated a potassium current in both κ-opioid-responding primary and μ-opioid-responding secondary RVM neurons, as reported previously (Pan et al. 2000). While previous studies have measured a potassium current to define primary and secondary neurons (Pan et al. 1990, 1997, 2000), the present study extends this functional description to include calcium channel currents. The differences in the proportion of μ- to κ-responding neurons measured by potassium (8:1) and calcium currents (4:3) may have been due to sampling differences between whole-cell recordings from brain slices and acutely dissociated neurons.
Nociceptin is likely to have acted via ORL1 receptors and not other opioid receptors within the RVM. The action of nociceptin was unaffected by the opioid receptor antagonist naloxone and was abolished by the recently described ORL1 antagonist J-113397 (Kawamoto et al. 1999; Bigoni et al. 2000; Corboz et al. 2000; Ozaki et al. 2000a,b). In contrast, the action of met-enkephalin was unaffected by J-113397 and was abolished by naloxone. The peptide analogue Phe-ψ-nociceptin acted as a partial antagonist at the ORL1 receptor. Phe-ψ-nociceptin has been reported to behave as either a partial antagonist or a pure antagonist in various assays of putative ORL1 function (Calo et al. 2000). These differences are likely to be due to variations in receptor reserve and receptor-effector coupling for ORL1. The potency of nociceptin in RVM neurons was similar to that previously reported in RVM (Pan et al. 2000) and other brain regions (Knoflach et al. 1996; Vaughan & Christie, 1996; Vaughan et al. 1997a; Connor & Christie, 1998; Meis & Pape, 1998; Wagner et al. 1998; Allen et al. 1999; Chiou, 1999; Connor et al. 1999; Emmerson & Miller, 1999; Pu et al. 1999). The differences in potency for calcium and potassium conductances (EC50 2 and 16 nm, respectively) in RVM may reflect stronger coupling of the ORL1 receptor to calcium conductances, as previously demonstrated for the μ-opioid receptor in acutely isolated neonatal locus coeruleus neurons (Ingram et al. 1997).
The outward current produced by nociceptin in RVM neurons within brain slices was the result of an increase in an inwardly rectifying potassium conductance (Kir), as has been demonstrated in RVM (Pan et al. 2000) and other brain regions (Connor et al. 1996; Vaughan & Christie, 1996; Lee et al. 1997; Vaughan et al. 1997a; Meis & Pape, 1998; Wagner et al. 1998; Allen et al. 1999; Chiou, 1999; Emmerson & Miller, 1999). Nociceptin produced an increase in conductance that was greater at more negative potentials. The current produced by nociceptin had a reversal potential similar to the Nernst potential predicted for a potassium conductance (-106 mV).
The inhibition of barium current through calcium channels (IBa) in acutely isolated RVM neurons by nociceptin was probably mediated by activation of heterotrimeric guanine nucleotide binding proteins (G-proteins). Similar G-protein-mediated inhibition of high voltage-activated IBa by nociceptin has been demonstrated within other regions of the nervous system (Knoflach et al. 1996; Abdulla et al. 1997; Connor & Christie, 1998; Connor et al. 1999). In the present study the inhibition of IBa by nociceptin was evident across a range of membrane potentials and was associated with a slowing of the activation of evoked currents, all characteristic features of the ubiquitous G-protein βγ-subunit-mediated pathway for inhibition of IBa (Herlitze et al. 1996; Ikeda, 1996). Further, inhibition of IBa by nociceptin was significantly reduced by a depolarising prepulse, which is thought to reflect a voltage-dependent dissociation of G-protein βγ-subunits from the calcium channels (Herlitze et al. 1996; Ikeda, 1996; Zamponi & Snutch, 1998). Inhibition of calcium channels by nociceptin is functionally important as this process may be involved in its presynaptic inhibitory effects on GABAA receptor-mediated synaptic transmission (see below).
