

Abstract
Rhythmic breathing during sleep requires that PCO2 be maintained above a sensitive hypocapnic apnoeic threshold. Hypoxia causes periodic breathing during sleep that can be prevented or eliminated with supplemental CO2. The purpose of this study was to determine the effect of hypoxia in changing the difference between the eupnoeic PCO2 and the PCO2 required to produce hypopnoea or apnoea (hypopnoea/apnoeic threshold) in sleeping humans.
The effect of hypoxia on eupnoeic end-tidal partial pressure of CO2 (PET,CO2) and hypopnoea/apnoeic threshold PET,CO2 was examined in seven healthy, sleeping human subjects. A bilevel pressure support ventilator in a spontaneous mode was used to reduce PET,CO2 in small decrements by increasing the inspiratory pressure level by 2 cmH2O every 2 min until hypopnoea (failure to trigger the ventilator) or apnoea (no breathing effort) occurred. Multiple trials were performed during both normoxia and hypoxia (arterial O2 saturation, Sa,O2 = 80 %) in a random order. The hypopnoea/apnoeic threshold was determined by averaging PET,CO2 of the last three breaths prior to each hypopnoea or apnoea.
Hypopnoeas and apnoeas were induced in all subjects during both normoxia and hypoxia. Hypoxia reduced the eupnoeic PET,CO2 compared to normoxia (42.4 ± 1.3 vs. 45.0 ± 1.1 mmHg, P < 0.001). However, no change was observed in either the hypopnoeic threshold PET,CO2 (42.1 ± 1.4 vs. 43.0 ± 1.2 mmHg, P > 0.05) or the apnoeic threshold PET,CO2 (41.3 ± 1.2 vs. 41.6 ± 1.0 mmHg, P > 0.05). Thus, the difference in PET,CO2 between the eupnoeic and threshold levels was much smaller during hypoxia than during normoxia (-0.2 ± 0.2 vs. -2.0 ± 0.3 mmHg, P < 0.01 for the hypopnoea threshold and -1.1 ± 0.2 vs. -3.4 ± 0.3 mmHg, P < 0.01 for the apnoeic threshold). We concluded that hypoxia causes a narrowing of the difference between the baseline PET,CO2 and the hypopnoea/apnoeic threshold PET,CO2, which could increase the likelihood of ventilatory instability.
Rhythmic breathing during sleep is critically dependent on the level of PCO2 in arterial blood (Fink, 1961; Skatrud & Dempsey, 1983; Datta et al. 1991). Small reductions of PCO2 to levels that are slightly below the awake, eupnoeic PCO2 consistently produce apnoea in sleeping humans. The importance of this sensitive apnoeic threshold for CO2 has been implicated in the periodic breathing with central apnoeas that is commonly observed in sleeping subjects at high altitudes. When supplemental CO2 sufficient to elevate the PCO2 a mere 1-2 mmHg was administered to subjects at high altitudes, and the hypoxaemic arterial PO2 (Pa,O2) was maintained via nitrogen administration, the apnoeas were prevented or eliminated, and the breathing pattern was stabilized (Berssenbrugge et al. 1983). Thus, hypocapnia plays an important role in the development and maintenance of breathing instability during hypoxia.
However, several observations suggest that hyperventilation and consequent reduction of eupnoeic PCO2 alone are insufficient to account for the breathing instability seen during hypoxia. For example, chronic hyperventilation does not cause breathing instability during sleep in disease states such as interstitial fibrosis (Perez-Padilla et al. 1985) or during the chronic administration of ventilatory stimulants, such as acetazolamide, theophylline and medroxyprogesterone acetate (Skatrud et al. 1981; White et al. 1982; DeBaker et al. 1995; Javaheri et al. 1996). On the contrary, these chronic ventilatory stimuli have been shown to have stabilizing effects on ventilation during sleep (Hudgel & Thanakitcharu, 1998). Likewise, during prolonged hypoxia, the susceptibility to periodic breathing decreases during the acclimatization process, such that periodic breathing during sleep is diminished despite an increased ventilation and there is an even greater reduction in arterial PCO2 (Pa,CO2) and alkalization of blood and cerebrospinal fluid (CSF). Administration of acetazolamide at high altitudes also reduces periodic breathing despite ventilatory stimulation (Hackett et al. 1987). These findings indicate that the lowering of the eupnoeic PCO2 by a ventilatory stimulant is not the only determinant of whether or not breathing instability will be initiated during sleep.
To reconcile these observations, we considered the possibility that the difference between the eupnoeic PCO2 and the apnoeic threshold PCO2, rather than the absolute eupnoeic PCO2, is the critical determinant of breathing instability during sleep. A smaller difference between the eupnoeic and apnoeic threshold would indicate an increase in the sensitivity of the ventilatory control system below eupnoea and thereby, increase the likelihood of ventilatory instability (Carley et al. 1988; Khoo et al. 1991). Consequently, a given transient reduction of PCO2 will cause a greater reduction in ventilation and result in apnoea during hypoxia. Accordingly, the present study was performed during both normoxia and hypoxia to determine precisely the eupnoeic PCO2 and the PCO2 required to cause hypopnoea or apnoea (hypopnoea/apnoeic threshold). We hypothesized that hypoxia reduces the difference between the eupnoeic PCO2 and hypopnoea/apnoeic threshold in sleeping humans.
METHODS
Subjects
Seven healthy, non-snoring volunteers (3 male and 4 females) with a mean age of 25 years (range, 19-35 years) and body mass index of 24 ± 2 served as subjects. Women were studied within 7-10 days of their last menstrual period. All were non-smokers and free from any cardiovascular, pulmonary and neurological diseases. This study was approved by the University of Wisconsin Health Sciences Human Subjects Committee. All experimental procedures were performed in accordance with the Declaration of Helsinki and all subjects gave their informed written consent to participate in the study.
