

Abstract
Various smooth muscles have unique contractile characteristics, such as the degree of Ca2+ sensitivity induced by physiological and pharmacological agents. Here we evaluated six different rabbit smooth muscle tissues for protein kinase C (PKC)-induced Ca2+ sensitization. We also examined the expression levels of myosin light chain phosphatase (MLCP), the MLCP inhibitor phosphoprotein CPI-17, and the thin filament regulator h-calponin.
Immunohistochemical and Western blot analyses indicated that CPI-17 was found primarily in smooth muscle, although expression varied among different tissues. Vascular muscles contained more CPI-17 than visceral muscles, with further distinction existing between tonic and phasic subtypes. For example, the tonic femoral artery possessed approximately 8 times the cellular CPI-17 concentration of the phasic vas deferens.
In contrast to CPI-17 expression patterns, phasic muscles contained more MLCP myosin-targeting subunit than tonic tissues. Calponin expression was not statistically different.
Addition of phorbol ester to α-toxin-permeabilized smooth muscle caused an increase in contraction and phosphorylation of both CPI-17 and myosin light chain (MLC) at submaximal [Ca2+]i. These responses were several-fold greater in femoral artery as compared to vas deferens.
We conclude that the expression ratio of CPI-17 to MLCP correlates with the Ca2+ sensitivities of contraction induced by a PKC activator. PKC stimulation of arterial smooth muscle with a high CPI-17 and low MLCP expression generated greater force and MLC phosphorylation than stimulation of visceral muscle with a relatively low CPI-17 and high MLCP content. This implicates CPI-17 inhibition of MLCP as an important component in modulating vascular muscle tone.
Smooth muscle is heterogeneous in both structure and function. Based on their electrophysiological and mechanical characteristics, smooth muscles can be broadly classified into tonic and phasic types (Somlyo & Somlyo, 1968). Tonic muscles such as the aorta, pulmonary and femoral arteries, along with the trachea, usually respond to excitatory agonists with a graded depolarization. Prolonged depolarization induced by high K+ typically evokes a slowly developing sustained contraction in tonic muscles. In contrast, phasic muscles, which include the portal vein, ileum, bladder and vas deferens, react to excitatory agonists by generating a spike-like action potential. In addition, depolarization with high K+ elicits an initial phasic contraction, followed by a decline to a low steady-state level (Himpens et al. 1989). The contractile diversities among different smooth muscles appear to be attributed, at least in part, to variations in cellular protein expression rather than to different time courses in [Ca2+]i (Himpens et al. 1988). Myosin heavy chain (see review by Adelstein & Sellers, 1996) and light chain isoforms (see Barany & Barany, 1996) are specifically distributed among smooth muscle tissues and the composition of these isoforms determines myosin motor activity. Moreover, variable expression of other regulatory contractile proteins possibly contributes to the differentiation of smooth muscle function (Khalil, 1992; North et al. 1994; Szymanski et al. 1998; Dirksen et al. 2000).
The primary mechanism that regulates actomyosin ATPase and contraction in all vertebrate smooth muscle is reversible phosphorylation of the 20 kDa myosin light chain (MLC) by MLC kinase (MLCK) and MLC phosphatase (MLCP) (Hartshorne,1987; Kamm & Stull, 1989). MLCK is a Ca2+-calmodulin-dependent kinase that is stimulated by an increase in [Ca2+]i. Although Ca2+ is the major contractile messenger for all types of smooth muscle, the Ca2+ sensitivity of MLC phosphorylation and contraction is an equally important factor for the regulation of smooth muscle contraction (see Somlyo & Somlyo, 1994). Tonic muscles (pulmonary and femoral arteries) have a 3-fold greater calcium sensitivity of MLC phosphorylation and contraction as compared to phasic muscles (portal vein and ileum) (Kitazawa et al. 1991a). The higher Ca2+ sensitivity of tonic muscle contraction is explained by a 4-fold lower dephosphorylation rate of MLC in tonic muscle, as compared to phasic muscle. This is evident even in phasic tissues with a higher MLCK content, suggesting that in situ, the primary determinant of smooth muscle Ca2+ sensitivity is MLCP rather than MLCK activity (Gong et al. 1992).
It is well documented that G protein-coupled receptor activation increases the Ca2+ sensitivity of MLC phosphorylation and contraction in all types of smooth muscle through the inhibition of MLCP (Kitazawa et al. 1991b; Kubota et al. 1992; see Somlyo & Somlyo, 1994). MLCP is a heterotrimeric enzyme with a 38 kDa type-1 phosphatase catalytic subunit δ-isoform (PP1C), a 110-130 kDa myosin-targeting subunit (MYPT1) and a 20-21 kDa subunit of unknown function (see Hartshorne et al. 1998). Two major pathways have been proposed for the regulation of MLCP. First, RhoA-activated kinase (Rho-kinase) phosphorylates Ser-696 of MYPT1, which inhibits the phosphatase activity (Kimura et al. 1996; Feng et al. 1999; Kawano et al. 1999; see Somlyo & Somlyo, 2000). In smooth muscle cells, this phosphorylation is enhanced by stimulation with the G protein activator GTPγS (Trinkle-Mulcahy et al. 1995; Swärd et al. 2000; Nagumo et al. 2000). In addition to MYPT1 phosphorylation, a second pathway involves an inhibitor protein for MLCP, called CPI-17, which was first isolated from pig aorta smooth muscle (Eto et al. 1995) and is only expressed in smooth muscle (Eto et al. 1997, 1999). Phosphorylation of CPI-17 at Thr-38 by PKC converts this protein to a potent inhibitor of the catalytic activity of not only PP1C, but also the MLCP holoenzyme (Eto et al. 1997; Senba et al. 1999) and MLCP anchored to myofibrils in situ (Li et al. 1998; Eto et al. 2000). The PKC δ-isoform has been isolated as a dominant CPI-17 kinase from pig aorta (Eto et al. 2001), and a recombinant PKC α-isoform can much more rapidly phosphorylate Thr-38 of CPI-17 as compared to calponin, caldesmon, MLC and myosin (Kitazawa et al. 1999). Furthermore, selective depletion of CPI-17 by skinning of smooth muscle cells diminishes PKC-induced Ca2+ sensitization of femoral artery strips and the response can be reconstituted by addition of PKC and CPI-17 together, but not by PKC alone (Kitazawa et al. 1999). These results indicate that CPI-17 is at least a key component in PKC-induced MLCP inhibition and calcium sensitization. It has recently been demonstrated that stimulation of the femoral artery by agonists, GTPγS and PKC activator induces Thr-38 phosphorylation of CPI-17 and contractile Ca2+ sensitization (Kitazawa et al. 2000). A PKC inhibitor (GF109203X) and a Rho-kinase inhibitor (Y27632) partially inhibited the CPI-17 phosphorylation and contraction induced by histamine, suggesting that possibly Rho-kinase and PKC phosphorylate CPI-17 in response to histamine signalling (Kitazawa et al. 2000; Koyama et al. 2000). In summary, these results imply that the CPI-17-MLCP signalling pathway may be as significant as the Rho-kinase-MLCP pathway in G protein-coupled agonist-induced Ca2+ sensitization of smooth muscle contraction.