Nociceptin inhibited evoked IPSCs as previously demonstrated for μ-opioids and CB1 cannabinoids in RVM (Pan et al. 1990; Vaughan et al. 1999). μ-Opioids and CB1 cannabinoids inhibit GABAergic synaptic transmission in RVM via a presynaptic mechanism because they reduce the rate of spontaneous mIPSCs and have no effect on their amplitude (Vaughan et al. 1999). In the present study, nociceptin had no effect on the rate or amplitude of spontaneous mIPSCs in RVM, unlike previous studies on mIPSCs in the periaqueductal grey (PAG) (Vaughan et al. 1997a) and on mEPSCs in the spinal cord (Liebel et al. 1997). However, nociceptin reduced the rate of mIPSCs at elevated external potassium concentrations (which activates voltage-dependent calcium channels) and this inhibition was abolished by blockade of calcium entry. While the functional significance of calcium channel modulation by nociceptin is not well established, these observations suggest that nociceptin inhibits transmitter release from GABAergic terminals in RVM by reducing presynaptic calcium entry. Thus nociceptin differs from μ-opioids and CB1 cannabinoids, both of which are associated with calcium-independent presynaptic processes in RVM and PAG (Vaughan et al. 1997b, 1999, 2000).
The precursor polypeptide for nociceptin, prepronociceptin, encodes up to four additional peptides (Houtani et al. 1996; Mollereau et al. 1996; Nothacker et al. 1996). Nocistatin (rat prepronociceptin125-132), which lies upstream of nociceptin, reduces the hyperalgesia and allodynia produced by nociceptin (Minami et al. 1998; Okuda-Ashitaka et al. 1998; Yamamoto & Sakashita, 1999; Zhao et al. 1999). The 28 amino acids carboxy-terminal to the nociceptin sequence (rat prepronociceptin154-181), unlike nocistatin, are strictly conserved across several species (Houtani et al. 1996; Mollereau et al. 1996; Nothacker et al. 1996; Okuda-Ashitaka et al. 1998). A fragment comprising the first 17 amino acids of rat prepronociceptin154-181 produces naloxone-sensitive analgesia in mice (Rossi et al. 1998). In the present study, nocistatin and rat prepronociceptin154-181 had no direct effects on the membrane properties and GABAergic and glutamatergic synaptic transmission in RVM neurons. In addition, nocistatin did not prevent the effects of a subsequent co-application of nociceptin. These results are consistent with the previously reported lack of effect of nocistatin and rat prepronociceptin154-181 on nociceptin binding to ORL1 (Nothacker et al. 1996; Okuda-Ashitaka et al. 1998). However, these results differ from those obtained from the spinal cord where nociceptin and nocistatin have functionally opposing actions of inhibiting glutamatergic and GABAergic synaptic transmission, respectively (Zeilhofer et al. 2000). These findings suggest that the modulation of analgesia by nocistatin and prepronociceptin154-181 does not occur within the RVM, at least via the cellular mechanisms described in the present study.
Microinjection of μ-opioids and CB1 cannabinoids but not nociceptin and κ-opioids into RVM produces analgesia (Heinricher et al. 1997; Pan et al. 1997; Meng et al. 1998). However, microinjection of nociceptin (Heinricher et al. 1997) and κ-opioids (Pan et al. 1997) into RVM reduces the analgesic actions of μ-opioids. While the role of primary and secondary RVM neurons in the descending antinociceptive actions of opioids has not been fully resolved, both μ-opioid and CB1 cannabinoid antinociception in RVM are associated with inhibition of on-cell activity and disinhibition of off-cell activity (which display increased and decreased action potential activity with tail-flick responses, respectively; Fields et al. 1991; Meng et al. 1998). In contrast, microinjection of nociceptin into RVM inhibits action potential activity of both on- and off-cells (Heinricher et al. 1997) and κ-opioid microinjection inhibits action potential activity of off-cells (Pan et al. 1997).