Polysomnographic methods
Overnight sleep studies were performed on each subject using standard polysomnographic techniques (Rechtschaffen & Kales, 1968), including electroencephalogram (C3/A2 and O2/A1), electrooculogram (EOG), electrocardiogram and submental electromyogram with surface electrodes to identify sleep stages and arousals. Ventilation was measured with a pneumotachograph (Hans Rudolph) coupled to a differential pressure transducer (Validyne) positioned between the nasal mask and the ventilator tubing. The flow signal was electronically integrated to provide inspiratory and expiratory volume. The airway pressure was measured from a side port on the mask. Respiratory effort was also monitored by respiratory inductive plethysmography (Respitrace, Ambulatory Monitoring Inc.), calibrated with an isovolume manoeuvre. Arterial oxyhaemoglobin saturation (Sa,O2) was measured by pulse oximetry (Ohmeda Oxicap no. 4700). End-tidal gas was sampled via a catheter in the nostril with a sampling tube passed through a side port of the nasal mask. End-tidal PCO2 and PO2 were measured by a gas analyser (Model CD-3A, AMETEK), which was calibrated by known gases before and after each experiment. All the variables were recorded continuously on a polygraph (Model 78D; Grass Instruments) and on videotape (Vetter).
Protocol
Subjects breathed through a sealed nasal mask attached to a mechanical ventilator (Hamilton Medical). The mouth was taped shut to prevent air leaks. The ventilator was set in the pressure support mode, which allowed an independent adjustment of the inspiratory and end-expiratory pressures. The trigger sensitivity of the ventilator was set at -2 cmH2O. To facilitate sleep and avoid sleep fragmentation, zolpidem (10 mg) was administered orally to all subjects 10 min prior to lights out. During stages 2-4, non-rapid eye movement (NREM) sleep, each trial was initiated with a 3-5 min period of spontaneous breathing obtained while the subject breathed with an inspiratory pressure and end-expiratory pressure of 2 cmH2O, i.e. continuous positive airway pressure (CPAP) at 2 cmH2O. The low level of CPAP was used to minimize the upper airway resistance.
Following the baseline CPAP period, the inspiratory pressure was increased to 6 cmH2O while maintaining end-expiratory pressure at 2 cmH2O. If no apnoea or hypopnoea occurred after 2 min, the inspiratory pressure was increased in 2 cmH2O increments at 2 min intervals with the end-expiratory pressure being maintained at 2 cmH2O. When periodic breathing occurred with recurrent apnoeas and/or hypopnoeas, the subject was returned to baseline CPAP and spontaneous breathing. At least 3-5 min of spontaneous breathing was required between trials to allow the Sa,O2 and PET,CO2 to return to their baseline levels.
Apnoea was defined as a delay in the perceptible inspiratory effort on the mask pressure, Respitrace and flow signals for a length of at least two baseline respiratory cycles, (arrow B in Fig. 1B). Hypopnoea was defined as two or more non-triggered efforts detected on the mask pressure tracing associated with a 50 % or greater reduction in tidal volume (arrow A in Fig. 1B). The pre-apnoea or pre-hypopnoea respiratory variables were determined by averaging data from the three breaths immediately prior to each apnoea or hypopnoea event. Trials that resulted in awakening or arousal were excluded from analysis.
Figure 1. Polygraph record of a single trial during normoxic (A) and hypoxic (B) breathing.
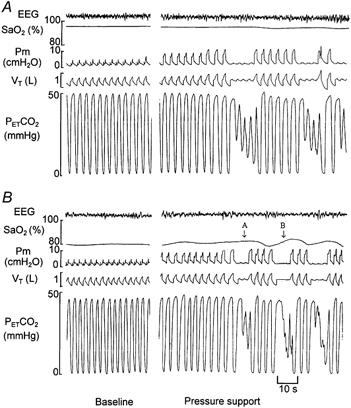
The baseline period shows spontaneous, stable breathing with continuous positive airway pressure (CPAP) of 2 cmH2O. During normoxia, the pressure-support level was increased to 8 cmH2O during inspiration with the end-expiratory level maintained at 2 cmH2O (8/2 cmH2O) and subsequently to 10/2 cmH2O. At 10/2 cmH2O, the reduction in PET,CO2 was sufficient to cause an initial hypopnoea and consequent periodic breathing. The hypopnoea threshold was determined as the mean PET,CO2 of the last three breaths immediately preceding the first hypopnoea. During hypoxia, arrow A indicates a hypopnoea with two successive untriggered breaths, and arrow B indicates a central apnoea with no respiratory effort.
Multiple trials were performed during both normoxia and hypoxia in a random order. For hypoxia trials, subjects inhaled a gas mixture with an inspired oxygen fraction (FI,O2) of 8-14 % to achieve an oxygen saturation (Sa,O2) of 80 %. When the Sa,O2 stabilized at 80 % for 5 min, the pressure-support protocol was initiated as described above. If a normoxia trial immediately followed a hypoxia trial, the pressure-support protocol was not initiated until the Sa,O2 returned to the normoxic baseline level.
Data analysis
Sleep stages were scored according to standard criteria (Rechtschaffen & Kales, 1968). Respiratory parameters including tidal volume (VT), breathing frequency (f), minute ventilation (), inspiratory time (TI), expiratory time (TE), end-tidal PO2 (PET,O2) and PET,CO2 were measured breath-by-breath and expressed as means ± s.e.m. Ventilatory measurements from stages 2-4 of non-rapid eye movement (NREM) sleep were combined for analysis.
Baseline values for respiratory parameters were determined by averaging all breaths during 3-5 min of stable, spontaneous breathing on CPAP during normoxia and hypoxia. At each level of pressure support, we averaged the respiratory parameters at each inspiratory pressure level for the periods before the occurrence of hypopnoea or apnoea. We then compared the respiratory parameters at each pressure-support level with each individual's baseline respiratory parameters using an ANOVA with repeated measures followed by Bonferroni t tests if necessary. We also used least squares linear regression analysis to examine the relationship between respiratory parameters and the pressure-support level. The mean pre-apnoea and pre-hypopnoea respiratory parameters were compared with each individual's baseline values as well as between normoxia and hypoxia using a two-way ANOVA with repeated measures. When the P value was significant, a Bonferroni t test was performed to localize the difference.
The reproducibility of the pre-apnoea and the pre-hypopnoea PET,CO2 was determined as the coefficient of variation for each subject using 3-7 trials per subject. The difference between the eupnoeic PET,CO2 and the pre-apnoea or the pre-hypopnoea PET,CO2 (ΔPET,CO2) was determined for each subject. We then compared ΔPET,CO2 between normoxia and hypoxia, as well as between hypopnoea and apnoea under the same condition (normoxia or hypoxia), using a two-way ANOVA with repeated measures. Another way of examining the relationship between the reduction of PET,CO2 and the development of apnoea was to calculate the change in ventilatory response to CO2 between eupnoea and the apnoea threshold. The ventilatory response was calculated by dividing the eupnoeic by the ΔPET,CO2 (eupnoeic PET,CO2 - apnoea threshold PET,CO2). The slope of the ventilatory response was compared between hypoxia and normoxia using Student's paired t test. Data are reported as means ± s.e.m.
