

Abstract
We tested the hypothesis that nitric oxide (NO) augments vagal neurotransmission and bradycardia via phosphorylation of presynaptic calcium channels to increase vesicular release of acetylcholine.
The effects of enzyme inhibitors and calcium channel blockers on the actions of the NO donor sodium nitroprusside (SNP) were evaluated in isolated guinea-pig atrial-right vagal nerve preparations.
SNP (10 μm) augmented the heart rate response to vagal nerve stimulation but not to the acetylcholine analogue carbamylcholine (100 nm). SNP also increased the release of [3H]acetylcholine in response to field stimulation. No effect of SNP was observed on either the release of [3H] acetylcholine or the HR response to vagal nerve stimulation in the presence of the guanylyl cyclase inhibitor 1H-(1,2,4)-oxadiazolo-(4,3-a)-quinoxalin-1-one (ODQ, 10 μm).
The phosphodiesterase 3 (PDE 3) inhibitor milrinone (1 μm) increased the release of [3H] acetylcholine and the vagal bradycardia and prevented any further increase by SNP. SNP was still able to augment the vagal bradycardia in the presence of the protein kinase G inhibitor KT5823 (1 μm) but not after protein kinase A (PKA) inhibition with H-89 (0.5 μm) or KT5720 (1 μm) had reduced the HR response to vagal nerve stimulation. Neither milrinone nor H-89 changed the HR response to carbamylcholine.
SNP had no effect on the magnitude of the vagal bradycardia after inhibition of N-type calcium channels with ω-conotoxin GVIA (100 nm).
These results suggests that NO acts presynaptically to facilitate vagal neurotransmission via a cGMP-PDE 3-dependent pathway leading to an increase in cAMP-PKA-dependent phosphorylation of presynaptic N-type calcium channels. This pathway may augment the HR response to vagal nerve stimulation by increasing presynaptic calcium influx and vesicular release of acetylcholine.
The role of the nitric oxide (NO)-cGMP pathway in the cholinergic modulation of cardiac pacemaking is controversial. Whilst some groups have shown that inhibition of endothelial nitric oxide synthase (eNOS), either pharmacologically (Han et al. 1994, 1995; Balligand, 1999) or via gene knockout (Han et al. 1998b), abolishes the reduction in L-type calcium current (ICa,L) in response to acetylcholine after prior adrenergic stimulation, others have not observed this (Vandecasteele et al. 1998, 1999; Belevych & Harvey, 2000). In addition, NO can directly stimulate heart rate (HR) by increasing the hyperpolarization-activated inward current (If) (Musialek et al. 1997). Postsynaptically, the functional significance of NO modulation of HR is therefore complicated by the interplay and opposing actions of the NO-cGMP pathway on these two pacemaking currents (Sears et al. 1998a).
Presynaptically, the neuronal isoform of NOS (nNOS) has been identified in parasympathetic ganglion innervating the sino-atrial node (Klimaschewski et al. 1992). Inhibition of nNOS (Conlon & Kidd, 1999; Herring et al. 2000) or guanylyl cyclase (Herring et al. 2000) reduces the HR response to vagal nerve stimulation. Some studies report no effect of NOS inhibition during vagal stimulation (Liu et al. 1996; Sears et al. 1998b), although this may be related to the developmental stage of the animal not expressing significant levels of nNOS (Herring et al. 2000). Amplification of the NO-cGMP pathway with NO donors or the membrane-permeant analogue of cGMP, 8-bromo-cGMP (8-Br-cGMP), increases the HR response to vagal nerve stimulation both in vitro and in vivo (Sears et al. 1999). However, no effect of nNOS inhibition (Herring et al. 2000) or 8-Br-cGMP (Sears et al. 1999) was observed on the HR response to the stable analogue of acetylcholine, carbamylcholine, suggesting that the main functional action of NO is presynaptic. One possibility is that the NO-cGMP pathway enhances vagal neurotransmission by increasing the release, or synthesis and vesicular storage, of acetylcholine. The rapid increase in the HR response to vagal nerve stimulation caused by NO donors (Sears et al. 1999) suggests that an effect on transmitter release is the more likely. NO has been implicated in the increase in the release of acetylcholine observed in the rat forebrain (Prast & Philippu, 1992) and in isolated synaptosomes (Morot Gaudry-Talarmain et al. 1997). It can also stimulate calcium currents in cardiac myocytes by raising cAMP via inhibition of phosphodiesterase 3 (PDE 3) (Ono & Trautwein, 1991). Moreover, both 8-Br-cAMP (Dawson et al. 1996) and pituitary adenylate cyclase-activating protein (PACAP) (Seebeck et al. 1996) can increase the release of radioactively labelled acetylcholine in isolated atria. This might conceivably occur via an increase in cAMP-dependent protein kinase A (PKA) phosphorylation of presynaptic calcium channels, thereby increasing calcium influx and promoting exocytotic release of transmitter.
Therefore we tested two hypotheses. Firstly, does NO, via guanylyl cyclase, augment vagal-induced bradycardia by facilitating the release of acetylcholine? Secondly, is this achieved via a presynaptic PDE 3 pathway leading to an increase in cAMP-PKA-dependent phosphorylation of presynaptic calcium channels?
METHODS
Experiments conformed with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health, USA (NIH Publication No. 85-23, revised 1996) and the Animals (Scientific Procedures) Act 1986 (UK). Experiments were performed under British Home Office Project Licence PPL 30/1133.
Isolated guinea-pig sino-atrial node-right vagus nerve preparation
Adult (500-700 g) female guinea-pigs (Cavia porcellus, Dunkin Hartley, n = 69) were killed by cervical dislocation and exsanguinated. The thorax was opened and the ventricles were removed so that heparinized (1000 U ml−1) Tyrode solution could be rapidly perfused into both atria. The heart was removed with the rib cage and mediasternum and placed in a dissection dish containing Tyrode solution aerated with 95 % O2-5 % CO2 at room temperature. Any remaining ventricle and both lungs were carefully removed and the atria and mediasternum dissected free from the thorax. The right vagus was carefully separated from the carotid artery and tied. Sutures (Ethicon 5/0 silk) were placed at the lateral edges of the two atria. The preparation was then transferred to a preheated (37 ± 0.2 °C), continuously oxygenated, water-jacketed organ bath containing 60 ml Tyrode solution. The atria were mounted vertically with the suture in the left atrium attached to a stainless-steel hook and that in the right atrium attached to an isometric force transducer (HDE F30) connected to an amplifier. Data were acquired on a Power Macintosh 8500 computer using a Biopac Systems MP100 data acquisition system and Acqknowledge 3.5 software. The beating rate was triggered from contraction, and the signals were displayed in real time. Data were stored on optical disk for off-line analysis.
Before the start of each protocol, the mounted atria were kept in Tyrode solution for at least 60 min until their beating rate stabilized (±5 beats min−1 over 20 min). The Tyrode solution in the organ bath was replaced approximately every 30 min throughout each protocol. The vagus nerve was stimulated at 3 or 5 Hz, 10-15 V, 1 ms pulse duration for 30 s, until three consistent responses were obtained. We have previously shown that all heart rate changes from vagal nerve stimulation are completely abolished by hyoscine in this preparation and are therefore due to the release of acetylcholine (Sears et al. 1998a). A control experiment showed that the HR response to vagal nerve stimulation remained constant (±1 beat min−1 at 5 Hz) over a 2 h period. Drugs were applied directly to the organ bath and incubation was continued until a consistent HR response to vagal nerve stimulation was obtained.