The present study suggests that the cellular actions of nociceptin are even more complex than a generalised inhibition of descending analgesic pathways, as suggested recently (Grisel & Mogil, 2000). The actions of nociceptin only partly overlap those of μ-opioids, κ-opioids and CB1 cannabinoids (Pan et al. 1990, 1997; Vaughan et al. 1999, 2000). Nociceptin activates a potassium current and inhibits calcium channel currents in both primary and secondary RVM neurons, which respond to κ- and μ-opioids, respectively. In contrast, CB1 cannabinoids have no postsynaptic actions in RVM. Like μ-opioids and CB1 cannabinoids, nociceptin inhibits GABAA receptor-mediated synaptic transmission in RVM neurons (disinhibition); however, the present findings indicate that this occurs via a different presynaptic mechanism. The differences in the cellular actions of nociceptin, κ-opioids, μ-opioids and CB1 cannabinoids suggest that their effects within RVM are likely to be dependent upon a complex balance between on- and off-cell activity. Further differences in the actions of these agents are likely to appear when the balance of RVM activity is altered by stress, during chronic drug treatment and in chronic pain states.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia and The Medical Foundation of The University of Sydney. E.A.J. was supported by the Wellcome Trust. We thank Dr R. Guerrini for the kind gift of Phe1ψ(CH2-NH)Gly2]-nociceptin-(1-13)NH2 and Banyu Pharmaceutical Co. Ltd for their kind gift of J-113397. We would also like to thank Dr G. Calo for his comments on the manuscript.
References
- Abdulla FA, Smith PA. Nociceptin inhibits T-type Ca2+ channel current in rat sensory neurons by a G-protein-independent mechanism. Journal of Neuroscience. 1997;17:8721–8728. doi: 10.1523/JNEUROSCI.17-22-08721.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen CN, Jiang Z-G, Teshima K, Darland T, Ikeda M, Nelson CS, Quigley DI, Yoshioka T, Allen RG, Rea MA, Grandy DK. Orphanin-FQ/nociceptin (OFQ/N) modulates the activity of suprachiasmatic nucleus neurons. Journal of Neuroscience. 1999;19:2152–2160. doi: 10.1523/JNEUROSCI.19-06-02152.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton B, Fein J, To T, Li X, Silberstein L, Evans CJ. Immunohistochemical localization of ORL-1 in the central nervous system of the rat. Journal of Comparative Neurology. 1996;368:229–251. doi: 10.1002/(SICI)1096-9861(19960429)368:2<229::AID-CNE5>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Bigoni R, Calo G, Rizzi A, Guerrini R, De Risi C, Hashimoto Y, Hashiba E, Lambert DG, Regoli D. In vitro characterization of J-113397, a non-peptide nociceptin/orphanin FQ receptor antagonist. Naunyn-Schmiedeberg's Archives of Pharmacology. 2000;361:565–568. doi: 10.1007/s002100000220. [DOI] [PubMed] [Google Scholar]
- Calo G, Guerrini R, Rizzi A, Salvadori S, Regoli D. Pharmacology of nociceptin and its receptor: a novel therapeutic target. British Journal of Pharmacology. 2000;129:1261–1283. doi: 10.1038/sj.bjp.0703219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou LC. [Phe1ψ(CH2-NH)Gly2]nociceptin-(1–13)-NH2 activation of an inward rectifier as a partial agonist of ORL1 receptors in rat periaqueductal gray. British Journal of Pharmacology. 1999;128:103–107. doi: 10.1038/sj.bjp.0702746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor M, Christie MJ. Modulation of the calcium channel currents of acutely dissociated rat periaqueductal grey neurons. Journal of Physiology. 