RESULTS
The baseline breathing pattern and the effect of pressure-support ventilation in producing hypopnoea and apnoea is illustrated during representative normoxia and hypoxia trials (Fig. 1). During normoxia, a stable breathing pattern was noted during the baseline period. A progressive increase in the pressure-support level (note increase in Pm) resulted in an increase in tidal volume and a reduction in PET,CO2, which eventually caused hypopnoea and subsequent periodic breathing. The hypopnoea threshold was determined as the mean PET,CO2 of the last three breaths, which immediately preceded the first hypopnoea. During hypoxic breathing, the periodic breathing following the initial hypopnoea included an episode of central apnoea.
Effect of hypoxia on baseline ventilation
During stable NREM sleep, hypoxia increased (5.4 ± 0.3 vs. 6.3 ± 0.4 l min−1, P < 0.05) and reduced PET,CO2 (45.0 ± 1.1 vs. 42.4 ± 1.3 mmHg, P < 0.001) compared to normoxia (Table 1). Spontaneously occurring sleep-disordered breathing was more common during hypoxia compared to normoxia (apnoea/hypopnoea index 9.8 ± 2.6 vs. 0.9 ± 0.5 events h−1, P < 0.05). Hypoxia increased the slope of the ventilatory response to CO2 in the range between eupnoeic PET,CO2 and the apnoeic threshold PET,CO2 (1.7 ± 0.1 vs. 5.5 ± 0.9 l min−1 mmHg−1, P < 0.05).
Table 1.
Baseline and pre-hypopnoea/apnoeic respiration
| Room air |
Hypoxia |
|||||
|---|---|---|---|---|---|---|
| Baseline | Pre-hypopnoea | Pre-apnoea | Baseline | Pre-hypopnoea | Pre-apnoea | |
| VT (ml) | 351 ± 15 | 479 ± 52† | 614 ± 63†‡ | 376 ± 24 | 402 ± 24† | 514 ± 64†‡ |
| f (breaths min−1) | 16 ± 1 | 14 ± 1† | 12 ± 1†‡ | 17 ± 1* | 17 ± 1*† | 16 ± 1*†‡ |
| (l min−1) | 5.4 ± 0.3 | 6.4 ± 0.6† | 7.1 ± 0.5† | 6.3 ± 0.4* | 6.5 ± 0.4† | 7.5 ± 0.6† |
| PET,CO2 (mmHg) | 45.0 ± 1.1 | 43.0 ± 1.2† | 41.6 ± 1.0†‡ | 42.4 ± 1.3* | 42.1 ± 1.4 | 41.3 ± 1.2†‡ |
| PET,O2 (mmHg) | 103 ± 3 | 108 ± 3 | 113 ± 3†‡ | 54 ± 2* | 59 ± 2* | 64 ± 2*†‡ |
P < 0.05, hypoxia vs. room air
P < 0.05, compared to the baseline value
P < 0.05, compared to hypopnoea. VT, tidal volume; , minute ventilation; PET,CO2, end-tidal partial pressure of CO2; PET,O2, end-tidal partial pressure of O2.
Effect of pressure support on ventilation prior to hypopnoea/apnoea
During room air breathing, prior to producing hypopnoea or apnoea, pressure support effectively increased and lowered PET,CO2 during normoxic breathing (Fig. 2). With the increase in the pressure-support level from 0-10 cmH2O, gradually increased (5.4 ± 0.3 to 7.3 ± 0.6 l min−1; r = 0.97, P < 0.05), and PET,CO2 gradually declined (45.0 ± 1.1 to 42.1 ± 1.3 mmHg; r = -0.94, P < 0.05). The pressure-support ventilation caused an increase in VT (0.35 ± 0.02 to 0.57 ± 0.07 l; r = 0.96, P < 0.05) and a decrease in breathing frequency (16 ± 0.7 to 14 ± 0.7 breaths min−1; r = -0.98, P < 0.01). The TI was prolonged (1.8 ± 0.1 to 2.2 ± 0.1 s; r = 0.90P < 0.05) with no clear effect on TE (2.2 ± 0.1 to 2.4 ± 0.2 s; r = 0.84, P > 0.05).
Figure 2. Effect of pressure-support levels on ventilation and respiratory cycle timing during normoxia (○) and hypoxia (•).
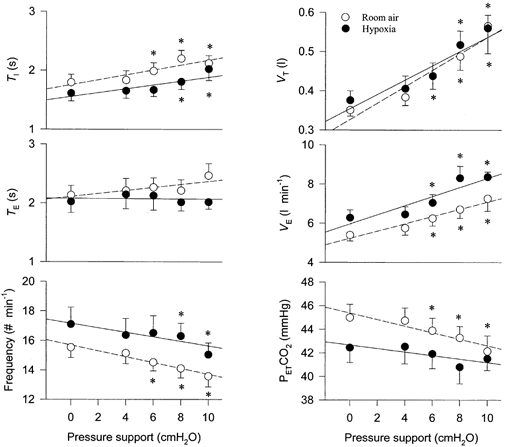
The increase of inspiratory pressure support caused an increase in tidal volume and minute ventilation and a decrease in breathing frequency. End-tidal PCO2 gradually decreased. Each point at pressure support levels of 0-8 cmH2O represents the mean data for the entire group of seven subjects. The pressure-support level of 10 cmH2O represents data from all seven subjects during normoxia but only four of seven subjects during hypoxia. During hypoxia, three of seven subjects developed apnoea at pressure support ≤ 10 cmH2O. TI, inspiratory time; TE, expiratory time; VT, tidal volume; , minute ventilation; PET,CO2, end-tidal PCO2. *P < 0.05 compared to the baseline value with zero pressure support.
During hypoxia, prior to producing hypopnoea or apnoea, pressure-support ventilation caused a similar increase in VT (0.37 ± 0.02 to 0.56 ± 0.03 l; r = 0.95, P < 0.05)and (6.3 ± 0.4 to 8.4 ± 0.3 l min−1; r = 0.92, P < 0.05), a reduction in breathing frequency (17 ± 1 to 15 ± 1 breaths min−1; r = -0.88, P = 0.05) and a prolonging of TI (1.6 ± 0.1 to 2.0 ± 0.2 s; r = 0.89, P < 0.05). A trend in reduction of PET,CO2 did not reach statistical significance until apnoea actually occurred (see below and Fig. 1B).