Measurement of [3H] acetylcholine release in response to field stimulation from isolated guinea-pig right atrial preparations
Adult (400-500 g) female guinea pigs (n = 12) were killed and the right atria removed as described above. The preparation was then transferred to a preheated (37 ± 0.2 °C), continuously oxygenated, water-jacketed organ bath containing 2 ml Tyrode solution where the atrium was pinned flat between two parallel silver stimulating electrodes 10 mm apart. The methods for radiolabelling cholinergic transmitter stores so that acetylcholine release could be quantified were similar to those originally devised by Wetzel and colleagues (Wetzel & Brown, 1983; Wetzel et al. 1985) and subsequently modified by Seebeck et al. (1996). After a 30 min equilibration period (during which the Tyrode solution was replaced every 15 min), the atrium was stimulated at 10 Hz (20 V, 1 ms pulse duration) for 1 min and then again after another minute to stimulate acetylcholine turnover. The preparation was incubated for 30 min with [3H] choline chloride (5 μCi, Amersham, UK), during which period the atrium was stimulated at 10 Hz for 10 s every 30 s to allow incorporation of the [3H] choline into the parasympathetic transmitter stores. Tyrode solution containing 50 μm hemicholinium 3 was used after the incubation period to reduce re-uptake of radioactively labelled transmitter. Excess [3H] choline was washed from the preparation by superfusion with Tyrode solution for 30 min at a rate of 2 ml min−1. Superfusion was then stopped and the bath solution replaced every 3 min, with a 0.5 ml sample being taken at each change of solution. Each sample was added to 4.5 ml scintillation fluid (Ecoscint A, National Diagnostics) and the amount of radioactivity (disintegrations per minute) was measured using a liquid scintillation counter (Tri-carb 2000CA, Packard). After 28 min and again after 43 min the atrium was stimulated at 10 Hz for 1 min. At the end of the experiment, the atrium was immersed overnight in ∼4 units ml−1 papain and the radioactivity contained in the extract determined. 3H outflow was expressed as a percentage of the total radioactivity in the atrium at the end of the experiment and that released after superfusion.
Solutions and drugs
Tyrode solution contained (mm): NaCl, 120; KCl, 4; MgCl2, 2; NaHCO3, 25; CaCl2, 2; Na2HPO4, 0.1; and glucose, 11. The solution was aerated with 95 % O2-5 % CO2 (pH 7.4) and the temperature was continuously monitored (Digitron 1408-K gauge) and kept at 37 ± 0.2 °C.
NOS inhibition produces only modest reductions in the magnitude of the vagal bradycardia depending on the expression of the nNOS enzyme (Herring et al. 2000). We therefore investigated the intracellular pathway by which NO acts by using increasing concentrations of a NO donor in the presence of various enzyme inhibitors. Sodium nitroprusside (SNP, 10 and 100 μm; Sigma) was used as the NO donor; it has previously been shown to increase the HR response to vagal nerve stimulation in a similar preparation (Sears et al. 1999). 1H-(1,2,4)-oxadiazolo-(4,3-a)-quinoxalin-1-one (ODQ, 10 μm, 40 min incubation; Tocris Cookson, UK) was used to inhibit guanylyl cyclase. The concentration of ODQ used has also been shown to produce full block of the isolated enzyme (Garthwaite et al. 1995) and to reduce the HR response to vagal nerve stimulation in vitro (Herring et al. 2000). Erythro-9-(2-hydroxy-3-nonyl)-adenine (EHNA, 10 μm, 20 min incubation; Sigma) was used to inhibit PDE 2, at a concentration that has previously been shown to be effective (Mery et al. 1995; Han et al. 1998a). Milrinone (1 μm, 20 min incubation; Calbiochem) was used as a potent and selective inhibitor of PDE 3. Since milrinone increases baseline heart rate by raising cAMP and stimulating If in sino-atrial node cells (DiFrancesco & Tortora, 1991), these experiments were repeated in the presence of the If blocker caesium chloride (Denyer & Brown, 1990) (2 mm, 20 min incubation; Sigma). PKA was inhibited using H-89 (0.5 μm, 20 min incubation; Calbiochem) or KT5720 (1 μm, 20 min incubation; Calbiochem) and protein kinase G (PKG) using KT5823 (1 μm, 20 min incubation; Calbiochem). The concentrations used were above the reported Ki values for the isolated enzymes (0.3 μm milrinone for PDE 3 (Harrison et al. 1986), 0.05 μm H-89 for PKA (Chijiwa et al. 1990), 0.06 μm KT5720 for PKA and 0.2 μm KT5823 for PKG (Kase et al. 1987)), but below those reported for non-specific actions of the drugs. The effects of these inhibitors on the HR response to vagal nerve stimulation were also compared with those of the stable analogue of acetylcholine, carbamylcholine chloride (100 nm; Sigma), to determine whether their likely effects on the vagal modulation of HR were pre- or postsynaptic. Presynaptic neuronal N-type calcium channels were blocked with ω-conotoxin GVIA (100 nm, 20 min incubation; Sigma) and P-type calcium channels with ω-agatoxin-TK (50 nm, 20 min incubation; Sigma). Both ω-conotoxin GVIA (Kerr & Yoshikami, 1984; Olivera et al. 1984) and ω-agatoxin-TK (Teramoto et al. 1993) produce block of their respective channels at nanomolar concentrations. The muscarinic M4 receptor was blocked using tropicamide (0.2 μm, 20 min incubation; Sigma). This concentration of tropicamide has previously been used to produce block of the receptor in isolated myocytes (Shi et al. 1999).
Drugs were dissolved in reagent grade water from an Elga purification system with the exception of ODQ, milrinone, EHNA, H-89, KT5720 and KT5823, which were dissolved in dimethylsulfoxide (DMSO). A control experiment showed that DMSO at the concentrations used did not effect the HR response to vagal nerve stimulation at 1, 3 or 5 Hz. As SNP is light sensitive, all experiments were carried out in a darkened room.
Statistical analysis
Data are presented as means ±s.e.m. One-way repeated measures ANOVA followed by Tukey's post hoc analysis was used to evaluate the effect of an intervention. Student's unpaired t test was used to evaluate the effect of an intervention between experimental groups. Statistical significance was accepted at P < 0.05. All data passed a normality test.
RESULTS
After the equilibration period, mean baseline HR stabilized at 170 ± 3 beats min−1 (n = 69) in the spontaneously beating double atrial preparation.