1998;509:47–58. doi: 10.1111/j.1469-7793.1998.047bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor M, Vaughan CW, Chieng B, Christie MJ. Nociceptin receptor coupling to a potassium conductance in rat locus coeruleus neurones in vitro. British Journal of Pharmacology. 1996;119:1614–1618. doi: 10.1111/j.1476-5381.1996.tb16080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor M, Vaughan CW, Jennings EA, Allen RG, Christie MJ. Nociceptin, Phe1ψ-nociceptin1–13, nocistatin and prepronociceptin154–181 effects on calcium channel currents and a potassium current in rat locus coeruleus in vitro. British Journal of Pharmacology. 1999;128:1779–1787. doi: 10.1038/sj.bjp.0702971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corboz MR, Rivelli MA, Egan RW, Tulshian D, Matasi J, Fawzi AB, Benbow L, Smith-Torhan A, Zhang HT, Hey JA. Nociceptin inhibits capsaicin-induced bronchoconstriction in isolated guinea pig lung. European Journal of Pharmacology. 2000;402:171–179. doi: 10.1016/s0014-2999(00)00505-7. [DOI] [PubMed] [Google Scholar]
- Emmerson PJ, Miller RJ. Pre- and postsynaptic actions of opioid and orphan opioid agonists in the rat arcuate nucleus and ventromedial hypothalamus in vitro. Journal of Physiology. 1999;517:431–445. doi: 10.1111/j.1469-7793.1999.0431t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields HL, Heinricher MM, Mason P. Neurotransmitters in nociceptive modulatory circuits. Annual Review of Neuroscience. 1991;14:219–245. doi: 10.1146/annurev.ne.14.030191.001251. [DOI] [PubMed] [Google Scholar]
- Grisel JE, Mogil JS. Effects of supraspinal orphanin FQ/nociceptin. Peptides. 2000;21:1037–1045. doi: 10.1016/s0196-9781(00)00236-9. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, McGaraughty S, Grandy DK. Circuitry underlying antiopioid actions of orphanin FQ in the rostral ventromedial medulla. Journal of Neurophysiology. 1997;78:3351–3358. doi: 10.1152/jn.1997.78.6.3351. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta-gamma subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Houtani T, Nishi M, Takeshima H, Nukada T, Sugimoto T. Structure and regional distribution of nociceptin/orphanin FQ precursor. Biochemical and Biophysical Research Communications. 1996;219:714–719. doi: 10.1006/bbrc.1996.0300. [DOI] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Ingram S, Wilding TJ, McCleskey EW, Williams JT. Efficacy and kinetics of opioid action on acutely dissociated neurons. Molecular Pharmacology. 1997;52:136–143. doi: 10.1124/mol.52.1.136. [DOI] [PubMed] [Google Scholar]
- Kawamoto H, Ozaki S, Itoh Y, Miyaji M, Arai S, Nakashima H, Kato T, Ohta H, Iwasawa Y. Discovery of the first potent and selective small molecule opioid receptor-like (ORL1) antagonist: 1. Journal of Medicinal Chemistry. 1999;42:5061–5063. doi: 10.1021/jm990517p. [DOI] [PubMed] [Google Scholar]
- Knoflach F, Reinscheid RK, Civelli O, Kemp JA. Modulation of voltage-gated calcium channels by orphanin FQ in freshly dissociated hippocampal neurons. Journal of Neuroscience. 1996;16:6657. doi: 10.1523/JNEUROSCI.16-21-06657.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachowicz JE, Shen Y, Monsma FJ, Jr, Sibley D R. Molecular cloning of a novel G protein-coupled receptor related to the opiate receptor family. Journal of Neurochemistry. 1995;64:34–40. doi: 10.1046/j.1471-4159.1995.64010034.x. [DOI] [PubMed] [Google Scholar]
- Lee K, Nicholson JR, McKnight AT. Nociceptin hyperpolarises neurones in the rat ventromedial hypothalamus. Neuroscience Letters. 1997;239:37–40. doi: 10.1016/s0304-3940(97)00875-6. [DOI] [PubMed] [Google Scholar]