Hypopnoea and apnoea produced by pressure-support ventilation
During normoxia, 42 trials of pressure-support ventilation produced apnoea or hypopnoea without an associated change in sleep stages (Fig. 1A). During hypoxia, 37 trials resulted in apnoea or hypopnoea (Fig. 1B). Apnoea length was slightly shorter during hypoxia than normoxia (12.1 ± 0.9 vs. 13.9 ± 0.9 s, P < 0.05). The hypoxic trials were associated with a greater decline of Sa,O2 at the end of hypopnoea or apnoea (hypopnoea, -3.8 ± 0.3 vs. -1.2 ± 0.2 %, P < 0.01; apnoea, -5.3 ± 0.3 vs. -2.1 ± 0.5 % P < 0.01). The time delay from the final change in pressure support to the onset of apnoea or hypopnoea was similar during normoxia and hypoxia in terms of either time (19 ± 4 vs. 22 ± 5 s,P > 0.05) or the number of breaths (4 ± 1 vs. 5 ± 1 breaths, P > 0.05).
Apnoeas were always preceded by a higher VT and and a lower PET,CO2 compared to the baseline levels in both normoxia and hypoxia (Table 1 and Fig. 1). Hypopnoeas, during normoxia, were also associated with a significant reduction in PET,CO2. However, during hypoxia, the reduction in PET,CO2 preceding hypopnoeas was small, inconsistent and insignificant. An example of the minimum reduction in PET,CO2 prior to a hypopnoea and the slight further reduction in PET,CO2 associated with an apnoea on the next cycle is shown in Fig. 1B.
For each individual, the pre-hypopnoea or pre-apnoea PET,CO2 was quite reproducible. The coefficient of variation was 2.7 ± 0.2 % for pre-hypopnoea and 1.2 ± 0.5 % for pre-apnoea during normoxia, and 3.2 ± 0.4 % for pre-hypopnoea and 2.8 ± 0.7 % for pre-apnoea during hypoxia. The occurrence of apnoea or hypopnoea during hypoxia was associated with a smaller increase in tidal volume above baseline levels compared to normoxia (VT, 26 ± 7 vs. 128 ± 39 ml, P < 0.05 for pre-hypopnoea; 138 ± 44 vs. 264 ± 57 ml, P < 0.05 for pre-apnoea). Likewise, the minimum pressure-support level which produced hypopnoea or apnoea was lower during hypoxia than during normoxia (5.1 ± 1.1 vs. 7.1 ± 0.6 cmH2O, P < 0.05).
The difference between the eupnoeic PET,CO2 and the apnoea or hypopnoea threshold PET,CO2 (ΔPET,CO2) was smaller during hypoxia compared to normoxia (-0.2 ± 0.2 vs. -2.0 ± 0.3 mmHg, P < 0.01 for hypopnoea and -1.1 ± 0.2 vs. -3.4 ± 0.3 mmHg, P < 0.01 for apnoea, Fig. 3). No difference was noted in the absolute level of PET,CO2 at the hypopnoeic threshold (43.0 ± 1.2 vs. 42.1 ± 1.4 mmHg, P > 0.05) or the apnoeic threshold (41.6 ± 1.0 vs. 41.3 ± 1.2 mmHg, P > 0.05) between the two conditions. Thus, hypoxia failed to lower the apnoeic threshold PET,CO2 to as great an extent as it lowered the eupnoeic PET,CO2. Moreover, the lower apnoeic threshold compared to the hypopnoeic threshold PET,CO2 during both normoxia and hypoxia suggests a dose-response phenomenon whereby a greater reduction of PET,CO2 is required to eliminate respiratory rhythm, as opposed to simply reducing amplitude (Wilson et al. 1999).
Figure 3. Eupnoeic and threshold PET,CO2 during normoxic and hypoxic trials.
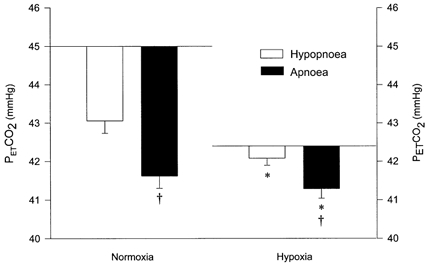
The horizontal line represents the eupnoeic PET,CO2 during stable breathing with no pressure support. □, the difference between the eupnoeic PET,CO2 and the hypopnoeic threshold PET,CO2. ▪, the difference between the eupnoeic PET,CO2 and the apnoeic threshold PET,CO2. *P < 0.05, ΔPET,CO2 (eupnoeic - threshold PET,CO2) during hypoxia compared to normoxia. †P < 0.05, ΔPET,CO2 required to produce apnoea (eupnoeic - apnoeic threshold PET,CO2) compared to the ΔPET,CO2 required to produce hypopnoea (eupnoeic - hypopnoeic threshold PET,CO2) during normoxia and hypoxia.
DISCUSSION
The major observation of this study is that the difference between the eupnoeic PET,CO2 and the hypopnoea/apnoeic threshold PET,CO2 was smaller during hypoxia than during normoxia. The closer proximity of the eupnoeic PET,CO2 to the hypopnoea/apnoeic threshold PET,CO2 was due to a disproportionate reduction in the eupnoeic PET,CO2 rather than a difference in the threshold PET,CO2 between normoxia and hypoxia. The close proximity between the eupnoeic PET,CO2 and the apnoeic threshold provides a potential explanation for why hypoxia causes breathing instability during sleep.
Mechanisms for failure of hypoxia to lower the hypopnoea/apnoeic threshold
Since all ventilatory stimulants lower the eupnoeic Pa,CO2, a fundamental difference in the effect of hypoxia may be its failure to reduce proportionally the hypopnoea/ apnoeic threshold. It is not completely understood why the apnoeic threshold is not lowered during hypoxia. However, compared to other non-hypoxic stimulants, hypoxia uniquely depresses the central nervous system, dilates cerebral vessels and impairs short-term potentiation. All these effects may work together in changing the overall excitability of the ventilatory control system, which is manifested by a relatively high hypopnoea/apnoeic threshold for PCO2.
