

Abstract
The production of the central inhibitory transmitter GABA (γ-aminobutyric acid) varies in response to different patterns of activity. It therefore seems possible that GABA metabolism can determine inhibitory synaptic strength and that presynaptic GABA content is a regulated parameter for synaptic plasticity.
We altered presynaptic GABA metabolism in cultured rat hippocampal slices using pharmacological tools. Degradation of GABA by GABA-transaminase (GABA-T) was blocked by γ-vinyl-GABA (GVG) and synthesis of GABA through glutamate decarboxylase (GAD) was suppressed with 3-mercaptopropionic acid (MPA). We measured miniature GABAergic postsynaptic currents (mIPSCs) in CA3 pyramidal cells using the whole-cell patch clamp technique.
Elevated intra-synaptic GABA levels after block of GABA-T resulted in increased mIPSC amplitude and frequency. In addition, tonic GABAergic background noise was enhanced by GVG. Electron micrographs from inhibitory synapses identified by immunogold staining for GABA confirmed the enhanced GABA content but revealed no further morphological alterations.
The suppression of GABA synthesis by MPA had opposite functional consequences: mIPSC amplitude and frequency decreased and current noise was reduced compared with control. However, we were unable to demonstrate the decreased GABA content in biochemical analyses of whole slices or in electron micrographs.
We conclude that the transmitter content of GABAergic vesicles is variable and that postsynaptic receptors are usually not saturated, leaving room for up-regulation of inhibitory synaptic strength. Our data reveal a new mechanism of plasticity at central inhibitory synapses and provide a rationale for the activity-dependent regulation of GABA synthesis in mammals.
The brain adapts to experience by changing synaptic efficacy. Extensively studied mechanisms of plasticity include changes in the probability of transmitter release and in postsynaptic responsiveness. However, evidence from aminergic synapses indicates that the biochemistry of neurotransmitters provides an additional tool to alter synaptic strength. For example, dopaminergic transmission can be boosted by the precursor l-DOPA, which enhances the transmitter content of individual vesicles (Pothos et al. 1996) and counteracts Parkinson's disease.
A similar principle may apply to one of the major central transmitters, the inhibitory amino acid GABA (γ-aminobutyric acid). Drugs enhancing the tissue content of GABA act as anticonvulsants (Löscher et al. 1989; Taylor et al. 1992), suggesting that inhibitory tone depends on GABA supply. Moreover, the production of endogenous GABA varies, depending on the functional needs of the network: gonadotropin secretion from hypothalamic neurons is regulated through the negative feedback of gonadal steroids on the activity of the GABA-producing enzyme glutamate decarboxylase (GAD; Grattan et al. 1996); in hippocampal interneurons, suppression of GAD expression by oestradiol results in decreased inhibition and subsequent changes in dendritic spine density (Murphy et al. 1998); GABA is down-regulated in sensory cortical areas following deafferentiation (Garraghty et al. 1991); and hippocampal interneurons react to repeated seizures with an up-regulation of GAD expression (Feldblum et al. 1990). These observations support the idea that presynaptic GABA concentration is varied in response to altered levels of activity in neuronal networks. It remains to be elucidated whether this adaptation constitutes a genuine (primary) mechanism of plasticity at mammalian inhibitory synapses or occurs secondarily in response to an altered synaptic use of GABA.
A decreased quantal size upon suppression of GAD activity (Murphy et al. 1998) or after non-specific block of vesicular transmitter loading (Zhou et al. 2000) can be well explained by a lower vesicular GABA content. An increased inhibitory strength arising from elevated GABA levels is more difficult to understand at the synaptic level. It is feasible that vesicular GABA content is enhanced in this situation and that the release of larger vesicles causes larger postsynaptic responses. This parsimonious explanation depends on two critical factors: (i) transport of GABA into vesicles is not saturated under normal conditions, i.e. there is room for bi-directional regulation of vesicular GABA content, and (ii) the postsynaptic GABAA receptors are not saturated by the transmitter content of single vesicles, i.e. there is a receptor reserve (Frerking et al. 1995; Hájos et al. 2000). Together, these features would allow for a biochemical gain control at inhibitory synapses.
In order to elucidate the potential for synaptic plasticity through alterations in GABA metabolism we used two opposite pharmacological tools to increase or decrease the presynaptic transmitter content of interneurons in cultured rat hippocampal brain slices. GABA degradation by GABA-transaminase (GABA-T) was blocked by treatment with γ-vinyl-GABA (GVG; Jung et al. 1977), whereas in other slices GABA synthesis was suppressed by the GAD-blocking agent 3-mercaptopropionic acid (MPA; Murphy et al. 1998). Analysis of spontaneous miniature inhibitory postsynaptic currents (mIPSCs) in CA3 pyramidal cells revealed that the efficacy of central GABAergic synapses can be up- and down-regulated by altering the production or degradation of GABA.