- Liebel JT, Swandulla D, Zeilhofer HU. Modulation of excitatory synaptic transmission by nociceptin in superficial dorsal horn neurones of the neonatal rat spinal cord. British Journal of Pharmacology. 1997;121:425–432. doi: 10.1038/sj.bjp.0701149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meis S, Pape H-C. Postsynaptic mechanisms underlying responsiveness of amygdaloid neurons to nociceptin/orphanin FQ. Journal of Neuroscience. 1998;18:8133–8144. doi: 10.1523/JNEUROSCI.18-20-08133.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng ID, Manning BH, Martin WJ, Fields HL. An analgesia circuit activated by cannabinoids. Nature. 1998;395:381–383. doi: 10.1038/26481. [DOI] [PubMed] [Google Scholar]
- Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, Butour JL, Guillemot JC, Ferrara P, Monsarrat B, Mazarguil H, Vassart G, Parmentier M, Constentin J. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- Minami T, Okuda-Ashitaka E, Nishiuchi Y, Kimura T, Tachibana S, Mori H, Ito S. Anti-nociceptive responses produced by human putative counterpart of nocistatin. British Journal of Pharmacology. 1998;124:1016–1018. doi: 10.1038/sj.bjp.0701995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollereau C, Parmentier M, Mailleux P, Butour JL, Moisand C, Chalon P, Caput D, Vassart G, Meunier JC. ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Letters. 1994;341:33–38. doi: 10.1016/0014-5793(94)80235-1. [DOI] [PubMed] [Google Scholar]
- Mollereau C, Simons M-J, Soularue P, Liners F, Vassart G, Meunier J-C, Parmentier M. Structure, tissue distribution, and chromosomal localization of the prepronociceptin gene. Proceedings of the National Academy of Sciences of the USA. 1996;93:8666–8670. doi: 10.1073/pnas.93.16.8666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal CR, Mansour A, Reinscheid R, Nothacker HP, Civelli O, Watson SJ. Localization of orphanin FQ (nociceptin) peptide and messenger RNA in the central nervous system of the rat. Journal of Comparative Neurology. 1999;406:503–547. [PubMed] [Google Scholar]
- Nothacker H-P, Reinscheid RK, Mansour A, Henningsen RA, Ardati A, Monsma FJ, Watson SJ, Civelli O. Primary structure and tissue distribution of the orphanin FQ precursor. Proceedings of the National Academy of Sciences of the USA. 1996;93:8677–8682. doi: 10.1073/pnas.93.16.8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda-Ashitaka E, Minami T, Tachibana S, Yoshihara Y, Nishiuchi Y, Kimura T, Ito S. Nocistatin, a peptide that blocks nociceptin action in pain transmission. Nature. 1998;392:286–289. doi: 10.1038/32660. [DOI] [PubMed] [Google Scholar]
- Ozaki S, Kawamoto H, Itoh Y, Miyaji M, Azuma T, Ichikawa D, Nambu H, Iguchi T, Iwasawa Y, Ohta H. In vitro and in vivo pharmacological characterization of J-113397, a potent and selective non-peptidyl ORL1 receptor antagonist. European Journal of Pharmacology. 2000a;402:45–53. doi: 10.1016/s0014-2999(00)00520-3. [DOI] [PubMed] [Google Scholar]
- Ozaki S, Kawamoto H, Itoh Y, Miyaji M, Iwasawa Y, Ohta H. A potent and highly selective nonpeptidyl nociceptin/orphanin FQ receptor (ORL1) antagonist: J-113397. European Journal of Pharmacology. 2000b;387:R17–18. doi: 10.1016/s0014-2999(99)00822-5. [DOI] [PubMed] [Google Scholar]
- Pan ZZ, Hirakawa N, Fields HL. A cellular mechanism for the bidirectional pain-modulating actions of orphanin FQ/nociceptin. Neuron. 2000;26:515–522. doi: 10.1016/s0896-6273(00)81183-6. [DOI] [PubMed] [Google Scholar]
- Pan ZZ, Tershner SA, Fields HL. Cellular mechanism for anti-analgesic action of agonists of the kappa-opioid receptor. Nature. 1997;389:382–385. doi: 10.1038/38730. [DOI] [PubMed] [Google Scholar]