The effects of NO on the HR response to vagal nerve stimulation and the evoked release of [3H] acetylcholine
The NO donor SNP (10 μm, n = 7) significantly increased the HR response to vagal nerve stimulation at 5 Hz (Fig. 1A and B) but not at 3 Hz (ΔHR: control, -32 ± 4 beats min−1; + 10 μm SNP, -37 ± 6 beats min−1; wash, -35 ± 5 beats min−1). However, higher concentrations of SNP (100 μm, n = 6) were able to significantly augment the vagal bradycardia at 3 Hz (ΔHR: control, -28 ± 1 beats min−1; + 100 μm SNP -40 ± 4 beats min−1; wash, -27 ± 3 beats min−1) and 5 Hz stimulation frequencies (ΔHR: control, -49 ± 3 beats min−1; + 100 μm SNP, -63 ± 4 beats min−1; wash, -44 ± 3 beats min−1). SNP (10 μm, n = 8) had no effect on the HR response to bath-applied carbamylcholine suggesting that the action of NO is presynaptic (ΔHR: control, -62 ± 3 beats min−1; 10 μm SNP, -67 ± 3 beats min−1; wash, -61 ± 3 beats min−1). In these experiments, SNP significantly increased baseline HR (ΔHR: 10 μm SNP, +41 ± 4 beats min−1, n = 15; 100 μm SNP, +57 ± 8 beats min−1, n = 6) due to postsynaptic stimulation of If (Musialek et al. 1997). We have previously shown that the increase in the magnitude of the vagal bradycardia to NO donors is independent of the shift in baseline HR, as it still occurs when the HR is held constant by blocking If (Sears et al. 1999). The fact that SNP had no effect on the HR response to carbamylcholine despite the increase in baseline HR supports this observation. To investigate whether NO augments vagal neurotransmission by increasing the release of acetylcholine, we radioactively labelled right atrial acetylcholine stores and measured the increase in 3H efflux following field stimulation. SNP (10 μm, n = 4) significantly increased the evoked release of [3H] acetylcholine in response to field stimulation (Fig. 1C and D).
Figure 1. SNP facilitates vagal bradycardia via an increase in acetylcholine release.
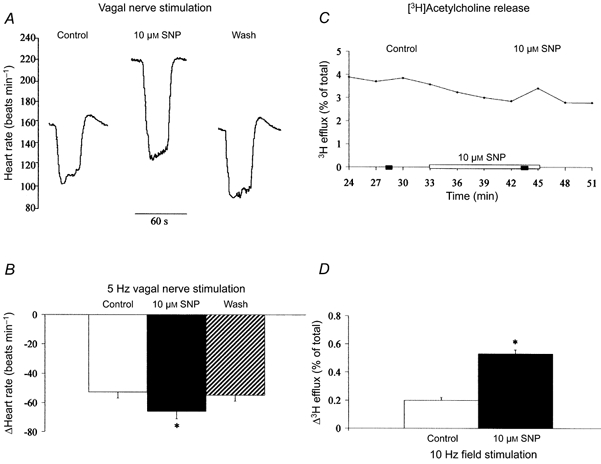
A and B, SNP (10 μm, n = 7) significantly increased (*P < 0.05) the heart rate response to right vagal nerve stimulation (5 Hz) and this effect was reversed on washout of the drug. We have previously shown that the increase in the magnitude of vagal bradycardia with SNP is independent of the postsynaptic stimulation of If and increase in baseline heart rate. C and D, SNP (10 μm, n = 4) also significantly (*P < 0.05) increased [3H] acetylcholine release (sampled every 3 min) in response to field stimulation (10 Hz). In C, and in subsequent figures, the horizontal filled bars indicate the periods during which field stimulation was applied.
The effects of NO on the HR response to vagal nerve stimulation and the evoked release of [3H] acetylcholine in the presence of guanylyl cyclase inhibition
Inhibition of guanylyl cyclase with ODQ (10 μm, n = 4) abolished the effect of SNP on the evoked release of [3H] acetylcholine following field stimulation (Fig. 2C and D). ODQ also significantly reduced the size of the vagal bradycardia at 3 and 5 Hz stimulation frequencies and prevented SNP increasing the HR response to vagal nerve stimulation at either 3 Hz (ΔHR: control, -37 ± 1 beats min−1; ODQ, -28 ± 2 beats min−1; + 10 μm SNP, -30 ± 2 beats min−1; + 100 μm SNP, -30 ± 2 beats min−1) or 5 Hz (Fig. 2A and B). The postsynaptic stimulation of If and baseline HR by SNP was also significantly reduced by guanylyl cyclase inhibition, from +41 ± 4 to +21 ± 5 beats min−1 at 10 μm and +57 ± 8 to +25 ± 4 beats min−1 at 100 μm SNP.
Figure 2. SNP augments vagal bradycardia and [3H] acetylcholine release via a guanylyl cyclase-dependent pathway.
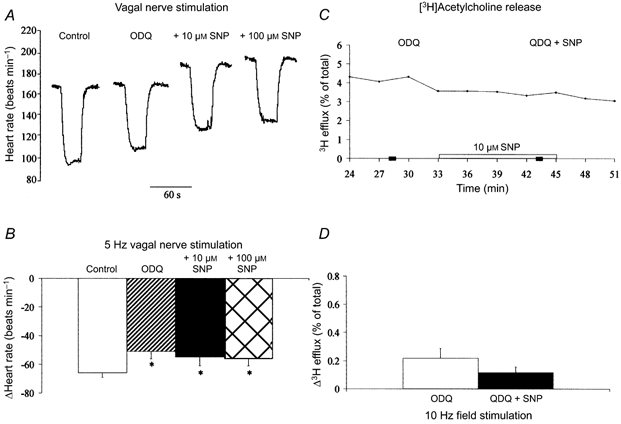
A and B, guanylyl cyclase inhibition (10 μm ODQ, n = 5) significantly reduced (*P < 0.05) the decrease in heart rate to right vagal nerve stimulation (5 Hz). However, SNP (10 and 100 μm) had no effect on the magnitude of the vagal bradycardia following guanylyl cyclase inhibition. C and D, the effect of SNP (10 μm) on the increase in [3H] acetylcholine release (sampled every 3 min) in response to field stimulation (10 Hz) was also abolished in the presence of ODQ (10 μm, n = 4), suggesting that SNP increases acetylcholine release via a NO-cGMP-dependent pathway.
These results show that SNP increases the release of acetylcholine and the magnitude of the HR response to vagal nerve stimulation via a presynaptic NO-guanylyl cyclase-dependent pathway.
The effects of NO on the HR response to vagal nerve stimulation and the evoked release of [3H] acetylcholine in the presence of PDE inhibition
Inhibition of PDE 3 with milrinone significantly increased the magnitude of the vagal bradycardia at 3 and 5 Hz stimulation frequencies, but in a separate set of experiments had no effect on the HR response to carbamylcholine (ΔHR: control, -62 ± 3 beats min−1; 1 μm milrinone, -65 ± 5 beats min−1; n = 7). The release of [3H] acetylcholine during field stimulation was also significantly increased from +0.199 ± 0.02 % (n = 4) to +0.462 ± 0.06 % (n = 4) in the presence of milrinone (P < 0.05, unpaired t test). This suggests that milrinone augments the vagal bradycardia via an increase in acetylcholine release.
In the presence of milrinone, SNP (10 μm, n = 7 or 100 μm, n = 6) had no further effect on the HR response to vagal nerve stimulation (Table 1).
Table 1.
SNP has no effect on the magnitude of vagal bradycardia (beats min−1) following inhibition of phosphodiesterase 3
| + SNP | |||||
|---|---|---|---|---|---|
| Stimulation frequency | Control | Milrinone | 10 μM | 100 μM | Milrinone |
| 3 Hz | −35 ± 5 | −78 ± 11 * | −73 ± 10 * | — | −79 ± 16 * |
| 5 Hz | −55 ± 4 | −104 ± 7 * | −102 ± 5 * | — | −108 ± 5 * |
| 3 Hz | −27 ± 3 | −64 ± 7 * | — | −61 ± 9 * | −59 ± 8 * |
| 5 Hz | −44 ± 3 | −92 ± 8 * | — | −93 ± 6 * | −85 ± 7 * |
Milrinone (1 μM) significantly increased the heart rate response to vagal nerve stimulation at 3 and 5 Hz
(P < 0.05 vs. control). However, SNP at 10 μM (n = 7) and 100 μM (n = 6) had no effect on the vagal bradycardia in the presence of milrinone.