CPI-17 expression is almost exclusively confined to smooth muscle tissues, and we believe that it is an endogenous mediator for the PKC signalling pathway that induces contractile Ca2+ sensitization. Yet between different smooth muscle tissues (aorta vs. urinary bladder), differential expression of CPI-17 is suggested by Northern and Western blot analyses (Eto et al. 1997, 1999). In preliminary experiments at submaximal [Ca2+] (Woodsome & Kitazawa, 2000), we observed that in the phasic vas deferens and urinary bladder, phorbol ester-induced contractions were several times smaller than those caused by GTPγS. In contrast, the tonic femoral artery displayed equal responses to both phorbol 12,13-dibutyrate (PDBu) and GTPγS (Masuo et al. 1994). Therefore we hypothesized that the variation in PDBu-induced Ca2+ sensitization may possibly be due to differential expression of CPI-17 and the phospho-CPI-17 target MLCP. In this study, we used immunoblotting techniques to measure the tissue-specific expression and concentration of CPI-17, MLCP and h-calponin in six different rabbit smooth muscle tissues, including vascular tonic or phasic and visceral tonic or phasic sub-phenotypes. These results were then compared to the contractile response and MLC phosphorylation induced by PKC or G protein activation at a given level of Ca2+ in the various smooth muscles. The preliminary results were presented at the February 2000 Biophysical Society Meeting in New Orleans (Woodsome & Kitazawa, 2000).
METHODS
Tissue preparation
The Animal Care and Use Committee of Georgetown University approved all procedures. White male albino rabbits (2-3 kg) were killed with an overdose of halothane, and the desired tissues were dissected and cleaned of non-smooth muscle media. The isolated smooth muscle was then cut into strips 750 μm wide and 3 mm long with natural wall thickness for the femoral artery (FA), aorta (AO) and portal vein (PV). Tissue strips were 250 μm in diameter and 2.5-3 mm long for the urinary bladder (UB), vas deferens (VD) and trachea (TR). Vascular endothelium was removed by gentle rubbing with a razor blade (Kitazawa et al. 1991a). Silk monofilaments were used to attach the strips to two tungsten needle tips, one of which was mounted to a force transducer (AM801, SensoNor, Horten, Norway). The strips were then immersed in a bubble plate (Horiuti, 1988) to allow fairly rapid solution exchange (< 1 s) and freezing for phosphorylation measurements.
Solutions
Normal external solution comprised (mm): 150 NaCl, 4 KCl, 2 calcium methanesulphonate (Ca-Ms), 2 magnesium ethanesulphonate, 5.6 glucose and 5 Hepes buffer. Depolarizing external solution had potassium methanesulphonate (K-Ms) substituted equally for NaCl with other chemicals in the same concentrations. Both solutions were neutralized by Tris buffer to a pH of 7.4. Standard intracellular relaxing solution used for the resting state was composed of (mm): 74.1 K-Ms, 2 Mg2+, 4.5 Mg-ATP, 1 EGTA, 30 Pipes buffer and 1 DTT (Kitazawa et al. 1991a). The activating solution was 10 mm EGTA, and an appropriate amount of Ca-Ms was added to adjust final [Ca2+] (Horiuti, 1988). All solutions had a pH of 7.1 and ionic strength was kept at 0.2 m.
Proteins
Pig aorta CPI-17 and His 6-tagged CPI-17 were prepared as described previously (Eto et al. 1995, 1997). Recombinant untagged CPI-17 was prepared from lysates of E. coli BL21 (DE3) including pig CPI-17 cDNA at Nde I/EcoR I sites of the pET30 vector (Novagen).
Antibodies
Anti-CPI-17 IgG and IgY antibodies were raised, respectively, in rabbit sera and chicken eggs, using His 6-tagged porcine CPI-17 as an antigen (Senba et al. 1999; Kitazawa et al. 2000). Both antibodies were further purified using Affigel-10 resin that was conjugated with untagged CPI-17. Monoclonal MYPT1 antibodies were from BAbCO. Monoclonal antibodies against α-actin and h-calponin were from Sigma. Secondary antibodies against chicken were from Promega. Anti-mouse secondary antibodies were from Sigma, and anti-rabbit secondary antibodies from Chemicon.
Measurement of force development
The force of contraction was first recorded with high [K+]o (154 mm), in the absence and presence of 100 μm phenylephrine, and the strips were then immersed in the relaxing solution for several minutes. The strips were then treated for 30 min at 30 °C with 20 μg ml−1 of Staphylococcus aureusα-toxin (List, Campbell, CA, USA) at pCa 6.3, buffered with 10 mm EGTA, in order to permeabilize the membrane whilst retaining cytosolic proteins (Masuo et al. 1994). The Ca2+ ionophore A23187 (10 μm; Calbiochem) was then applied to the strips for 20 min at 20 °C in order to remove Ca2+ from the sarcoplasmic reticulum. The force of contraction was then recorded at pCa 6.3 for the PV, TR, UB and VD, and at pCa 6.7 for the FA and AO, for 10 min, and then with either 1 μm PDBu (Gibco BRL) or 30 μm GTPγS (Boehringer Mannheim).
Measurement of MLC phosphorylation
Muscle strips were prepared as described above, and subjected to the same experimental conditions. Permeabilized strips were initially immersed in the appropriate pCa solution and then subjected to PDBu or GTPγS. The tissues were then rapidly frozen in liquid chlorodifluoromethane, cooled by liquid N2. The strips were then placed on top of frozen acetone containing 10 % trichloroacetic acid (TCA) overnight at -80 °C. Then the strips and the TCA-acetone solution were gradually warmed to -20 °C, then 4 °C, and finally to room temperature. TCA was removed by washing with acetone. After air drying the strips, the tissue was homogenized in a buffer containing 0.1 % SDS, 20 mm DTT, 10 % glycerol and 0.1 mg ml−1 bovine serum albumin (BSA). The samples were then subjected to 2-dimensional gel electrophoresis as described previously (Kitazawa et al. 1991a). The isoelectric focusing tube gel was run overnight until a constant current was reached. The tube gels were then run on SDS-PAGE gels, and the proteins transferred to nitrocellulose membrane using a wet transfer technique. The membranes were then washed in phosphate-buffered saline solution (containing 137 mm NaCl, 2.7 mm KCl, 8 mm Na2HPO4 and 1.5 mm KH2PO4) overnight at room temperature. The membranes were rinsed with de-ionized H2O, and then stained with colloidal gold (BioRad). The blots were subsequently scanned and analysed (software from Signal Analytics Co., Vienna, VA, USA). The percentage phosphorylation (PP) was evaluated using the following equation:
where U is unphosphorylated, P1 is monophosphorylated, and P2 is diphosphorylated MLC.
Measurement of protein expression
Three of the selected tissues (FA, PV and VD) possessed smooth muscle layers that could be microscopically dissected from the extracellular non-smooth muscle media. However, as a result of their anatomy, tissue from the aorta (AO), trachea (TR) and urinary bladder (UB) prohibit the removal of the non-smooth muscle component. Thus, the tissues from AO, TR and UB contain significant amounts of non-smooth muscle cells and the measured values for concentrations of h-calponin, CPI-17 and MYPT1, all three of which are smooth muscle specific, are underestimated when total protein content is matched. Our experiments on the first three tissues (FA, PV and VD) indicated an approximate equality concerning smooth muscle-specific h-calponin expression (see Results). Therefore we assumed that h-calponin was equally expressed in smooth muscle tissues of different types and thus we compared CPI-17 and MYPT1 expression using the h-calponin-matched AO, FA, PV, TR, UB and VD samples in an attempt to equalize the smooth muscle cell content in the Western blots. The tissues were cleaned of connective tissue and immersed intact in 10 % TCA-acetone overnight. After TCA treatment, the tissues were washed in acetone and allowed to dry. Homogenization buffer (10 mm Tris, 1 % SDS, pH 7.4) was then added to the tissues in a volume equal to 5 times the wet weight, and the mixture was ground in a glass hand homogenizer. The resultant homogenates were then centrifuged and the supernatants collected. Protein concentration was measured using a BCA protein assay (Pierce). Laemmli sample buffer (LSB; with final concentrations 62.5 mm Tris, 1 % SDS, 15 % glycerol, 30 mm DTT and 0.005 % Bromophenol Blue) was then added to dilute and equalize the protein concentrations. For h-calponin and α-smooth muscle actin measurement, the tissue samples were further diluted with LSB. CPI-17, h-calponin and α-smooth muscle actin were run on 15 %, and MYPT1 on 8 % polyacrylamide gels. Proteins were then transferred to nitrocellulose membranes (Amersham Pharmacia Biotech) using a wet transfer method. For CPI-17, the membranes were blocked in Tris-buffered saline (TBS) solution containing 0.05 % Tween-20, 5 % non-fat milk and 1 % BSA for 3 h at room temperature. For MYPT1, h-calponin and α-smooth muscle actin, the membranes were blocked overnight at room temperature in TBS-Tween-20 with 5 % non-fat milk. After primary antibody treatment, the membranes were incubated with alkaline phosphatase-conjugated secondary antibodies, and the bands were developed with alkaline phosphatase substrate (Sigma). The bands were analysed as described above.