Although hypoxia-induced respiratory depression has not been observed in all preparations (Neubauer et al. 1990; Curran et al. 2000), when hypoxia lasts longer than 5 min, as it did in our subjects, it may impair neuronal function in the central nervous system, including the bulbar network of respiratory neurons (Natsui & Kuwana, 1992). Extreme hypoxia is associated with the elimination of respiratory neural activity and cessation of respiratory drive (Trippenbach et al. 1990; LaManna et al. 1996). The mechanism underlying the depressant effect of hypoxia on ventilation is complicated at the cellular and molecular levels and may be related to metabolic impairment causing ATP depletion (LaManna et al. 1996; Mironov et al. 1998), reduction of synaptic interaction among respiratory neurons (Ramires et al. 1998), increased membrane hyperpolarization of respiratory neurons (Neubauer et al. 1990) or hypoxia-induced production of neuro-inhibitory substances (Schaeffer et al. 1988; Runold et al. 1989; Schmidt et al. 1995; Richter et al. 1999). This hypoxia-produced central neural dysfunction may impair the neural switching mechanism from expiration to inspiration and weaken the resistance of the respiratory system to instability (Younes, 1989). Therefore, more input from other sources is needed to initiate inspiration. A modest reduction of CO2-related drive could more easily lead to hypopnoea/apnoea and would be manifested as a higher PCO2 threshold.
Hypoxic respiratory depression may also be related to central alkalosis due to cerebral vasodilatation and increased cerebral blood flow (Cherniack et al. 1970; Berkenbosch & DeGoede, 1988). Since central chemoreceptors have a stabilizing effect on respiratory activity, any reduction in the activity of the central chemoreceptors tends to increase the susceptibility to periodic breathing (Khoo et al. 1991). Peripheral hypocapnia alone can cause a reduction in tidal volume amplitude, but is not sufficient to trigger apnoea as long as the central chemoreceptors are active (Smith et al. 1997). The hypoxia-induced cerebral vessel dilatation may lower the brain PCO2 and thereby, reduce the difference between brain and arterial PCO2. As a result, a higher arterial and end-tidal PCO2 would be required during hypoxia to maintain the same level of cerebrospinal fluid pH and PCO2 as during normoxia. Consequently, a higher hypopnoea/apnoea threshold would be observed during hypoxia. Given the relatively mild level of hypoxaemia imposed in our study (PET,O2≈54 mmHg), we think it unlikely that cerebral blood flow or the arterial-to-CSF PCO2 change was significantly affected (Dempsey et al. 1974).
Finally, the higher hypopnoea/apnoeic threshold during hypoxia may reflect a hypoxia-induced defect in short-term potentiation in the respiratory control system (Badr et al. 1992). Short-term potentiation stabilizes ventilation following the withdrawal of any ventilatory stimuli. Hypocapnia has a powerful inhibitory influence even in the presence of a normal level of short-term potentiation. During hypoxia, the impairment in short-term potentiation may cause hypopnoea/apnoea to occur following a smaller reduction in PCO2, which is manifested by a higher hypopnoea/apnoea threshold.
All the aforementioned effects of hypoxia will make the ventilatory control system more sensitive to inhibitory afferents in general, including the effect of neuromechanical inhibition. As a result, the respiratory control system would be expected to become unstable and to demonstrate excessive reductions in tidal volume at levels of PCO2 that would not produce instability during other types of ventilatory stimulation. This effect would be manifested as the failure to lower the apnoeic threshold in proportion to the lowering of the eupnoeic PCO2. This enhanced effect of non-PCO2-related inhibitory influences may also be the reason why we did not see the statistically significant reduction in the pre-hypopnoeic PCO2 during hypoxia that we did during normoxia.
Consequences of elevated hypopnoea/apnoeic threshold
The close proximity of the eupnoeic PCO2 and the hypopnoea/apnoeic threshold has important implications for the development of periodic breathing during hypoxia. Our findings confirmed the destabilizing effect of hypoxia on breathing pattern by showing the development of periodic breathing at a lower pressure- support level, a smaller increase in VT, and a smaller reduction in PET,CO2. Even during spontaneous breathing, hypoxia was associated with a greater degree of breathing instability compared to normoxia. How can the observed change in the relationship between eupnoea and the threshold PCO2 account for the greater degree of ventilatory instability?
The mechanism whereby the narrowed difference between the eupnoeic and threshold PCO2 can increase the likelihood of periodic breathing during hypoxia can be considered as an increase in the sensitivity of the ventilatory response to chemical stimuli below the eupnoeic range of PCO2. Control system theory indicates that ventilatory instability is enhanced under conditions of increased sensitivity of the peripheral chemoreceptors (Lahiri et al. 1983; Carley et al. 1988; Khoo et al. 1991). An appropriate sensitivity is important in maintaining blood gas homeostasis and ventilatory stability. Too high a sensitivity may be associated with central apnoeas because a small reduction in chemical stimuli produces a large reduction in ventilation (Khoo et al. 1991; Xie et al. 1995; Solin et al. 2000). Previous studies have shown that hypoxia increases the ventilatory response to CO2 above eupnoea (Cunningham et al. 1986). Our study provides unique data concerning the ventilatory response to reductions in PCO2 below eupnoea during normoxia and hypoxia. During hypoxia, the smaller difference in PCO2 between eupnoea and apnoeic threshold indicates a steeper slope of the ventilatory response to CO2 below eupnoea (see Fig. 4). Few previous studies have investigated this response in sleeping humans. Our value of 1.7 l min−1 mmHg−1 during normoxia is similar to the 1.56-1.87 l min−1 mmHg−1 previously reported (Henke et al. 1988; Meza et al. 1998). During hypoxia, our data showed that the slope was increased substantially (5.5 l min−1 mmHg−1). A similar effect of hypoxia on the ventilatory response to CO2 below eupnoea was also found in our previous work where the slope increased from 1.48 l min−1 mmHg−1 during normoxia to 3.10 l min−1 mmHg−1 during hypoxia (Skatrud & Dempsey, 1983). As a direct result of this increase in slope below eupnoea, a similar decrease in PCO2 will produce a much greater reduction in ventilation during hypoxia compared to normoxia. Thus, hypoxia enhances the susceptibility to periodic breathing by decreasing the amount of hyperventilation required to produce apnoea.
Figure 4. Ventilatory response to CO2 below the eupnoeic PCO2.