In these experiments, milrinone significantly increased baseline HR by 60 ± 5 beats min−1 (n = 20) due to postsynaptic elevation of cAMP and stimulation of If (DiFrancesco & Tortora, 1991). Milrinone also significantly reduced the HR response to SNP, to 18 ± 2 beats min−1 at 10 μm and 15 ± 4 beats min−1 at 100 μm SNP, as has been previously reported (Musialek et al. 2000). To show that the effects of milrinone on the vagal bradycardia were independent of these changes in baseline HR, the experiment was repeated in the presence of 2 mm Cs+, a blocker of If. Cs+ significantly reduced baseline HR (ΔHR: -46 ± 5 beats min−1, n = 6) and the vagal bradycardia (ΔHR: -35 ± 4 to -23 ± 3 beats min−1 at 3 Hz; -58 ± 4 to -35 ± 4 beats min−1 at 5 Hz). In the presence of Cs+, milrinone still increased the size of the vagal bradycardia but SNP (10 μm) had no further effect at 3 Hz (ΔHR: control, -23 ± 3 beats min−1; milrinone, -52 ± 5 beats min−1; + SNP, -55 ± 5 beats min−1) and 5 Hz stimulation (Fig. 3A and B). In the presence of milrinone, SNP (10 μm, n = 4) also had no effect on the release of [3H] acetylcholine (Fig. 3C and D).
Figure 3. SNP augments vagal bradycardia and [3H] acetylcholine release via a PDE 3-dependent pathway.
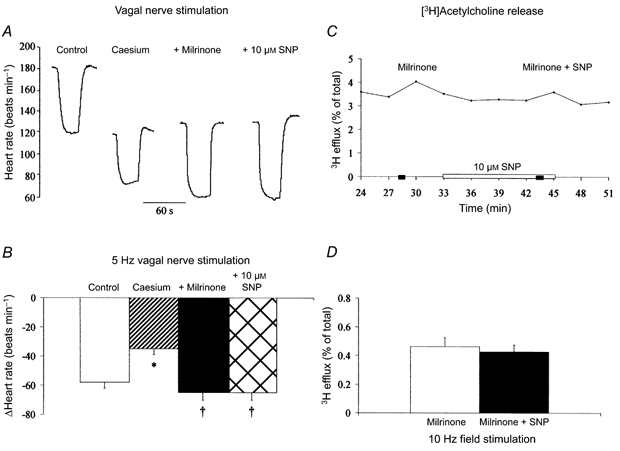
A and B, PDE 3 inhibition (1 μm milrinone, n = 6) significantly increased (†P < 0.05) vagal bradycardia at 5 Hz. However, SNP (10 μm) had no effect on the decrease in heart rate to vagal stimulation following PDE 3 inhibition. The experiment was performed in the presence of 2 mm Cs+ to block the hyperpolarization-activated current, If, and prevent changes in baseline heart rate. Cs+ itself significantly reduced (*P < 0.05) the magnitude of the vagal bradycardia. C and D, the increase in [3H] acetylcholine release (sampled every 3 min) in response to field stimulation (10 Hz) was significantly augmented (P < 0.05, unpaired t test) after inhibition of PDE 3 (1 μm milrinone, n = 4). However, the effect of SNP (10 μm) was abolished in the presence of PDE 3 inhibition.
In contrast, inhibition of PDE 2 with EHNA (10 μm) had no effect on the HR response to vagal nerve stimulation (n = 5). In the presence of EHNA, SNP increased baseline HR by +57 ± 4 and +71 ± 5 beats min−1 at concentrations of 10 and 100 μm, respectively, and was still able to augment the vagal bradycardia (ΔHR: control, -58 ± 4 beats min−1; EHNA, -54 ± 5 beats min−1; + 10 μm SNP, -70 ± 6 beats min−1; + 100 μm SNP, -77 ± 8 beats min−1, at 5 Hz).
The effects of NO on the HR response to vagal nerve stimulation in the presence of protein kinase inhibition
The PKA inhibitor H-89 (0.5 μm) significantly reduced the magnitude of the vagal bradycardia at 3 and 5 Hz stimulation frequencies (n = 5), but in a separate set of experiments had no effect on the HR response to carbamylcholine (ΔHR: control, -55 ± 6 beats min−1; H-89, -53 ± 5 beats min−1; n = 5). In the presence of H-89, SNP was unable to increase the HR response to vagal nerve stimulation at 3 Hz (ΔHR: control, -32 ± 2 beats min−1; H-89, -23 ± 3 beats min−1; + 10 μm SNP, -21 ± 3 beats min−1; + 100 μm SNP, -25 ± 5 beats min−1) or 5 Hz (Fig. 4A and B). SNP still increased baseline HR in the presence of H-89 by 39 ± 11 beats min−1 at 10 μm SNP and 57 ± 8 beats min−1 at 100 μm SNP. Similar results were observed with another PKA inhibitor KT5720, which significantly reduced the magnitude of vagal bradycardia and prevented augmentation of the response with SNP (ΔHR: control, -59 ± 5 beats min−1; 1 μm KT5720, -51 ± 5 beats min−1; + 10 μm SNP, -47 ± 6 beats min−1; + 100 μm SNP, -50 ± 5 beats min−1, at 5 Hz, n = 5). The increase in baseline HR to SNP was also intact in the presence of KT5720 (ΔHR: 10 μm SNP, +40 ± 5 beats min−1; 100 μm SNP, +55 ± 4 beats min−1).
Figure 4. SNP augments vagal bradycardia via a mechanism involving protein kinase A but not protein kinase G.
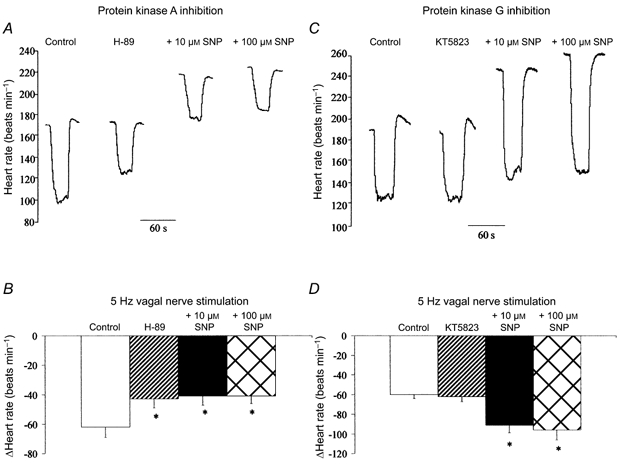
A and B, inhibition of PKA (0.5 μm H-89, n = 5) significantly reduced (*P < 0.05) the heart rate response to right vagal nerve stimulation (5 Hz). However, SNP (10 and 100 μm) had no effect on the magnitude of the vagal bradycardia following PKA inhibition. C and D, inhibition of PKG (1 μm KT5823, n = 6) had no effect on the heart rate response to right vagal nerve stimulation. SNP (10 and 100 μm) was still able to significantly increase (*P < 0.05) the vagal bradycardia following PKG inhibition.