Calibration of staining for CPI-17 and MYPT1
Quantitative immunoassays for comparing the protein content in various smooth muscles were established. For CPI-17 (Fig. 1A), we applied 0-45 ng of recombinant purified protein, which produced a range of staining densities that encompassed the protein band staining from actual tissue samples. For MYPT1 (Fig. 1B), due to the lack of stability of the recombinant protein, we instead used 0-45 μg of total VD protein, which rendered a range spanning the staining densities of the tissue samples. For all Western blotting, we applied between 25 and 37.5 μg of total protein, which fell on the more linear portion of the curve. However, as some of the samples stained on the lower end of the curve, we calibrated our results to reflect the non-linear staining of the MYPT1 antibody. MYPT1 in muscles consisted of two bands, but often the separation was not distinct. MYPT1 has been shown previously to consist of two isoforms (Shimizu et al. 1994). When these were clearly evident as shown here, the staining densities of the bands were added together. These assays allowed comparison of the relative expression of CPI-17 and MYPT1 in multiple tissue samples.
Figure 1. Relationship between amount of samples and density of Western blots for CPI-17 and MYPT1.
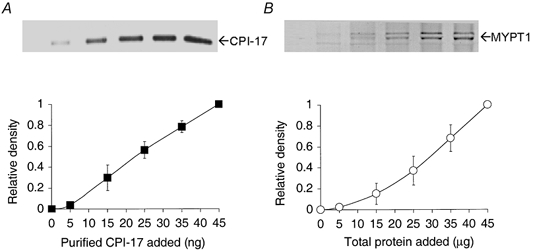
A shows a representative Western blot using polyclonal anti-CPI-17 antibodies against purified recombinant CPI-17; B shows a blot using monoclonal anti-MYPT1 antibodies against a control sample extracted from VD (n = 3 for both). The Western blots were prepared with 0-45 ng of purified protein for CPI-17 and 0-45 μg of total VD protein for MYPT1. Values for no protein were indiscernible from background values.
Estimation of CPI-17 content
Samples with known tissue wet weight and total protein concentration were subjected to Western blotting and their bands were compared to that of samples of purified CPI-17. For each tissue of each animal, at least three blots were carried out to determine the correlating amount of CPI-17. The amount of cellular CPI-17 in the various tissues was then estimated by multiplying by the dilution factor of the original wet sample and then dividing by the molecular weight (16 700) of CPI-17.
Immunohistochemistry
Immunohistochemical analysis was essentially performed as described previously (Everett et al. 2000). Briefly, individual tissues from male rats were fixed in 4 % paraformaldehyde solution for 20 min at room temperature then paraffin embedded for sectioning. Separate tissue sections were incubated with the CPI-17 affinity-purified rabbit polyclonal antibody. Control slides were incubated with non-specific mouse IgG (Vector Laboratories, Burlingame, CA, USA) using the same concentration as the primary antibody. Sections were blocked with horse serum, followed by incubation with the primary antibody. Primary antibody binding was visualized using the ABC method with a red alkaline phosphatase substrate as a fluorescent substrate (Vector Laboratories), according to the manufacturer's protocol. The sections were then mounted between a glass slide and a coverslip with Vectorshield mounting medium (Vector Laboratories). Images were acquired with a Nikon Microphoto-FXA/SA fluorescence microscope attached to a Hamamatsu CCD camera (Eto et al. 2000).
Statistics
Results shown are the means ±s.e.m. of n experiments. The statistical significance was evaluated with Student's two-tailed t test with P < 0.05 considered significant.
RESULTS
Smooth muscle-specific localization of CPI-17
To examine the specific expression of CPI-17, various tissues were harvested from rats for immunohistochemical analysis. Figure 2 shows immunolocalization of CPI-17 in aorta (A), heart (B) and lung (C) as compared to their phase-contrast images. In confirmation of previous Northern analysis of pig RNA (Eto et al. 1997) and Western blot analysis of rat protein (Eto et al. 1999), CPI-17 was expressed in smooth muscle cells of the aortic media but neither in endothelial nor in adventitial cells (Fig. 2A). It should be noted that CPI-17 also appears to be highly expressed in small blood vessels of the peri-adventitial fat (as indicated by arrows in Fig. 2A). Immunostaining of the heart (Fig. 2B) detected CPI-17 with specific localization within the cross-section of the medial wall of a blood vessel, while no CPI-17 was seen in the surrounding cardiac myocytes. In the lung (Fig. 2C), CPI-17 was highly expressed in the smooth muscle layer of a blood vessel and also in smooth muscle along the walls of the alveolar ducts. In Western blotting analysis, small but significant CPI-17 bands were seen for the lung and brain, but not in the heart, liver, kidney, testes, spleen, thymus, or skeletal muscle when total protein was matched (Eto et al. 1999). In the lung, the observation of small amounts of CPI-17 is probably due to the large amount of smooth muscle within the organ (Fig. 2C). This is supported by the fact that the lung also had higher levels of h-calponin and α-smooth muscle actin expression (not shown). However, the brain did not express similar levels of these two smooth muscle-specific proteins (not shown). Western blotting patterns were reproducible using either rabbit or chicken anti-CPI-17 in both the rat and rabbit (not shown).
Figure 2. Smooth muscle-specific expression of CPI-17.
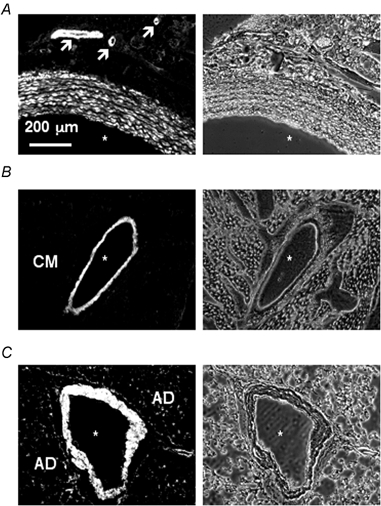
Tissue sections of adult rat aorta (A), heart (B) and lung (C) are shown with both the immunostained image (left) and the corresponding phase contrast image (right). Asterisks mark blood vessel lumens. Arrows in A indicate small blood vessels that stain positively for CPI-17. CM stands for cardiac muscle (B), which does not contain CPI-17. AD stands for alveolar ducts (C), which show some positive staining. Scale bar represents 200 μm.
Expression of CPI-17, MYPT1, h-calponin and α-actin in smooth muscle tissues
Using quantitative Western analysis, the expression levels of several proteins known to be important for PKC-induced calcium sensitization were first examined when total protein concentration was matched in tissue extracts. Figure 3A illustrates the representative Western blots of α-smooth muscle actin, h-calponin, CPI-17, and MYPT1 in extracts from smooth muscle layers of three selected tissues: tonic FA, phasic PV and phasic VD. In further agreement with earlier studies (Fatigati & Murphy, 1984; Szymanski et al. 1998), we found that the expression of α-smooth muscle actin was slightly but significantly greater in tonic as compared to phasic smooth muscles. However, calponin expression did not differ significantly (P > 0.05) among the three different smooth muscles (Fig. 3A and B). In contrast, CPI-17 expression varied greatly among the tissues (Fig. 3A). The CPI-17 content was significantly higher in the tonic FA as compared to the phasic PV, and markedly greater than the phasic visceral VD. When the densities of the FA blots were normalized to 1, the relative value for the PV was 0.70 ± 0.07 and that for the VD was 0.16 ± 0.08 (n = 4). For the evaluation of MLCP content, we determined that the relative expression of MYPT1 among smooth muscle tissues was an appropriate measure of MLCP content as MYPT1 interaction with PP1C is critical to MLCP function (Hartshorne et al. 1998). In contrast to CPI-17 expression, MYPT1 content was significantly higher in the phasic PV and VD as compared to the tonic FA, with values of 3.77 ± 0.47 for the PV and 2.65 ± 0.35 for the VD (n = 4).