- Pan ZZ, Williams JT, Osborne PB. Opioid actions on single nucleus raphe magnus neurons from rat and guinea-pig in vitro. Journal of Physiology. 1990;427:519–532. doi: 10.1113/jphysiol.1990.sp018185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu L, Bao GB, Ma L, Pei G. Acute desensitization of nociceptin/orphanin FQ inhibition of voltage-gated calcium channels in freshly dissociated hippocampal neurons. European Journal of Neuroscience. 1999;11:3610–3616. doi: 10.1046/j.1460-9568.1999.00776.x. [DOI] [PubMed] [Google Scholar]
- Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, Grandy DK, Langen H, Monsma FJ, Jr, Civelli O. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270:792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- Rossi GC, Mathis JP, Pasternak GW. Analgesic activity of orphanin FQ2, murine prepro-orphanin FQ141–157, in mice. NeuroReport. 1998;9:1165–1168. [PubMed] [Google Scholar]
- Vaughan CW, Christie MJ. Increase by the ORL1 receptor (opioid receptor-like1) ligand, of inwardly rectifying conductance in dorsal raphe nucleus neurons. British Journal of Pharmacology. 1996;17:996–1003. doi: 10.1111/j.1476-5381.1996.tb15329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Connor M, Bagley EE, Christie MJ. Actions of cannabinoids on membrane properties and synaptic transmission in rat periaqueductal gray neurons in vitro. Molecular Pharmacology. 2000;57:288–295. [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Christie MJ. Actions of the ORL1 receptor ligand nociceptin on membrane properties of rat periaqueductal gray neurons in vitro. Journal of Neuroscience. 1997a;17:996–1003. doi: 10.1523/JNEUROSCI.17-03-00996.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Connor MA, Christie MJ. How opioids inhibit GABA-mediated neurotransmission. Nature. 1997b;390:611–614. doi: 10.1038/37610. [DOI] [PubMed] [Google Scholar]
- Vaughan CW, McGregor IS, Christie MJ. Cannabinoid receptor activation inhibits GABAergic neurotransmission in rostral ventromedial medulla neurons in vitro. British Journal of Pharmacology. 1999;127:935–940. doi: 10.1038/sj.bjp.0702636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EJ, Ronnekleiv OK, Grandy DK, Kelly MJ. The peptide orphanin FQ inhibits beta-endorphin neurons and neurosecretory cells in the hypothalamic arcuate nucleus by activating an inwardly-rectifying K+ conductance. Neuroendocrinology. 1998;67:73–82. doi: 10.1159/000054301. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Sakashita Y. Effect of nocistatin and its interaction with nociceptin orphanin FQ on the rat formalin test. Neuroscience Letters. 1999;262:179–182. doi: 10.1016/s0304-3940(99)00073-7. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Snutch TP. Decay of prepulse facilitation of N type calcium channels during G Protein inhibition is consistent with binding of a single G(βγ) subunit. Proceedings of the National Academy of Sciences of the USA. 1998;95:4035–4039. doi: 10.1073/pnas.95.7.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilhofer HU, Selbach UM, Guhring H, Erb K, Ahmadi S. Selective suppression of inhibitory synaptic transmission by nocistatin in the rat spinal cord dorsal horn. Journal of Neuroscience. 2000;20:4922–4929. doi: 10.1523/JNEUROSCI.20-13-04922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao CS, Li BS, Zhao GY, Liu HX, Luo F, Wang Y, Tian JH, Chang JK, Han JS. Nocistatin reverses the effect of orphanin FQ/nociceptin in antagonizing morphine analgesia. NeuroReport. 1999;10:297–299. doi: 10.1097/00001756-199902050-00017. [DOI] [PubMed] [Google Scholar]