Inhibition of PKG with KT5823 (1 μm) had no effect on the magnitude of the vagal bradycardia (n = 6) and SNP was still able to augment the HR response to vagal nerve stimulation in the presence of KT5823 at 3 Hz (ΔHR: control, -35 ± 1 beats min−1; KT5823, -37 ± 4 beats min−1; + 10 μm SNP, -58 ± 10 beats min−1; + 100 μm SNP, -52 ± 10 beats min−1) and 5 Hz (Fig. 4C and D). SNP still increased baseline HR in the presence of KT5823, by 56 ± 7 beats min−1 at 10 μm SNP and 67 ± 6 beats min−1 at 100 μm SNP.
These results suggest that NO facilitates the vagal bradycardia via a presynaptic pathway involving PDE 3 and PKA but not PDE 2 or PKG.
The effects of NO on the HR response to vagal nerve stimulation in the presence of presynaptic neuronal calcium channel blockers
Inhibition of either N-type calcium channels with ω-conotoxin GVIA (100 nm, n = 6) or P-type calcium channels with ω-agatoxin-TK (50 nm, n = 5) reduced but did not abolish the vagal bradycardia. Both ω-conotoxin GVIA and ω-agatoxin-TK have previously been shown to have no effect on the HR response to carbamylcholine (Hong & Chang, 1995). SNP was unable to facilitate vagal neurotransmission following block of N-type calcium channels at 5 (Fig. 5A and B), 7 or 9 Hz stimulation frequencies (e.g. ΔHR: 9 Hz: control, -106 ± 6 beats min−1; ω-conotoxin GVIA, -32 ± 2 beats min−1; + 10 μm SNP, -32 ± 2 beats min−1; + 100 μm SNP, -28 ± 3 beats min−1).
Figure 5. SNP facilitates vagal bradycardia via N-type but not P-type calcium channels.
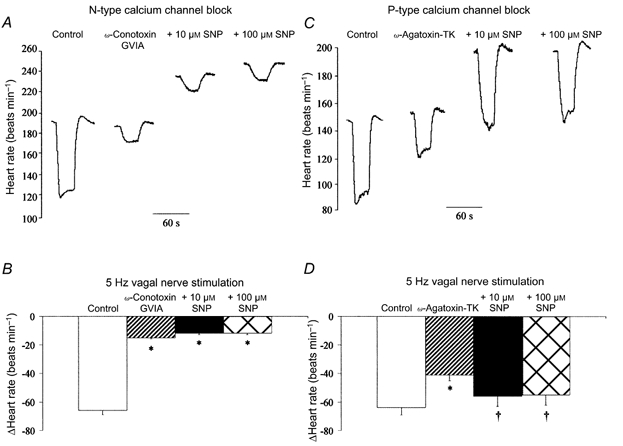
A and B, block of N-type calcium channels (100 nmω-conotoxin GVIA, n = 6) significantly reduced (*P < 0.05) the heart rate response to right vagal nerve stimulation (5 Hz). However, SNP (10 and 100 μm) had no effect on the magnitude of the vagal bradycardia following block of N-type calcium channels. This can be compared with block of P-type calcium channels (C and D; 50 nmω-agatoxin-TK, n = 5), which also significantly reduced (*P < 0.05) the heart rate response to right vagal nerve stimulation. However, SNP (10 and 100 μm) was still able to significantly increase (†P < 0.05) the magnitude of the vagal bradycardia following block of P-type calcium channels.
However, following inhibition of P-type calcium channels, SNP still increased the HR response to vagal nerve stimulation (Fig. 5C and D). These results suggest that NO facilitates the vagal bradycardia via a presynaptic pathway involving N- but not P-type calcium channels (see Fig. 6).
Figure 6. A presynaptic pathway for NO-cGMP-dependent modulation of acetylcholine release.
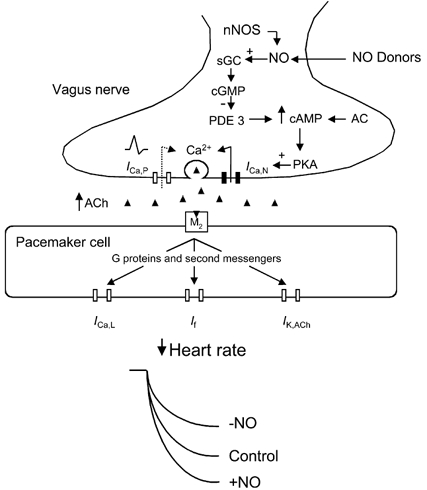
Summary diagram showing the proposed mechanism by which nitric oxide (NO) generated by NO donors or neuronal nitric oxide synthase (nNOS) increases the heart rate response to vagal nerve stimulation. NO stimulates presynaptic soluble guanylyl cyclase (sGC) to produce cyclic guanine monophosphate (cGMP) which inhibits phosphodiesterase 3 (PDE 3). This elevates intracellular cyclic adenine monophosphate (cAMP, synthesized by adenylyl cyclase, AC) levels and increases the protein kinase A (PKA)-dependent phosphorylation of N-type calcium channels. Both N- and P-type calcium currents (ICa,N and ICa,P) control the exocytotic release of acetylcholine (ACh). Postsynaptically, ACh binds to muscarinic cholinergic (M2) receptors on sino-atrial node pacemaker cells and via several second messenger pathways reduces the L-type calcium current (ICa,L) and hyperpolarization-activated current (If), and increases the acetylcholine-dependent potassium current (IK,Ach) to decrease heart rate.
SNP increased baseline HR by 30 ± 2 and 47 ± 3 beats min−1 at 10 and 100 μm, respectively, in the presence of ω-conotoxin GVIA and by 44 ± 6 and 58 ± 6 beats min−1 at 10 and 100 μm, respectively, in the presence of ω-agatoxin-TK. These results also demonstrate that the increase in the vagal bradycardia due to SNP is dependent on the pharmacological intervention rather than on postsynaptic stimulation of pacemaking by NO and increases in baseline HR.
Experiments measuring [3H] acetylcholine release were performed in the presence of the choline uptake inhibitor hemicholinium 3, so it is unlikely that SNP increases transmitter release via this route. To test whether NO acts via modulation of the presynaptic autoinhibitory muscarinic M4 receptor, we added SNP in the presence of tropicamide. Tropicamide (0.2 μm) significantly enhanced the heart rate response to vagal nerve stimulation (n = 8), consistent with block of M4 receptors. However, SNP (100 μm) was still able to augment the vagal-induced bradycardia (ΔHR, 5 Hz: control, -60 ± 2 beats min−1; tropicamide, -76 ± 4 beats min−1; + 10 μm SNP, -82 ± 7 beats min−1; + 100 μm SNP, -97 ± 8 beats min−1). Again SNP increased baseline HR by 46 ± 7 and 64 ± 7 beats min−1 at 10 and 100 μm, respectively. These results suggests that SNP does not act via inhibition of presynaptic M4 receptors.
DISCUSSION
The main new findings of this study are that (1) NO increases the evoked release of acetylcholine and the HR response to vagal nerve stimulation via a presynaptic guanylyl cyclase-dependent pathway, and (2) NO augments the vagal bradycardia by cGMP stimulation of PDE 3, increasing cAMP-PKA-dependent phosphorylation of presynaptic N-type calcium channels (see Fig. 6).