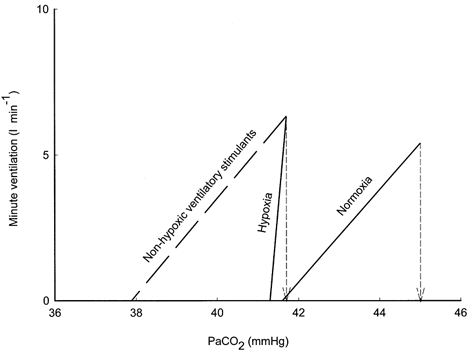
The continuous lines were drawn between the point of eupnoeic ventilation at eupnoeic PCO2 and the point of PCO2 at the apnoeic threshold PCO2 using data from the present study during normoxia and hypoxia. The slope of the ventilatory response below eupnoeic PCO2 was greater during hypoxia compared to normoxia. The dashed line represents a hypothetical non-hypoxic ventilatory stimulant that lowered the eupnoeic PCO2 to the same extent as we observed during hypoxia, but did not change the slope of the ventilatory response below eupnoea compared to room air breathing. Note that a much smaller reduction in PCO2 is required to produce apnoea during hypoxia compared to the non-hypoxic ventilatory stimulant.
The arterial PCO2 represents only one of the many inputs to respiratory rhythm generation. Thus, the apnoeic threshold is changeable and varies according to other afferent inputs (Boden et al. 1998). This may explain why other ventilatory stimulants that lower the eupnoeic PCO2 do not enhance ventilatory instability but may actually stabilize the breathing pattern. For example, patients with pulmonary fibrosis have a low Pa,O2 and Pa,CO2 but do not have an abnormal degree of periodic breathing during sleep (Perez-Padilla et al. 1985). Pharmacological ventilatory stimulants such as theophylline, acetazolamide and progesterone have a stabilizing effect on ventilation during sleep despite a reduction in eupnoeic Pa,CO2 (DeBaker et al. 1995; Javaheri et al. 1996; Hudgel & Thankitcharu, 1998). The propensity of these ventilatory stimuli to stabilize the breathing pattern during sleep may be due to a proportional reduction in the eupnoeic PCO2 and apnoeic threshold. For example, if we assume that the non-hypoxic ventilatory stimulants have a sensitivity to the reduction of PCO2 below eupnoea that is similar to normoxia, then the unchanged slope is associated with an increased difference between eupnoea and the apnoeic threshold (Fig. 4). Thus, the stabilizing influence of non-hypoxic ventilatory stimuli may be related to the decreased propensity to lower PCO2 to the apnoeic threshold following periods of transient hyperpnoea. Alternatively, an unusually low level of eupnoeic PCO2 would require a large change in alveolar ventilation to further reduce the PCO2 towards the apnoeic threshold. In any case, our hypoxic data indicate the importance of a reduced difference between the eupnoeic and threshold PCO2 rather than the absolute level of the eupnoeic PCO2 in predicting the susceptibility to breathing instability.
Critique of methods
The use of mechanical ventilation to determine the apnoea/hypopnoeic threshold is associated with several pitfalls. First, the premature delivery of a ventilator breath could pre-empt spontaneous respiratory rhythm and cause TE prolongation independently of PCO2 (Knox & King, 1976; Satoh et al. 2001). However, our use of the subject-assisted, pressure-support mode, assured that the subject initiated the inspiratory activity and precluded the possibility of pre-emption of spontaneous breaths. Second, the downregulation of inspiratory muscle activity by neuromechanical inhibition can occur independently of hypocapnic inhibition (Wilson et al. 1999). Vagal or other inhibitory influences related to the mechanical ventilation could have contributed to the observed hypopnoea and apnoea, especially during hypoxia, when the Pa,CO2 of our subjects was already close to the hypocapnic apnoeic threshold. Our methodology minimized this effect since the use of pressure-support ventilation, with a gradual increase in the pressure levels, ensured that only the minimal increase in tidal volume was used to cause a reduction in PCO2 and that only the minimal positive pressure was supplied to produce hyperventilation. Since plots of ventilation measurements against pressure show that ventilation responses at a given level of pressure support are similar in hypoxia and normoxia trials, comparable pressure-support effects and neuromechanical influences are expected in hypoxia vs. normoxia trials. In fact, pressure-support ventilation may have produced even less neuromechanical inhibition during hypoxia compared to normoxia, since lower pressures were used and smaller increases in tidal volume were required during hypoxia. Therefore, the greater susceptibility to hypopnoea and apnoea during hypoxia cannot be attributed to a disproportionate degree of neuromechanical inhibition during hypoxia, unless hypoxia increased the subject's sensitivity to neuromechanical inhibition as discussed above.
End-tidal PCO2 was used as a non-invasive surrogate for arterial PCO2. We have previously demonstrated a predictable relationship between end-tidal and arterial PCO2 during hypoxia (Badr et al. 1992) and mechanical ventilation (Wilson et al. 1999). Since PET,CO2 can only be measured during the ventilatory phase, we used the three breaths immediately preceding the hypopnoea or apnoea as indicative of the hypopnoea/apnoeic threshold PCO2. We did not find a difference in PET,CO2 among the three breaths prior to the hypopnoea/apnoea. The reproducibility of hypopnoea/apnoeic threshold within a subject confirmed that the PET,CO2 was a good representation of the threshold PCO2. Although a systematic error is still possible, it should not affect the difference between eupnoeic PCO2 and threshold PCO2, which was the measurement of interest.
The use of zolpidem was necessary to facilitate sleep and reduce arousability in the laboratory. Zolpidem has been shown to have no effect on ventilation, oxygen saturation or breathing stability during normoxic or hypoxic sleep (Beaumont et al. 1996). However, its effect on the apnoeic threshold is unknown. Even if zolpidem did change the threshold, the effect would be present in both normoxic and hypoxic conditions and thus, would not change our conclusions.
Conclusions
Hypoxia distinguishes itself from other respiratory stimulants by showing a decrease in the proximity between the eupnoeic PCO2 and the hypopnoea/apnoeic threshold PCO2 as a result of the failure to reduce the threshold PCO2 in a parallel manner to the reduction in eupnoeic PCO2. The relatively high apnoeic threshold may reflect impairment of the ventilatory control system by hypoxia. Consequently, a greater level of afferent input from the periphery, as manifested by a higher apnoeic threshold for PCO2, is required to maintain rhythmic breathing. The combination of a low PCO2 and a relatively high threshold makes it easier for transient reductions of Pa,CO2 to reach the apnoeic threshold and cause breathing instability during sleep.