Figure 3. Relative expression of α-smooth muscle actin, h-calponin, CPI-17 and MYPT1.
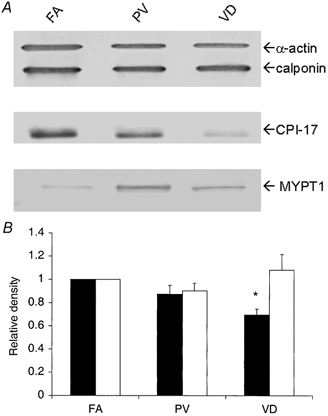
Contractile proteins from three rabbit smooth muscle tissues (FA, femoral artery; PV, portal vein; and VD, vas deferens) were probed. For α-actin and h-calponin, 1.25 μg of total protein was subjected to SDS-PAGE and Western blotting (upper lane in A). For anti-CPI-17 (middle lane) and anti-MYPT1 (lower lane), 25 μg of total protein was used. In B, filled bars are staining intensity for α-actin and open bars are for h-calponin. The densities for the FA were set to 1 and the relative densities for the PV and VD are plotted (n = 4). * Significant difference from the value for FA.
As h-calponin appeared to be equally expressed in smooth muscle regardless of phenotype (Fig. 3B), we applied this observation to experiments for the tonic AO, phasic UB and tonic TR smooth muscle. These were tissues that did not allow for complete removal of the non-smooth muscle component (see Methods). These three new tissues, in addition to those previously discussed (FA, PV and VD), were subjected to Western immunoblotting as shown in Fig. 4. Calponin content was matched to within ±10 % across the six different tissues (shown in the upper panel of Fig. 4A). Although the total protein concentration was not equal, the representative h-calponin-matched Western blots of CPI-17 and MYPT1 are shown in the middle and lower panel of Fig. 4A. Figure 4B (CPI-17) and C (MYPT1) gives a graphical representation of the data. The AO contained significantly more CPI-17 than the FA, while the TR, UB and VD expressed significantly less CPI-17 (n = 3). When the staining density of the FA blot was normalized to 1, the relative values for the AO, PV, TR, UB and VD were, respectively, 1.32 ± 0.11, 0.74 ± 0.15, 0.52 ± 0.20, 0.28 ± 0.06 and 0.24 ± 0.04. For MYPT1 expression, the FA was again normalized to 1 (n = 3). The phasic muscles PV, UB and VD were found to contain more MYPT1 (2.22 ± 0.21, 1.77 ± 0.33 and 1.98 ± 0.23, respectively), whereas the tonic AO, TR and FA expressed significantly less (respectively, 0.98 ± 0.28, 1.23 ± 0.02 and 1). It is worth mentioning that the phasic PV expressed a relatively high level of CPI-17 with significantly elevated MYPT1 expression. The tonic visceral TR contained less CPI-17 than the tonic arteries, and less MYPT1 than the phasic visceral UB and VD. These results show that the amounts of MLCP and its specific inhibitor CPI-17 vary independently in different smooth muscle tissues.
Figure 4. Relative expression of CPI-17 and MYPT-1 in six smooth muscle tissues.
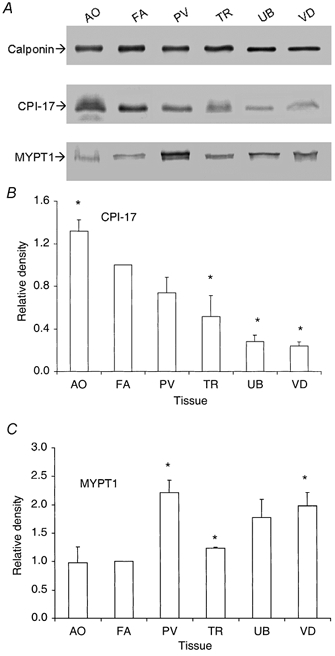
The Western blots of h-calponin, CPI-17 and MYPT1 for six different smooth muscle tissues are shown in A. To better compare CPI-17 and MYPT1 expression among different tissues, the samples were first h-calponin matched using the h-calponin density values. The volumes of the tissue sample for subsequent CPI-17 and MYPT1 blots (n = 3) were adjusted appropriately to give equal amounts of h-calponin in each tissue lane as shown in A. B (CPI-17) and C (MYPT1) are graphical representations of the h-calponin-matched blots. * Significant difference from the FA, which was normalized to 1. AO, aorta; TR, trachea; and UB, urinary bladder.
Concentration of CPI-17 in smooth muscle cells
To estimate the intracellular concentration of CPI-17 in different smooth muscles, samples from the FA, PV and VD were probed along with purified CPI-17 protein. As before, these three tissues were carefully cleaned of non-muscle tissue and the samples were then matched for total protein. A volume equal to 25 μg of total protein was applied for the FA and PV and 50 μg for the VD. The greater volume for the VD was necessary to ensure that the blot fell on the linear portion of the curve of the densities for the purified protein (see Fig. 1). The amount of smooth muscle cell volume as compared to the total smooth muscle layer was allowed for in the dilution factor. We assumed muscle cell volume to be 39 % for the FA (Gong et al. 1992) and 69 % for the PV (Todd et al. 1983). The VD was also assumed to be 69 % as this was the volume previously found for the ileum (Gong et al. 1992). Our results found CPI-17 concentrations in the FA, PV and VD samples to be 6.7 ± 1.1, 4.4 ± 0.7 and 0.8 ± 0.1 μm, respectively.
Tissue-specific PDBu-induced contractions and correlation with CPI-17 expression
To compare the calcium sensitization caused by activation of the PKC-CPI-17 pathway alone as compared to stimulation of all types of G protein, which may activate multiple calcium-sensitizing pathways including RhoA-Rho-kinase-MYPT1 as well as PKC-CPI-17 pathways, we used the PKC activator PDBu and the G protein agonist GTPγS in α-toxin-permeabilized smooth muscle. The use of an α-toxin preparation, in contrast to other permeabilization methods such as β-escin, saponin and Triton X-100, had the advantage of retaining endogenous CPI-17 and other small regulatory proteins, whilst allowing control of the intracellular composition and concentration of low molecular weight solutes (Kitazawa et al. 1999). In the permeabilized tissue strips, [Ca2+] was buffered with 10 mm EGTA and intracellular Ca2+ stores were eliminated with A23187. The higher Ca2+ sensitivity of the FA and AO mandated using a lower [Ca2+] to produce a submaximal level of control contraction near the threshold, similar to the control contractions of the other smooth muscle strips at higher [Ca2+] (Kitazawa et al. 1991a). Use of the same pCa for all tissues would not allow for a clear indication of the Ca2+-sensitizing effects of these agonists. Figure 5 depicts the responses of the various smooth muscle tissues to the addition of PDBu and GTPγS (n = 3-11). As GTPγS markedly potentiated a small force of contraction at a submaximal [Ca2+] in all α-toxin-permeabilized tissues, the control and PDBu-induced contractions were normalized to the force induced by GTPγS for each tissue type. The AO and FA had the most potent responses to PDBu (96 ± 14 and 95 ± 4 % of the contraction induced by GTPγS, respectively), and these contractions did not significantly differ from those caused by GTPγS. However, in the PV, TR, UB and VD, the contraction induced by PDBu was significantly less than that caused by GTPγS (76 ± 2, 66 ± 10, 44 ± 7 and 15 ± 3 %, respectively). In summary, we found the vascular tonic muscles showed the greatest response to PDBu and the visceral phasic muscles displayed the weakest reactions to PDBu as compared to GTPγS.