NO augments vagal neurotransmission by facilitating acetylcholine release
We have shown that SNP facilitates the HR response to vagal nerve stimulation at physiological stimulation frequencies (3-5 Hz), an effect previously shown to be independent of changes in baseline HR and mimicked by the membrane-permeant analogue of cGMP, 8-Br-cGMP (Sears et al. 1999). The NO donor molsidamine has also been shown to increase the heart rate response to vagal nerve stimulation in the rabbit in vivo (Sears et al. 1999). We also show that SNP (like 8-Br-cGMP) has no effect on the HR response to carbamylcholine, suggesting that, functionally, SNP is acting predominantly via a presynaptic pathway to modulate HR. Other evidence is also consistent with this idea. The nNOS knockout mouse has a higher resting basal HR than its wild-type control, and a blunted HR response to atropine, suggesting a lower vagal tone (Jumrussirikul et al. 1998). In isolated atria-vagal preparations from this mouse, the HR response to vagal nerve stimulation, but not to carbamylcholine, is reduced compared with its wild-type control (Choate et al. 2000), further suggesting that knockout of the nNOS gene inhibits vagal neurotransmission via a presynaptic pathway. Our data support this hypothesis, as we also show that SNP increases the release of 3H-labelled acetylcholine in response to field stimulation in isolated atria. This has not been shown before in the heart. Functionally, SNP was also unable to augment the vagal bradycardia or increase the evoked release of acetylcholine in the presence of guanylyl cyclase inhibition, suggesting that release of NO and stimulation of guanylyl cyclase is responsible for the effects of SNP (rather than nitrosylation or generation of superoxide radicals caused by some NO donors; Sarkar et al. 2000). Similarly, others have shown that NO increases the release of acetylcholine in rat forebrain (Prast & Philippu, 1992) and isolated synaptosomes (Morot Gaudry-Talarmain et al. 1997), and that NOS inhibition decreases the release of acetylcholine in guinea-pig gastric fundus (Sotirov et al. 1999).
A presynaptic pathway for NO in the vagal control of heart rate
Inhibition of PDE 3 increased the HR response to vagal nerve stimulation, but not to carbamylcholine, an effect that is independent of changes in baseline HR, and increased the release of [3H] acetylcholine. PKA inhibition also reduced the HR response to vagal nerve stimulation but not to carbamylcholine. This suggests that the presynaptic cAMP-PKA system can modulate vagal neurotransmission. Both the membrane-permeant cAMP analogue 8-Br-cAMP (Dawson et al. 1996) and stimulation of adenylate cyclase with PACAP (Seebeck et al. 1996) increase the release of radioactively labelled acetylcholine in isolated atria. Given the fact that inhibition of PDE 2 had no effect on the HR response to vagal nerve stimulation, the basal activity of PDE 3 appears to be higher than that of PDE 2 presynaptically. In our study NO was unable to augment the vagal bradycardia or release of [3H] acetylcholine in the presence of PDE 3 inhibition. Therefore, NO stimulation of presynaptic guanylyl cyclase may produce cGMP that inhibits PDE 3 to raise presynaptic cAMP and activate PKA. Inhibition of PKA also abolished the augmentation of vagal bradycardia by SNP whilst inhibition of PDE 2 or PKG had no effect. In cardiac myocytes, NO can stimulate calcium currents via inhibition of PDE 3 to raise cAMP and increase the activity of PKA (Ono & Trautwein, 1991). As PKA phosphorylates presynaptic N- and P-type calcium channels in hippocampal neurons (Hell et al. 1995), we investigated whether NO augments the vagal bradycardia via presynapatic calcium channels.
Inhibition of either N- or P-type calcium channels reduced but did not abolish the HR response to vagal nerve stimulation. Previous reports have indicated that cardiac acetylcholine release is controlled at least in part by N-type calcium channels (Akiyama & Yamazaki, 2000). SNP was still able to augment the vagal bradycardia following P-type calcium channel inhibition but not after N-type calcium channel inhibition. This suggests that the facilitatory action of NO on acetylcholine release is mediated by cGMP stimulation of PDE 3, increasing cAMP-PKA-dependent phosphorylation of presynaptic N-type calcium channels (see Fig. 6). An effect of NO on choline uptake or autoinhibitory muscarinic receptors is unlikely since we observed an increase in the evoked release of [3H] acetylcholine in the presence of the inhibitor of choline uptake, hemicholinium 3. Furthermore, SNP was still able to augment the vagal bradycardia in the presence of the M4 receptor antagonist tropicamide, and muscarinic autoinhibition of acetylcholine release is not mediated via the adenylyl cyclase-cAMP system in mouse atria (Dawson et al. 1996).
The role of NO in the vagal control of heart rate
Postsynaptically, NO can directly stimulate pacemaking via a cGMP-dependent increase in the hyperpolarization-activated current (If) in sino-atrial node cells (Musialek et al. 1997). NO generated by muscarinic receptor stimulation of eNOS has also been implicated in the cholinergic inhibition of adrenergically stimulated ICa,L (Han et al. 1994, 1995). Prior adrenergic stimulation is thought to be essential for this mechanism, as cAMP levels need to be sufficiently high so that NO-dependent stimulation of PDE 2 can then reduce cAMP and ICa,L (Han et al. 1998a). Whilst some groups report that inhibition of eNOS (Han et al. 1994, 1995) or knockout of the eNOS gene (Han et al. 1998b) abolishes the effect of acetylcholine on pre-stimulated ICa,L, others have not observed this (Vandecasteele et al. 1998, 1999; Belevych & Harvey, 2000). Reasons for these differences have been discussed in detail elsewhere (see Balligand, 1999 for a review). When all data are taken together, there appears to be an important interplay between ICa,L and If in the M2-receptor-coupled NO-cGMP-dependent modulation of HR (Sears et al. 1998a). A comparison of studies is complicated because of the different experimental approaches used: nerve stimulation versus applied transmitter, necessity for adrenergic prestimulation, developmental stage of tissue and species. All appear to significantly influence the outcome. Nevertheless, there appears to be a clear picture emerging that suggests an important role for NO in the facilitation of vagal neurotransmission and the resulting bradycardia.
Interestingly, the effect of NO on the HR response to sympathetic nerve stimulation mirrors the vagal response. NO has been shown to decrease the release of noradrenaline and reduce the heart rate response to sympathetic nerve stimulation in vitro (Schwarz et al. 1995; Choate & Paterson, 1999). This may be due to an action of cGMP on PDE 2 that will reduce cAMP and PKA-dependent phosphorylation.
Functional implications
It is well established that cholinergic activation antagonizes the potentially pro-dysrhythmic effects of high sympathetic tone on the heart (Kienzle, 1995). It is not surprising therefore that high vagal tone is a good prognostic indicator against sudden cardiac death (Cole et al. 1999). The NO-cGMP pathway may be important in pathophysiological states where vagal signalling is impaired, e.g. hypertension (Murphy et al. 1991; Petretta et al. 1995a, b). Guanylyl cyclase is downregulated in vasculature tissue from spontaneously hypertensive rats (Ruetten et al. 1999). However, it is not known whether a reduction in neuronal guanylyl cyclase causes impaired release of acetylcholine in hypertension.
Acknowledgments
We are grateful to the British Heart Foundation for supporting this study. N.H. is supported by a Wellcome Trust Prize studentship as part of the Wellcome Trust Cardiovascular Research Initiative at Oxford University and is a Phizackerley Senior Scholar at Balliol College, Oxford.