Acknowledgments
This work was supported by the VA Research Service, and NIH R01 HL62561-02. Dr Skatrud is the recipient of an NIH Sleep Academic Award. Technical assistance was provided by Dominic S. Puleo and Kristine J. Kimmler.
References
- Badr M S, Skatrud J B, Dempsey J. Determinants of poststimulus potentiation in humans during NREM sleep. Journal of Applied Physiology. 1992;73:1958–1971. doi: 10.1152/jappl.1992.73.5.1958. [DOI] [PubMed] [Google Scholar]
- Beaumont M, Goldenberg F, Lejeune D, Marotte H, Harf A, Lofaso F. Effect of zolpidem on sleep and ventilatory patterns at simulated altitude of 4,000 meters. American Journal of Respiratory and Critical Care Medicine. 1996;153:1864–1869. doi: 10.1164/ajrccm.153.6.8665047. [DOI] [PubMed] [Google Scholar]
- Berkenbosch A, Degoede J. Effects of brain hypoxia on ventilation. European Respiratory Journal. 1988;1:184–190. [PubMed] [Google Scholar]
- Berssenbrugge A, Dempsey J, Iber C, Skatrud J, Wilson P. Mechanisms of hypoxia-induced periodic breathing during sleep in humans. Journal of Physiology. 1983;343:507–524. doi: 10.1113/jphysiol.1983.sp014906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden A G, Harris M C, Parkes M J. Apnoeic threshold for CO2 in the anaesthetized rat: fundamental properties under steady-state conditions. Journal of Applied Physiology. 1998;85:898–907. doi: 10.1152/jappl.1998.85.3.898. [DOI] [PubMed] [Google Scholar]
- Carley D W, Shannon D C. A minimal mathematical model of human periodic breathing. Journal of Applied Physiology. 1988;65:1400–1409. doi: 10.1152/jappl.1988.65.3.1400. [DOI] [PubMed] [Google Scholar]
- Cherniack N S, Edelman N H, Lahiri S. Hypoxia and hypercapnia as respiratory stimulants and depressants. Respiratory Physiology. 1970;11:113–126. doi: 10.1016/0034-5687(70)90107-6. [DOI] [PubMed] [Google Scholar]
- Cunningham D J C, Robbins P A, Wolff C B. Integration of respiratory responses to changes in alveolar partial pressures of CO2 and O2 and in arterial pH. In: Fishman A P, Cherniack N A, Widdicombe J G, Geiger S R, editors. Handbook of Physiology, section 3, The Respiratory System, vol. II, Control of Breathing. Bethseda: MD USA American Physiological Society; 1986. pp. 475–528. [Google Scholar]
- Curran A K, Rodman J R, Eastwood P R, Henderson K S, Dempsey J A, Smith C A. Ventilatory responses to specific CNS hypoxia in sleeping dogs. Journal of Applied Physiology. 2000;88:1840–1852. doi: 10.1152/jappl.2000.88.5.1840. [DOI] [PubMed] [Google Scholar]
- Datta A K, Shea S A, Horner R L, Guz A. The influence of induced hypocapnia and sleep on the endogenous respiratory rhythm in humans. Journal of Physiology. 1991;440:17–33. doi: 10.1113/jphysiol.1991.sp018693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debacker W A, Verbraecken J, Willemen M, Wittesaele W, Decock W, Vande Heyning P. Central apnoea index decreases after prolonged treatment with acetazolamide. American Journal of Respiratory and Critical Care Medicine. 1995;151:87–91. doi: 10.1164/ajrccm.151.1.7812578. [DOI] [PubMed] [Google Scholar]
- Dempsey J A, Forster H V, Dopico G A. Ventilatory acclimatization to moderate hypoxemia in man. The role of spinal fluid (H+) Journal of Clinical Investigation. 1974;53:1091–1100. doi: 10.1172/JCI107646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink B R. Influence of cerebral activity in wakefulness on regulation of breathing. Journal of Applied Physiology. 1961;16:15–29. doi: 10.1152/jappl.1961.16.1.15. [DOI] [PubMed] [Google Scholar]
- Hackett P H, Roach R C, Harrison G L, Schoene R B, Mills W J JR. Respiratory stimulants and sleep periodic breathing at high altitude. Almitrine versus acetazolamide. American Journal of Respiratory and Critical Care Medicine. 1987;135:896–898. doi: 10.1164/arrd.1987.135.4.896. [DOI] [PubMed] [Google Scholar]
- Henke K G, Arias A, Skatrud J B, Dempsey J A. Inhibition of inspiratory muscle activity during sleep. Chemical and nonchemical influences. American Review of Respiratory Disease. 1988;138:8–15. doi: 10.1164/ajrccm/138.1.8. [DOI] [PubMed] [Google Scholar]
- Hudgel D W, Thanakitcharu S. Pharmacologic treatment of sleep-disordered breathing. American Journal of Respiratory and Critical Care Medicine. 1998;158:691–699. doi: 10.1164/ajrccm.158.3.9802019. [DOI] [PubMed] [Google Scholar]
- Javaheri S, Parker T J, Wexler L, Liming L D, Lindower P, Roselle G A. Effect of theophylline on sleep disordered breathing in heart failure. New England Journal of Medicine. 1996;335:562–567. doi: 10.1056/NEJM199608223350805. [DOI] [PubMed] [Google Scholar]
- Khoo M C K, Gottschalk A, Pack A I. Sleep-induced breathing and apnoea: a theoretical study. Journal of Applied Physiology. 1991;70:2014–2024. doi: 10.1152/jappl.1991.70.5.2014. [DOI] [PubMed] [Google Scholar]
- Knox C K, King G W. Changes in the Breuer-Hering reflexes following rostral pontine lesion. Respiratory Physiology. 1976;28:189–206. doi: 10.1016/0034-5687(76)90038-4. [DOI] [PubMed] [Google Scholar]
- Lahiri S, Maret K, Sherpa M G. Dependence of high altitude sleep apnoea on ventilatory sensitivity to hypoxia. Respiratory Physiology. 1983;52:281–301. doi: 10.1016/0034-5687(83)90086-5. [DOI] [PubMed] [Google Scholar]
- LaManna J C, Haxhiu M A, Kutina-Nelson K L, Pundik S, Erokwu B, Yeh E R, Lust W D, Cherniack N S. Decreased energy metabolism in brain stem during central respiratory depression in response to hypoxia. Journal of Applied Physiology. 1996;81:1772–1777. doi: 10.1152/jappl.1996.81.4.1772. [DOI] [PubMed] [Google Scholar]