Figure 5. Calcium sensitization of contractile response of various rabbit smooth muscle tissues.
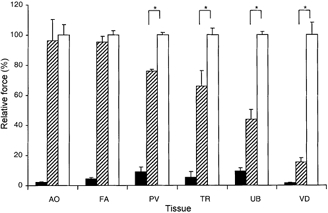
Contractions for each tissue (n = 3-11) were normalized to each tissue's contractile force in response to GTPγS treatment (set as 100 %). Control contractions were measured at pCa 6.7 for the FA and AO and pCa 6.3 for the PV, TR, UB and VD. The temperature for each condition was 20 °C. Either 1 μm PDBu or 30 μm GTPγS was added. * Significant difference between the PDBu- and GTPγS-induced contractions in each smooth muscle tissue. Filled bars are Ca2+ alone, hatched are Ca2++ PDBu and open bars are Ca2++ GTPγS. The results show different PDBu-induced contractions in different smooth muscles. The GTPγS-induced contractions were 55 ± 3.8 % for AO, 64 ± 1.8 % for FA, 64 ± 1.0 % for PV, 69 ± 2.9 % for TR, 82 ± 1.7 % for UB and 70 ± 2.0 % for VD when compared to contractions induced by pCa 4.5 + GTPγS. Note that in phasic tissues, due to high phosphatase activity, the contractions at pCa 4.5 + GTPγS are still less than full maximal contraction (Kitazawa et al. 1991a).
Figure 6 illustrates the relationship between the CPI-17/MYPT1 expression ratios and PDBu-potentiated contractions for the various tissues. The values used for contraction (PDBu-induced contraction - control)/ (GTPγS-induced contraction - control) were normalized to the FA, the result being the relative PDBu-potentiated contractions for each tissue. The PDBu-potentiated contractions tended to increase as the CPI-17/MYPT1 ratio increased.
Figure 6. Correlation between relative expression ratio of CPI-17/MYPT1 and PDBu-potentiated contraction.
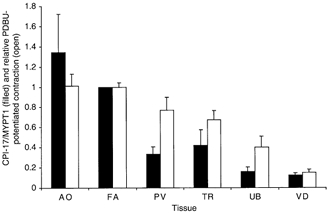
PDBu-potentiated contractions (open bars) were plotted along with relative expression of CPI-17 to MYPT1 (filled bars) obtained from data in Fig. 4. PDBu-potentiated contractions were normalized with the FA = 1. Units assigned to the ratio are arbitrary and do not indicate a molar ratio.
Phosphorylation of MLC and CPI-17 by PDBu and GTPγS
To further examine the PKC-CPI-17 signalling pathway leading to inhibition of MLCP, we measured the phosphorylation of MLC and CPI-17 at constant submaximal [Ca2+] (pCa 6.7 for the FA, 6.3 for the PV and VD) in α-toxin-permeabilized smooth muscle. MLC phosphorylation levels were significantly increased by GTPγS in the three different tissues (Fig. 7A). As was found in the experiments with the contractile force (Fig. 5), MLC phosphorylation in response to the PKC activator PDBu varied among these smooth muscles (n = 3-6). In the FA, PDBu nearly doubled the MLC phosphorylation, from 35 ± 1 % to a level of 59 ± 4 % of total MLC, which was not significantly different from that induced by GTPγS (60 ± 3 %). In the PV, PDBu elevated the level of MLC phosphorylation significantly from 20 ± 3 % to a value of 34 ± 4 %, which was somewhat less than the GTPγS-induced phosphorylation (47 ± 6 %). PDBu did not significantly increase phosphorylation of MLC in the VD (17 ± 2 % as compared to 20 ± 4 %). In addition, the PDBu-induced MLC phosphorylation in the VD was significantly less than that induced by GTPγS (31 ± 2 %).
Figure 7. Phosphorylation levels of MLC and CPI-17 induced by Ca2+, Ca2++ PDBu and Ca2++ GTPγS.
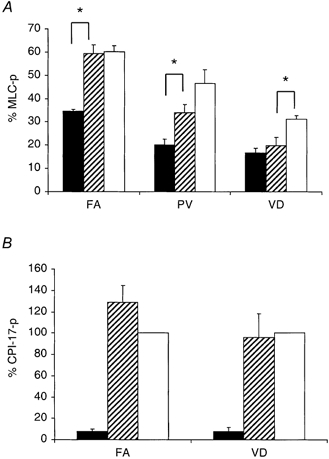
A illustrates MLC phosphorylation expressed as a percentage of total MLC (% MLC-p: n = 3-6) with conditions identical to those of the contraction experiments in Fig. 5. Filled bars are Ca2+ alone control, hatched bars are control + 1 μm PDBu and open bars are control + 30 μm GTPγS. * Significant difference between various treatments. B shows phosphorylation of CPI-17 (CPI-17-p) at Thr-38 in the FA and VD, at control pCa 6.7 (FA) or pCa 6.3 (VD) alone, control + PDBu and control + GTPγS. Values were normalized with GTPγS-induced phosphorylation (100 %).
Using an antibody that specifically recognized only CPI-17 phosphorylated at Thr-38 (Kitazawa et al. 2000), we found that PDBu and GTPγS both greatly increased levels of phosphorylated CPI-17 in the FA to, respectively, 18- and 14-fold the control phosphorylation level of CPI-17 at pCa 6.7 alone (Fig. 7B; n = 3). In the phasic VD, PDBu and GTPγS both increased phosphorylation of CPI-17 at pCa 6.3 by a factor of 13 (Fig. 7B, n = 3). For these experiments the blot densities for the GTPγS-treated samples were normalized to 100 %. The increase in FA CPI-17 phosphorylation closely paralleled both MLC phosphorylation (Fig. 7A) and force development (Fig. 5). However, in the VD, despite a large increase in relative CPI-17 phosphorylation in response to both PDBu and GTPγS (Fig. 7B), which mirrored the effects seen in the FA, contraction and MLC phosphorylation were significantly increased only with GTPγS but not with PDBu (Fig. 7A).
DISCUSSION
This study clearly demonstrates singular expression of CPI-17, a smooth muscle MLCP inhibitor protein. CPI-17 is specifically localized in smooth muscle, yet expression is highly heterogeneous among individual smooth muscle tissues. CPI-17 is generally expressed more in vascular than visceral tissues regardless of contractile characteristics. Within vascular or visceral tissues, the tonic muscle phenotype appears to contain more CPI-17 than phasic muscles, e.g. the tonic AO and FA contain more CPI-17 than the phasic PV, and the tonic TR more CPI-17 than the phasic UB and VD. The relative order of CPI-17 expression found in the different tissues was identical using either calponin-matched or total protein-matched samples. It is also noteworthy that high expression of CPI-17 was observed, using immunohistochemical analyses, in small blood vessels supplying several organs including the aorta (Fig. 2A), brain, heart, lung and small intestine (data not shown). Therefore, expression of CPI-17 is strictly controlled in smooth muscle. CPI-17 is also expressed in cultured aortic smooth muscle cells, but the degree of expression is markedly decreased upon cell passages (M. Eto, unpublished data), suggesting a dependence on contractile phenotype. It is not detected in cultured fibroblasts including REF52, NIH3T3 and COS7 cells (Eto et al. 1999, 2000). CPI-17 expression contrasts with that of another MLCP inhibitor, Rho-kinase, which is ubiquitously expressed in vivo and also in various cultured cells (see Somlyo & Somlyo, 2000).