References
- Akiyama T, Yamazaki T. Adrenergic inhibition of endogenous acetylcholine release on postganglionic cardiac vagal nerve terminals. Cardiovascular Research. 2000;46:531–538. doi: 10.1016/s0008-6363(00)00027-4. [DOI] [PubMed] [Google Scholar]
- Balligand JL. Regulation of cardiac beta-adrenergic response by nitric oxide. Cardiovascular Research. 1999;43:607–620. doi: 10.1016/s0008-6363(99)00163-7. [DOI] [PubMed] [Google Scholar]
- Belevych AE, Harvey RD. Muscarinic inhibitory and stimulatory regulation of the L-type calcium current is not altered in cardiac ventricular myocytes from mice lacking endothelial nitric oxide synthase. Journal of Physiology. 2000;528:279–289. doi: 10.1111/j.1469-7793.2000.00279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. Journal of Biological Chemistry. 1990;265:5267–5272. [PubMed] [Google Scholar]
- Choate JK, Danson EJF, Morris JF, Paterson DJ. Vagal modulation of heart rate in the neuronal nitric oxide synthase knock-out mouse in-vitro. Circulation. 2000;102:S619. abstract. [Google Scholar]
- Choate JK, Paterson DJ. Nitric oxide inhibits the positive chronotropic and inotropic responses to sympathetic nerve stimulation in the isolated guinea-pig atria. Journal of the Autonomic Nervous System. 1999;75:100–108. doi: 10.1016/s0165-1838(98)00173-8. [DOI] [PubMed] [Google Scholar]
- Cole CR, Blackstone EH, Pashkow FJ, Snader CE, Lauer MS. Heart-rate recovery immediately after exercise as a predictor of mortality. New England Journal of Medicine. 1999;341:1351–1357. doi: 10.1056/NEJM199910283411804. [DOI] [PubMed] [Google Scholar]
- Conlon K, Kidd C. Neuronal nitric oxide facilitates vagal chronotropic and dromotropic actions on the heart. Journal of the Autonomic Nervous System. 1999;75:136–146. doi: 10.1016/s0165-1838(98)00185-4. [DOI] [PubMed] [Google Scholar]
- Dawson JJ, Iannazzo L, Majewski H. Muscarinic autoinhibition of acetylcholine release in mouse atria is not transduced through cyclic AMP or protein kinase C. Journal of Autonomic Pharmacology. 1996;16:79–85. doi: 10.1111/j.1474-8673.1996.tb00415.x. [DOI] [PubMed] [Google Scholar]
- Denyer JC, Brown HF. Pacemaking in rabbit isolated sino-atrial node cells during Cs+ block of the hyperpolarization-activated current if. Journal of Physiology. 1990;429:401–409. doi: 10.1113/jphysiol.1990.sp018264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco D, Tortora P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature. 1991;351:145–147. doi: 10.1038/351145a0. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo [4,3-a]quinoxalin-1-one. Molecular Pharmacology. 1995;48:184–188. [PubMed] [Google Scholar]
- Han X, Kobzik L, Severson D, Shimoni Y. Characteristics of nitric oxide-mediated cholinergic modulation of calcium current in rabbit sino-atrial node. Journal of Physiology. 1998a;509:741–754. doi: 10.1111/j.1469-7793.1998.741bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Kubota I, Feron O, Opel DJ, Arstall MA, Zhao YY, Huang P, Fishman MC, Michel T, Kelly RA. Muscarinic cholinergic regulation of cardiac myocyte ICa-L is absent in mice with targeted disruption of endothelial nitric oxide synthase. Proceedings of the National Academy of Sciences of the USA. 1998b;95:6510–6515. doi: 10.1073/pnas.95.11.6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Shimoni Y, Giles WR. An obligatory role for nitric oxide in autonomic control of mammalian heart rate. Journal of Physiology. 1994;476:309–314. doi: 10.1113/jphysiol.1994.sp020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Shimoni Y, Giles WR. A cellular mechanism for nitric oxide-mediated cholinergic control of mammalian heart rate. Journal of General Physiology. 1995;106:45–65. doi: 10.1085/jgp.106.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SA, Reifsnyder DH, Gallis B, Cadd GG, Beavo JA. Isolation and characterization of bovine cardiac muscle cGMP-inhibited phosphodiesterase: a receptor for new cardiotonic drugs. Molecular Pharmacology. 1986;29:506–514. [PubMed] [Google Scholar]
- Hell JW, Yokoyama CT, Breeze LJ, Chavkin C, Catterall WA. Phosphorylation of presynaptic and postsynaptic calcium channels by cAMP-dependent protein kinase in hippocampal neurons. EMBO Journal. 1995;14:3036–3044. doi: 10.1002/j.1460-2075.1995.tb07306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring N, Golding S, Paterson DJ. Pre-synaptic NO-cGMP pathway modulates vagal control of heart rate in isolated adult guinea pig atria. Journal of Molecular and Cellular Cardiology. 2000;32:1795–1804. doi: 10.1006/jmcc.2000.1214. [DOI] [PubMed] [Google Scholar]
- Hong SJ, Chang CC. Calcium channel subtypes for the sympathetic and parasympathetic nerves of guinea-pig atria. British Journal of Pharmacology. 1995;116:1577–1582. doi: 10.1111/j.1476-5381.1995.tb16375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumrussirikul P, Dinerman J, Dawson TM, Dawson VL, Ekelund U, Georgakopoulos D, Schramm LP, Calkins H, Snyder SH, Hare JM, Berger RD. Interaction between neuronal nitric oxide synthase and inhibitory G protein activity in heart rate regulation in conscious mice. Journal of Clinical Investigation. 1998;102:1279–1285. doi: 10.1172/JCI2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M. K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochemical and Biophysical Research Communications. 1987;142:436–440. doi: 10.1016/0006-291x(87)90293-2. [DOI] [PubMed] [Google Scholar]
- Kerr LM, Yoshikami D. A venom peptide with a novel presynaptic blocking action. Nature. 1984;308:282–284. doi: 10.1038/308282a0. [DOI] [PubMed] [Google Scholar]
- Kienzle ML. Parasympathetic influence in cardiac electrophysiology. In: Podrid PJ, Kowey PR, editors. Cardiac Arrhythmia - Mechanisms, Diagnosis, and Management. Baltimore, USA: Williams and Wilkins; 1995. pp. 168–181. [Google Scholar]
- Klimaschewski L, Kummer W, Mayer B, Couraud JY, Preissler U, Philippin B, Heym C. Nitric oxide synthase in cardiac nerve fibers and neurons of rat and guinea pig heart. Circulation Research. 1992;71:1533–1537. doi: 10.1161/01.res.71.6.1533. [DOI] [PubMed] [Google Scholar]
- Liu JL, Murakami H, Zucker IH. Effects of NO on baroreflex control of heart rate and renal nerve activity in conscious rabbits. American Journal of Physiology. 1996;270:R1361–1370. doi: 10.1152/ajpregu.1996.270.6.R1361. [DOI] [PubMed] [Google Scholar]