- Meza S, Mendez M, Ostrowski M, Younes M. Susceptibility to periodic breathing with assisted ventilation during sleep in normal subjects. Journal of Applied Physiology. 1998;85:1929–1940. doi: 10.1152/jappl.1998.85.5.1929. [DOI] [PubMed] [Google Scholar]
- Mironov S L, Langohr K, Haller M, Richter D W. Hypoxia activates ATP-dependent potassium channels in inspiratory neurones of neonatal mice. Journal of Physiology. 1998;509:755–766. doi: 10.1111/j.1469-7793.1998.755bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsui T, Kuwana S. Respiratory depression following acute hypoxia in vagotomized and carotid sinus nerve denervated cats. In: Honda Y, Mimamoto Y, Konno K, Widdicombe J G, editors. Control of Breathing and its Modelling Perspective. New York and London: Plenum Press; 1992. pp. 311–314. [Google Scholar]
- Neubauer J A, Melton J E, Edelman H H. Modulation of respiration during brain hypoxia. Journal of Applied Physiology. 1990;68:441–449. doi: 10.1152/jappl.1990.68.2.441. [DOI] [PubMed] [Google Scholar]
- Perez-Padilla R, West P, Lertzman M, Kryger M H. Breathing during sleep in patients with interstitial lung disease. American Review of Respiratory Disease. 1985;132:224–229. doi: 10.1164/arrd.1985.132.2.224. [DOI] [PubMed] [Google Scholar]
- Ramirez J M, Quellmalz U J, Wilken B, Richter D W. The hypoxic response of neurones within the in vitro mammalian respiratory network. Journal of Physiology. 1998;507:571–582. doi: 10.1111/j.1469-7793.1998.571bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechtschaffen A, Kales A. A Manual for Standardized Terminology, Techniques and Scoring System for Sleep Stages of Human Subjects. Washington, DC: National Institutes of Health; 1968. [Google Scholar]
- Richter D W, Schmidt-Garcon P, Pierrefiche O, Bischoff A M, Lalley P M. Neurotransmitters and neuromodulators controlling the hypoxic respiratory response in anaesthetized cats. Journal of Physiology. 1999;514:567–578. doi: 10.1111/j.1469-7793.1999.567ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runold M H, Lagercrantz H, Prabhakar N R, Fredholm B B. Role of adenosine in hypoxic ventilatory depression. Journal of Applied Physiology. 1989;65:541–546. doi: 10.1152/jappl.1989.67.2.541. [DOI] [PubMed] [Google Scholar]
- Satoh M, Eastwood P R, Smith C A, Dempsey J A. Non-chemical elimination of inspiratory motor output via mechanical ventilation in sleep. American Journal of Respiratory and Critical Care Medicine. 2001;163:1356–1364. doi: 10.1164/ajrccm.163.6.2004169. [DOI] [PubMed] [Google Scholar]
- Schaeffer J I, Haddad G G. Ventilatory response to moderate and severe hypoxia in adult dogs: Role of endorphins. Journal of Applied Physiology. 1988;65:1383–1388. doi: 10.1152/jappl.1988.65.3.1383. [DOI] [PubMed] [Google Scholar]
- Schmidt C, Bellingham M C, Richter D W. Adenosinergic modulation of respiratory neurons and hypoxic responses in the anaesthetized cat. Journal of Physiology. 1995;483:769–781. doi: 10.1113/jphysiol.1995.sp020621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skatrud J B, Dempsey J A. Interaction of sleep state and chemical stimuli in sustaining rhythmic ventilation. Journal of Applied Physiology. 1983;55:813–822. doi: 10.1152/jappl.1983.55.3.813. [DOI] [PubMed] [Google Scholar]
- Skatrud J B, Dempsey J A, Iber C, Berssenbrugge A. Correction of CO2 retention during sleep in patients with chronic obstructive pulmonary diseases. American Review of Respiratory Disease. 1981;124:260–268. doi: 10.1164/arrd.1981.124.3.260. [DOI] [PubMed] [Google Scholar]
- Smith C A, Harms C A, Henderson K S, Dempsey J A. Ventilatory effects of specific carotid body hypoxia and hypocapnia in the awake dog. Journal of Applied Physiology. 1997;82:791–798. doi: 10.1152/jappl.1997.82.3.791. [DOI] [PubMed] [Google Scholar]
- Solin P, Roebuck T, Johns D P, Haydn Walters E, Naughton M T. Peripheral and central ventilatory responses in central sleep apnea with and without congestive heart failure. American Journal of Respiratory and Critical Care Medicine. 2000;162:2194–2200. doi: 10.1164/ajrccm.162.6.2002024. [DOI] [PubMed] [Google Scholar]
- Trippenbach T, Richter D W, Acker H. Hypoxia and ion activities within the brain stem of newborn rabbits. Journal of Applied Physiology. 1990;68:2494–2503. doi: 10.1152/jappl.1990.68.6.2494. [DOI] [PubMed] [Google Scholar]
- White D P, Zwillich C W, Pickett C K, Douglas N J, Fidley M D, Weil J V. Central sleep apnoea: improvement with acetazolamide therapy. Archives of Internal Medicine. 1982;142:1816–1819. [PubMed] [Google Scholar]
- Wilson C R, Satoh M, Skatrud J B, Dempsey J A. Non-chemical inhibition of respiratory motor output during mechanical ventilation in sleeping humans. Journal of Physiology. 1999;518:605–618. doi: 10.1111/j.1469-7793.1999.0605p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie A, Rutherford R, Rankin F, Wong B, Bradley T D. Hypocapnia and increased ventilatory responsiveness in patients with idiopathic central sleep apnoea. American Journal of Respiratory and Critical Care Medicine. 1995;152:1950–1955. doi: 10.1164/ajrccm.152.6.8520761. [DOI] [PubMed] [Google Scholar]
- Younes M. The physiologic basis of central apnoea and periodic breathing. Current Pulmonology. 1989;10:265–326. [Google Scholar]