One major downstream effect of the PKC pathway is inhibition of MLCP with a resultant increase in the calcium sensitivity of MLC phosphorylation and contraction (Itoh et al. 1993; Masuo et al. 1994; Gailly et al. 1997). The amount of MLCP might therefore be one of the modifying factors of PKC activator-induced Ca2+ sensitization of contraction. This study indicates differential expression of MYPT1 (for MLCP) in various smooth muscle tissues. Relative expression correlates to the mode of contraction and relaxation with higher MLCP content in phasic compared to tonic smooth muscle (Kitazawa et al. 1991a; Gong et al. 1992; this study). The rate of MLC dephosphorylation was about 4 times slower in the α-toxin-permeabilized FA as compared to the PV (Gong et al. 1992), consistent with several times less MLCP content in the former as compared to the latter. However, the correlation between MLCP content and PKC-induced contraction is very low, which suggests little association between PKC-induced Ca2+ sensitization and MLCP content alone. Since MLCP activity can be regulated by CPI-17, the balance between CPI-17 and MLCP expression may be related to the various levels of PKC-induced Ca2+ sensitization in different tissues. We estimated the cellular concentration of CPI-17 to be from 0.8 μm in the phasic visceral VD up to 6-7 μm in the tonic arterial FA. Attempts to estimate the concentration of MLCP were frustrated by the instability of recombinant MYPT1. As reported previously, the concentration of MLCP in smooth muscle has been estimated, based on specific activity, to be ‘at least’ sub-micromolar in myofibrils from phasic chicken gizzard muscle (Alessi et al. 1992), suggesting a similar order of magnitude in cellular concentration as compared to CPI-17. In the FA, the majority of CPI-17 was phosphorylated by PDBu (Kitazawa et al. 2000), and the rate of MLC dephosphorylation was inhibited to half of control by a saturated concentration of PDBu (Masuo et al. 1994). Thus we propose that the functional concentration of MLCP is roughly as much as functional CPI-17 in tonic arterial smooth muscle. This results in a large calcium sensitization of MLC phosphorylation and contraction by a PKC activator. Therefore the concentration of CPI-17 is sufficient to act as a physiological inhibitor for MLCP in tonic arterial smooth muscle. Yet in the VD, relative CPI-17 expression is several times less and MYPT1 expression several times more than in the tonic arteries, suggesting that the CPI-17 content is significantly less than the MLCP content. Furthermore, despite a similar relative increase in the amount of CPI-17 phosphorylation in the FA and VD, only the FA had a concurrent increase in MLC phosphorylation and contraction by PDBu. This implies that CPI-17 content limits the effects of PKC activation, and that other G protein pathways are responsible for the GTPγS-induced calcium sensitization in phasic visceral tissue. Application of 5 μm thiophosphorylated exogenous CPI-17 to Triton X-100-demembranated TR, VD and UB visceral smooth muscle also caused a large calcium sensitization (data not shown), as seen in demembranated FA smooth muscle (Li et al. 1998; Kitazawa et al. 1999). This suggests that endogenous CPI-17 in visceral smooth muscle has the same Ca2+-sensitizing effect as in the FA, and that both the cellular concentration of CPI-17 and the ratio between CPI-17 and MLCP are important factors in the regulation of Ca2+ sensitivity. Therefore tonic arterial smooth muscle with high CPI-17 and low MLCP content responds to PKC stimulation with a large increase in Ca2+ sensitivity. In contrast, phasic smooth muscle with low CPI-17 and high MLCP content has a small to insignificant response to a PKC activator at constant [Ca2+]. A tissue such as the phasic PV with relatively high CPI-17 and high MLCP content, or the tonic TR with relatively low CPI-17 and low MLCP content should have a moderate increase in calcium sensitivity due to PKC stimulation. We thus propose that the unique order of the CPI-17/MLCP ratio correlates to the degree of Ca2+ sensitization of contraction and MLC phosphorylation induced by the PKC activator phorbol ester.
In addition to expression levels of CPI-17 and MLCP, protein kinase action on CPI-17 is another factor in the regulation of MLCP under stimulation by Ca2+-sensitizing agonists. Phorbol ester, GTPγS and excitatory agonists, but not high K+ alone, significantly increase the level of phospho-CPI-17 found in α-toxin-permeabilized FA (Kitazawa et al. 2000; this study). Histamine-induced contraction and phosphorylation of CPI-17 is blocked by the G- protein inhibitor GDPβS, and is partially inhibited by either the Rho-kinase inhibitor Y27632 or the PKC inhibitor GF109203X. These results suggest the existence of complex pathways for calcium sensitization. PKC-δ has been isolated as a dominant kinase for CPI-17 from pig AO, and this kinase is also partially inhibited by Y27632 (Eto et al. 2001). Thus, CPI-17 could be phosphorylated by PKC-δ in histamine-stimulated arterial smooth muscles, even though this response is sensitive to different kinase inhibitors. Recently it has been reported that CPI-17 can be phosphorylated at Thr-38 by Rho-kinase and another RhoA-activated protein kinase (PKN) in vitro (Koyama et al. 2000; Hamaguchi et al. 2000). In spite of a low specific activity compared to PKC, phosphorylation of CPI-17 by RhoA-associated kinases might be physiological. We propose that CPI-17 is a point of mutual convergence, where various Ca2+-sensitizing pathways meet for inhibition of MLCP in a particular smooth muscle type.
In tonic arterial smooth muscle, the G protein activator GTPγS increases phosphorylation of in situ CPI-17 to a level similar to that invoked by PDBu (this study), inhibits MLCP activity by about 50 %, which is also similar to PDBu action (Kitazawa et al. 1991b; Masuo et al. 1994), and increases the calcium sensitivity of MLC phosphorylation and contraction to a level close to that induced by PDBu (Masuo et al. 1994; this study). These results imply that in vascular smooth muscle with a high CPI-17/MLCP expression ratio, the G protein-mediated inhibition of MLCP is mainly through the CPI-17-MLCP signalling pathway. Contractile Ca2+ sensitivity in response to a PKC activator appears higher in the arterial smooth muscle of spontaneously hypertensive rats (Silver et al. 1992; Sasajima et al. 1997) and in coronary artery vasospasm in a swine model (Kadokami et al. 1996). These findings suggest that CPI-17 may be involved in vascular tone disorders, although RhoA/Rho-kinase signalling is another strong candidate for these smooth muscle abnormalities (Kandabashi et al. 2000; Sato et al. 2000).
In contrast, the expression of CPI-17 in phasic visceral smooth muscle is significantly less as compared to MYPT1, and the PDBu-induced MLC phosphorylation and contraction are much lower than those caused by GTPγS. This indicates that another mechanism(s) for the G protein-mediated inhibition of MLCP exists in phasic visceral smooth muscle. MYPT1 phosphorylation is another crucial factor involved in the regulation of MLCP. Several G protein-coupled receptor agonists and the direct G protein-activator GTPγS significantly increase MLC phosphorylation through inhibition of MLCP in phasic as well as tonic smooth muscle, and even in cultured smooth muscle and fibroblast cells (see review by Somlyo & Somlyo, 2000). It has recently been shown that MYPT1 is phosphorylated in response to GTPγS in permeabilized portal vein (Trinkle-Mulcahy et al. 1995), ileum (Swärd et al. 2000), cultured aortic smooth muscle cells (Nagumo et al. 2000) and in response to lysophosphatidic acid in Swiss 3T3 cells (Feng et al. 1999). MYPT1 phosphorylation is associated with inhibition of the catalytic activity of MLCP in vitro (Hartshorne et al. 1998), and this phosphorylation is inhibited in situ by Y27632 and HA1077, another Rho-kinase inhibitor (Feng et al. 1999; Swärd et al. 2000; Nagumo et al. 2000). These results support the hypothesis that activation of the RhoA/Rho-kinase pathway leads to phosphorylation of MYPT1 and is the predominant route in phasic muscle for inhibition of MLCP and the concomitant increase in MLC phosphorylation and contraction.