- Méry PF, Pavoine C, Pecker F, Fischmeister R. Erythro-9-(2-hydroxy-3-nonyl)adenine inhibits cyclic GMP-stimulated phosphodiesterase in isolated cardiac myocytes. Molecular Pharmacology. 1995;48:121–130. [PubMed] [Google Scholar]
- Morot Gaudry-Talarmain Y, Moulian N, Meunier FA, Blanchard B, Angaut-Petit D, Faille L, Ducrocq C. Nitric oxide and peroxynitrite affect differently acetylcholine release, choline acetyltransferase activity, synthesis, and compartmentation of newly formed acetylcholine in Torpedo marmorata synaptosomes. Nitric Oxide. 1997;1:330–345. doi: 10.1006/niox.1997.0141. [DOI] [PubMed] [Google Scholar]
- Murphy CA, Sloan RP, Myers MM. Pharmacologic responses and spectral analyses of spontaneous fluctuations in heart rate and blood pressure in SHR rats. Journal of the Autonomic Nervous System. 1991;36:237–250. doi: 10.1016/0165-1838(91)90047-7. [DOI] [PubMed] [Google Scholar]
- Musialek P, Lei M, Brown HF, Paterson DJ, Casadei B. Nitric oxide can increase heart rate by stimulating the hyperpolarization-activated inward current, I(f) Circulation Research. 1997;81:60–68. doi: 10.1161/01.res.81.1.60. [DOI] [PubMed] [Google Scholar]
- Musialek P, Rigg L, Terrar DA, Paterson DJ, Casadei B. Role of cGMP-inhibited phosphodiesterase and sarcoplasmic calcium in mediating the increase in basal heart rate with nitric oxide donors. Journal of Molecular and Cellular Cardiology. 2000;32:1831–1840. doi: 10.1006/jmcc.2000.1216. [DOI] [PubMed] [Google Scholar]
- Olivera BM, McIntosh JM, Cruz LJ, Luque FA, Gray WR. Purification and sequence of a presynaptic peptide toxin from Conus geographus venom. Biochemistry. 1984;23:5087–5090. doi: 10.1021/bi00317a001. [DOI] [PubMed] [Google Scholar]
- Ono K, Trautwein W. Potentiation by cyclic GMP of β-adrenergic effect on Ca2+ current in guinea-pig ventricular cells. Journal of Physiology. 1991;443:387–404. doi: 10.1113/jphysiol.1991.sp018839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petretta M, Bianchi V, Marciano F, Themistoclakis S, Canonico V, Sarno D, Iovino G, Bonaduce D. Influence of left ventricular hypertrophy on heart period variability in patients with essential hypertension. Journal of Hypertension. 1995a;13:1299–1306. doi: 10.1097/00004872-199511000-00012. [DOI] [PubMed] [Google Scholar]
- Petretta M, Marciano F, Bianchi V, Migaux ML, Valva G, De Luca N, Salemme L, Berardino S, Bonaduce D. Power spectral analysis of heart period variability in hypertensive patients with left ventricular hypertrophy. American Journal of Hypertension. 1995b;8:1206–1213. doi: 10.1016/0895-7061(95)00252-9. [DOI] [PubMed] [Google Scholar]
- Prast H, Philippu A. Nitric oxide releases acetylcholine in the basal forebrain. European Journal of Pharmacology. 1992;216:139–140. doi: 10.1016/0014-2999(92)90223-q. [DOI] [PubMed] [Google Scholar]
- Ruetten H, Zabel U, Linz W, Schmidt HH. Downregulation of soluble guanylyl cyclase in young and aging spontaneously hypertensive rats. Circulation Research. 1999;85:534–541. doi: 10.1161/01.res.85.6.534. [DOI] [PubMed] [Google Scholar]
- Sarkar D, Vallance P, Amirmansour C, Harding SE. Positive inotropic effects of NO donors in isolated guinea-pig and human cardiomyocytes independent of NO species and cyclic nucleotides. Cardiovascular Research. 2000;48:430–439. doi: 10.1016/s0008-6363(00)00202-9. [DOI] [PubMed] [Google Scholar]
- Schwarz P, Diem R, Dunn NJ, Fostermann U. Endogenous and exogenous nitric oxide inhibits norepinephrine release from rat heart sympathetic nerves. Circulation Research. 1995;77:841–848. doi: 10.1161/01.res.77.4.841. [DOI] [PubMed] [Google Scholar]
- Sears CE, Choate JK, Paterson DJ. Inhibition of nitric oxide synthase slows heart rate recovery from cholinergic activation. Journal of Applied Physiology. 1998a;84:1596–1603. doi: 10.1152/jappl.1998.84.5.1596. [DOI] [PubMed] [Google Scholar]
- Sears CE, Choate JK, Paterson DJ. Effect of nitric oxide synthase inhibition on the sympatho-vagal control of heart rate. Journal of the Autonomic Nervous System. 1998b;73:63–73. doi: 10.1016/s0165-1838(98)00123-4. [DOI] [PubMed] [Google Scholar]
- Sears CE, Choate JK, Paterson DJ. NO-cGMP pathway accentuates the decrease in heart rate caused by cardiac vagal nerve stimulation. Journal of Applied Physiology. 1999;86:510–516. doi: 10.1152/jappl.1999.86.2.510. [DOI] [PubMed] [Google Scholar]
- Seebeck J, Schmidt WE, Kilbinger H, Neumann J, Zimmermann N, Herzig S. PACAP induces bradycardia in guinea-pig heart by stimulation of atrial cholinergic neurones. Naunyn-Schmiedeberg's Archives of Pharmacology. 1996;354:424–430. doi: 10.1007/BF00168432. [DOI] [PubMed] [Google Scholar]
- Shi H, Wang H, Wang Z. M3 muscarinic receptor activation of a delayed rectifier potassium current in canine atrial myocytes. Life Sciences. 1999;64:251–257. doi: 10.1016/s0024-3205(99)00142-3. [DOI] [PubMed] [Google Scholar]
- Sotirov E, Papasova M, Santha E. Nitric oxide (NO) increases acetylcholine release from and inhibits smooth muscle contraction of guinea-pig gastric fundus. Brain Research Bulletin. 1999;49:297–302. doi: 10.1016/s0361-9230(99)00019-2. [DOI] [PubMed] [Google Scholar]
- Teramoto T, Kuwada M, Niidome T, Sawada K, Nishizawa Y, Katayama K. A novel peptide from funnel web spider venom, omega-Aga-TK, selectively blocks, P-type calcium channels. Biochemical and Biophysical Research Communications. 1993;196:134–140. doi: 10.1006/bbrc.1993.2225. [DOI] [PubMed] [Google Scholar]
- Vandecasteele G, Eschenhagen T, Fischmeister R. Role of the NO-cGMP pathway in the muscarinic regulation of the L-type Ca2+ current in human atrial myocytes. Journal of Physiology. 1998;506:653–663. doi: 10.1111/j.1469-7793.1998.653bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandecasteele G, Eschenhagen T, Scholz H, Stein B, Verde I, Fischmeister R. Muscarinic and beta-adrenergic regulation of heart rate, force of contraction and calcium current is preserved in mice lacking endothelial nitric oxide synthase. Nature Medicine. 1999;5:331–334. doi: 10.1038/6553. [DOI] [PubMed] [Google Scholar]
- Wetzel GT, Brown JH. Relationships between choline uptake, acetylcholine synthesis and acetylcholine release in isolated rat atria. Journal of Pharmacology and Experimental Therapeutics. 1983;226:343–348. [PubMed] [Google Scholar]
- Wetzel GT, Goldstein D, Brown JH. Acetylcholine release from rat atria can be regulated through an alpha 1-adrenergic receptor. Circulation Research. 1985;56:763–766. doi: 10.1161/01.res.56.5.763. [DOI] [PubMed] [Google Scholar]