In conclusion, we describe the signalling pathway for calcium sensitization as consisting of several branches and possible parallel pathways. All tissues respond to GTPγS, which would activate heterotrimeric and monomeric G proteins. When using a specific activator (PDBu) of the PKC portion of G protein-induced calcium sensitization, different smooth muscles showed variable responses. The sensitization of contraction in each smooth muscle is individually modulated, not only by variation in agonist receptors, but also by tissue-specific expression of regulatory/contractile proteins and possibly disease state. Yet this is logical in that smooth muscle is highly specialized in a multitude of ways, and the G- protein pathway for calcium sensitization is another example of the intricacies involved in smooth muscle function.
Acknowledgments
We thank Matthew R. Lee for valuable discussion on the manuscript and Mallappa Anitha for technical assistance. This work was supported by National Institutes of Health grants HL51824 to T.K. and CA40042 to D.L.B., and a postdoctoral fellowship from the AHA Mid-Atlantic Affiliate to M.E.
References
- Adelstein RS, Sellers JR. Myosin structure and function. In: Barany M, editor. Biochemistry of Smooth Muscle Contraction. San Diego CA USA: Academic Press; 1996. pp. 3–19. [Google Scholar]
- Alessi D, MacDougall LK, Sola MM, Ikebe M, Cohen P. The control of protein phosphatase-1 by targeting subunits: The major myosin phosphatase in avian smooth muscle is a novel form of protein phosphatase-1. European Journal of Biochemistry. 1992;210:1023–1035. doi: 10.1111/j.1432-1033.1992.tb17508.x. [DOI] [PubMed] [Google Scholar]
- Barany K, Barany M. Myosin light chains. In: Barany M, editor. Biochemistry of Smooth Muscle Contraction. San Diego CA USA: Academic Press; 1996. pp. 21–35. [Google Scholar]
- Dirksen WP, Vladic F, Fisher SA. A myosin phosphatase targeting subunit isoform transition defines a smooth muscle developmental phenotypic switch. American Journal of Physiology. 2000;278:C589–600. doi: 10.1152/ajpcell.2000.278.3.C589. [DOI] [PubMed] [Google Scholar]
- Eto M, Karginov A, Brautigan DL. A novel phosphoprotein inhibitor of protein type-1 phosphatase holoenzymes. Biochemistry. 1999;38:16952–16957. doi: 10.1021/bi992030o. [DOI] [PubMed] [Google Scholar]
- Eto M, Kitazawa T, Yazawa M, Mukai H, Ono Y, Brautigan DL. Histamine-induced vasoconstriction involves phosphorylation of a specific inhibitor protein for myosin phosphatase by protein kinase C alpha and delta isoforms. Journal of Biological Chemistry. 2001 doi: 10.1074/jbc.M103206200. in the Press. [DOI] [PubMed] [Google Scholar]
- Eto M, Ohmori T, Suzuki M, Furuya K, Morita F. A novel protein phosphatase-1 inhibitory protein potentiated by protein kinase C. Isolation from porcine aorta media and characterization. Journal of Biochemistry. 1995;118:1104–1107. doi: 10.1093/oxfordjournals.jbchem.a124993. [DOI] [PubMed] [Google Scholar]
- Eto M, Senba S, Morita F, Yazawa M. Molecular cloning of a novel phosphorylation-dependent inhibitory protein of protein phosphatase-1 (CPI17) in smooth muscle: specific location in smooth muscle. FEBS Letters. 1997;410:356–360. doi: 10.1016/s0014-5793(97)00657-1. [DOI] [PubMed] [Google Scholar]
- Eto M, Wong L, Yazawa M, Brautigan DL. Inhibition of myosin/moesin phosphatase by expression of the phosphoinhibitor protein CPI-17 alters microfilament organization and retards cell spreading. Cell Motility and the Cytoskeleton. 2000;46:222–234. doi: 10.1002/1097-0169(200007)46:3<222::AID-CM6>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Everett AD, Lobe DR, Matsumura ME, Nakamura H, McNamara CA. Hepatoma-derived growth factor stimulates smooth muscle cell growth and is expressed in vascular development. Journal of Clinical Investigation. 2000;105:567–575. doi: 10.1172/JCI7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatigati V, Murphy RA. Actin and tropomyosin variants in smooth muscles. Dependence on tissue type. Journal of Biochemistry. 1984;259:14383–14388. [PubMed] [Google Scholar]
- Feng JH, Ito M, Kureishi Y, Ichikawa K, Amano M, Isaka N, Okawa K, Iwamatsu A, Kaibuchi K, Hartshorne DJ, Nakano T. Rho-associated kinase of chicken gizzard smooth muscle. Journal of Biological Chemistry. 1999;274:37385–37390. doi: 10.1074/jbc.274.6.3744. [DOI] [PubMed] [Google Scholar]
- Gailly P, Gong MC, Somlyo AV, Somlyo AP. Possible role of atypical protein kinase C activated by arachidonic acid in Ca2+ sensitization of rabbit smooth muscle. Journal of Physiology. 1997;500:95–109. doi: 10.1113/jphysiol.1997.sp022002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong MC, Cohen P, Kitazawa T, Ikebe M, Masuo M, Somlyo AP, Somlyo AV. Myosin light chain phosphatase activities and the effects of phosphatase inhibitors in tonic and phasic smooth muscle. Journal of Biological Chemistry. 1992;267:14662–14668. [PubMed] [Google Scholar]
- Hamaguchi T, Ito M, Feng J, Seko T, Koyama M, Machida H, Takase K, Amano M, Kaibuchi K, Hartshorne DJ, Nakano T. Phosphorylation of CPI-17, an inhibitor of myosin phosphatase, by protein kinase N. Biochemical and Biophysical Research Communications. 2000;274:825–830. doi: 10.1006/bbrc.2000.3225. [DOI] [PubMed] [Google Scholar]
- Hartshorne DJ. Biochemistry of the contractile process in smooth muscle. In: Johnson LR, editor. Physiology of the Gastrointestinal Tract. NY USA: Raven Press; 1987. pp. 423–482. [Google Scholar]
- Hartshorne DJ, Ito M, Erdodi F. Myosin light chain phosphatase: subunit composition, interactions and regulation. Journal of Muscle Research and Cell Motility. 1998;19:325–341. doi: 10.1023/a:1005385302064. [DOI] [PubMed] [Google Scholar]
- Himpens B, Matthijs G, Somlyo AP. Desensitization to cytoplasmic Ca2+ and Ca2+ sensitivities of guinea-pig ileum and rabbit pulmonary artery smooth muscle. Journal of Physiology. 1989;413:489–503. doi: 10.1113/jphysiol.1989.sp017665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himpens B, Matthijs G, Somlyo AV, Butler TM, Somlyo AP. Cytoplasmic free calcium, myosin light chain phosphorylation, and force in phasic and tonic smooth muscle. Journal of General Physiology. 1988;92:713–729. doi: 10.1085/jgp.92.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuti K. Mechanism of contracture on cooling of caffeine-treated frog skeletal muscle fibres. Journal of Physiology. 1988;398:131–148. doi: 10.1113/jphysiol.1988.sp017034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh H, Shimomura A, Okubo S, Ichikawa K, Ito M, Konishi T, Nakano T. Inhibition of myosin light chain phosphatase during Ca2+-independent vasocontraction. American Journal of Physiology. 1993;265:C1319–1324. doi: 10.1152/ajpcell.1993.265.5.C1319. [DOI] [PubMed] [Google Scholar]
- Kadokami T, Shimokawa H, Fukumoto Y, Ito A, Takayanagi T, Egashira K, Takeshita A. Coronary artery spasm does not depend on the intracellular calcium store but is substantially mediated by the protein kinase C-mediated pathway in a swine model with interleukin-1β in vivo. Circulation. 1996;94:190–196. doi: 10.1161/01.cir.94.2.190. [DOI] [PubMed] [Google Scholar]
- Kamm KE, Stull JT. Regulation of smooth muscle contractile elements by second messengers. Annual Review of Physiology. 1989;51:299–313. doi: 10.1146/annurev.ph.51.030189.001503. [DOI] [PubMed] [Google Scholar]
- Kandabashi T, Shimokawa H, Miyata K, Kunihiro I, Kawano Y, Fukata Y, Higo T, Egashira K, Takahashi S, Kaibuchi K, Takeshita A. Inhibition of myosin phosphatase by upregulated Rho-kinase plays a key role for coronary artery spasm in a porcine model with interleukin-1β. Circulation. 2000;101:1319–1323. doi: 10.1161/01.cir.101.11.1319. [DOI] [PubMed] [Google Scholar]
- Kawano Y, Fukata Y, Oshiro N, Amano M, Nakamura T, Ito M, Matsumura F, Inagaki M, Kaibuchi K. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. Journal of Cell Biology. 1999;147:1023–1037. doi: 10.1083/jcb.147.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil RA, Lajoie C, Resnick MS, Morgan KG. Ca2+-independent isoforms of protein kinase-C differentially translocate in smooth muscle. American Journal of Physiology. 1992;263:C714–719. doi: 10.1152/ajpcell.1992.263.3.C714. [DOI] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng JH, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-Associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Eto M, Woodsome TP, Brautigan DL. Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. Journal of Biological Chemistry. 2000;275:9897–9900. doi: 10.1074/jbc.275.14.9897. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Gaylinn BD, Denney GH, Somlyo AP. G-protein-mediated Ca2+ sensitization of smooth muscle contraction through myosin light chain phosphorylation. Journal of Biological Chemistry. 1991a;266:1708–1715. [PubMed] [Google Scholar]
- Kitazawa T, Masuo M, Somlyo AP. G protein-mediated inhibition of myosin light chain phosphatase in vascular smooth muscle. Proceedings of the National Academy of Sciences of the USA. 1991b;88:9307–9310. doi: 10.1073/pnas.88.20.9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa T, Takizawa N, Ikebe M, Eto M. Reconstitution of protein kinase C-induced contractile calcium sensitization in Triton X-100-demembranated rabbit arterial smooth muscle. Journal of Physiology. 1999;520:139–152. doi: 10.1111/j.1469-7793.1999.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama M, Ito M, Feng J, Seko T, Shiraki K, Takase K, Hartshorne DJ, Nakano T. Phosphorylation of CPI-17, an inhibitory phosphoprotein of smooth muscle myosin phosphatase, by Rho-kinase. FEBS Letters. 2000;475:197–200. doi: 10.1016/s0014-5793(00)01654-9. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Nomura M, Kamm KE, Mumby MC, Stull JT. GTPγS-dependent regulation of smooth muscle contractile elements. American Journal of Physiology. 1992;262:C405–410. doi: 10.1152/ajpcell.1992.262.2.C405. [DOI] [PubMed] [Google Scholar]
- Li L, Eto M, Lee MR, Morita F, Yazawa M, Kitazawa T. Possible involvement of the novel CPI-17 protein in protein kinase C signal transduction of rabbit arterial smooth muscle. Journal of Physiology. 1998;508:871–881. doi: 10.1111/j.1469-7793.1998.871bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuo M, Reardon S, Ikebe M, Kitazawa T. A novel mechanism for the calcium sensitizing effect of protein kinase C on vascular smooth muscle: Inhibition of myosin light chain phosphatase. Journal of General Physiology. 1994;104:265–286. doi: 10.1085/jgp.104.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagumo H, Sasaki Y, Ono Y, Okamoto H, Seto M, Takuwa Y. Rho kinase inhibitor HA-1077 prevents Rho-mediated myosin phosphatase inhibition in smooth muscle cells. American Journal of Physiology. 2000;278:C57–65. doi: 10.1152/ajpcell.2000.278.1.C57. [DOI] [PubMed] [Google Scholar]
- North AJ, Gimona M, Lando Z, Small JV. Actin isoform compartments in chicken gizzard smooth muscle cells. Journal of Cell Science. 1994;107:445–455. doi: 10.1242/jcs.107.3.445. [DOI] [PubMed] [Google Scholar]
- Sasajima H, Shima H, Toyoda Y, Kimura K, Yoshikawa A, Hano T, Nishio I. Increased Ca2+ sensitivity of contractile elements via protein kinase C in alpha-toxin permeabilized SMA from young spontaneously hypertensive rats. Cardiovascular Research. 1997;36:86–91. doi: 10.1016/s0008-6363(97)00131-4. [DOI] [PubMed] [Google Scholar]
- Sato M, Tani E, Fujikawa H, Kaibuchi K. Involvement of Rho-kinase-mediated phosphorylation of myosin light chain in enhancement of cerebral vasospasm. Circulation Research. 2000;87:195–200. doi: 10.1161/01.res.87.3.195. [DOI] [PubMed] [Google Scholar]
- Senba S, Eto M, Yazawa M. Identification of trimeric myosin phosphatase (PP1M) as a target for a novel PKC-potentiated protein phosphatase-1 inhibitory protein (CPI17) in porcine aorta smooth muscle. Journal of Biochemistry. 1999;125:354–362. doi: 10.1093/oxfordjournals.jbchem.a022294. [DOI] [PubMed] [Google Scholar]
- Shimizu H, Ito M, Miyahara M, Ichikawa K, Okubo S, Konishi T, Naka M, Tanaka T, Hirano K, Hartshorne DJ, Nakano T. Characterization of the myosin-binding subunit of smooth muscle myosin phosphatase. Journal of Biological Chemistry. 1994;269:30407–30411. [PubMed] [Google Scholar]
- Silver PJ, Cumiskey WR, Harris AL. Vascular protein kinase C in Wistar-Kyoto and spontaneously hypertensive rats. European Journal of Pharmacology. 1992;212:143–149. doi: 10.1016/0014-2999(92)90322-u. [DOI] [PubMed] [Google Scholar]
- Somlyo AV, Somlyo AP. Electromechanical and pharmacomechanical coupling in vascular smooth muscle. Journal of Pharmacology and Experimental Therapeutics. 1968;159:129–145. [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Signal transduction by G-proteins, Rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. Journal of Physiology. 2000;522:177–185. doi: 10.1111/j.1469-7793.2000.t01-2-00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swärd K, Dreja K, Susnjar M, Hellstrand P, Hartshorne DJ, Walsh MP. Inhibition of Rho-associated kinase blocks agonist-induced calcium sensitization of myosin phosphorylation and force in guinea-pig ileum. Journal of Physiology. 2000;522:33–49. doi: 10.1111/j.1469-7793.2000.0033m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski PT, Chacko TK, Rovner AS, Goyal RK. Differences in contractile protein content and isoforms in phasic and tonic smooth muscles. American Journal of Physiology. 1998;275:C684–692. doi: 10.1152/ajpcell.1998.275.3.C684. [DOI] [PubMed] [Google Scholar]
- Todd ME, Laye CG, Osborne DN. The dimensional characteristics of smooth muscle in rat blood vessels. A computer-assisted analysis. Circulation Research. 1983;53:319–331. doi: 10.1161/01.res.53.3.319. [DOI] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L, Ichikawa K, Hartshorne DJ, Siegman MJ, Butler TM. Thiophosphorylation of the 130 kDa subunit is associated with a decreased activity of myosin light chain phosphatase in alpha-toxin-permeabilized smooth muscle. Journal of Biological Chemistry. 1995;270:18191–18194. doi: 10.1074/jbc.270.31.18191. [DOI] [PubMed] [Google Scholar]
- Woodsome T, Kitazawa T. CPI-17 and myosin light chain phosphatase expression in smooth muscle. Biophysical Journal. 2000;78:111A. doi: 10.1074/jbc.275.14.9897. [DOI] [PubMed] [Google Scholar]