METHODS
Hippocampal slice culture
Young (6- to 9-day-old) Wistar rats of either sex were anaesthetized with ether, decapitated and the brain removed. Animal procedures were approved by the local ethics committee and concord with national law. The hippocampus was dissected and slices were cut at a thickness of 400 μm on a McIlwain tissue chopper and cultured on porous membranes (Millicell-CM, 0.4 μm pores) using the method of Stoppini et al. (1991). The culture medium contained minimal essential medium (MEM, Life Technologies, Karlsruhe, Germany; including 1 mg l−1 pyridoxal HCl), Hanks’ buffered saline solution (HBSS, Life Technologies) and 25 % horse serum (Life Technologies). GVG (γ-vinyl-GABA; 40 μm) was added to the medium beginning at day 9 in vitro for the next 4 days in order to reach a saturating effect on GABA levels (Rimvall & Martin, 1992) and to outlast any possible initial hyperexcitability (Löscher et al. 1989). MPA (3-mercaptopropionic acid; 10 μm) was added overnight before the measurements (between days 12 and 15 in vitro), similar to Murphy et al. (1998). For electrophysiological or biochemical analysis, controls were always taken from the same preparations as drug-treated slices.
Biochemical studies
Tissue GABA content was measured by HPLC in three to four slices from three independent cultures after homogenization of the tissue in 0.1 n HCl (1-3 slices 100 μl−1) by ultrasound sonification. After centrifugation (12 000 g, 10 min, 4 °C) the supernatant was rapidly frozen. The pellet was resuspended in 50 μl Triton X-100 (1 %) and the protein content was determined using the BCA method. Values were expressed as picomoles of GABA per microgram of protein (means ±s.d).
Electrophysiological recordings
Prior to experiments, the slices were removed from the incubator and drugs were washed out with artificial cerebrospinal fluid (ACSF, see below) for 30-120 min. Whole-cell patch clamp recordings (Blanton et al. 1989) were performed at room temperature (20-25 °C) using an EPC-7 amplifier (List Medical, Darmstadt, Germany). Pipette resistance was 2-5 MΩ, leading to series resistances between 4 and 17 MΩ, which were regularly controlled throughout the experiment. The intracellular solution contained (mm): CsCl 125, MgCl2 2, CaCl2 2, Hepes 10 and EGTA 10, pH 7.3; the extracellular ACSF was composed of (mm): NaCl 129, NaHCO3 21, KCl 3, NaH2PO4 1.25, CaCl2 1.6, MgSO4 1.8 and glucose 10, saturated with 95 % O2-5 % CO2, pH 7.4. Miniature IPSCs were recorded at a holding potential of -60 mV in the presence of 10 μm 6-nitro-7-sulphamoylbenzo(f)quinoxlaine-2,3-dione (NBQX), 30 μmd-aminophosphonovalerate (APV) and 0.5 μm TTX. Currents were amplified (5-10 mV pA−1), filtered at 3 kHz and stored on videotape.
Data analysis and statistics
Synaptic events were analysed off-line after filtering at 1 kHz and redigitized at 8 kHz with a CED 1401 interface (Cambridge Electronic Design, Cambridge, UK). Individual mIPSCs were detected with the CDR program (Strathclyde Electrophysiology Software, courtesy of Dr J. Dempster) using a detection trigger level of 5 pA and were separated from noise through inspection by eye. The SCAN program of the same software package was used to create amplitude distribution histograms (one bin corresponding to 2 pA) and interevent interval histograms (one bin corresponding to 20 ms).
The statistical significance of differences in mean values was tested with the non-parametric Mann-Whitney U test and differences between amplitude or interevent interval distributions were tested with the non-parametric Kolmogorov-Smirnov test, with P < 0.05 taken as significant.
Electron microscopy
For electron microscopy, slice cultures were fixed for 30 min in 4 % formaline, 0.05 % glutaraldehyde and 0.2 % picric acid in 0.1 m phosphate buffer, pH 7.4. They were dehydrated and flat-embedded in Araldite. Ultrathin sections were subjected to the immunogold post-embedding technique using a rabbit antiserum against GABA (Sigma, A 2052) at 1:1000 dilution. Synapses on CA3 neurons showing clear GABA immunoreactivity were quantitatively analysed for the number of gold grains per square area.
Drugs were purchased from Sigma (Deisenhofen, Germany) and Tocris (Cologne, Germany). γ-Vinyl-GABA was a kind gift from Hoechst Marion Roussel (Frankfurt, Germany).
RESULTS
Inhibitory transmission is up- or down-regulated by blockers of GABA degradation or synthesis
We recorded spontaneous TTX-resistant mIPSCs from CA3 pyramidal layer neurons in the absence of GVG or MPA. Currents from cells in which GABA degradation had been suppressed with GVG (40 μm, 4 days) appeared much less steady, displaying an increased amplitude and frequency of spontaneous synaptic events (Fig. 1) as well as an apparently increased baseline noise in event-free intervals (Fig. 2). Current decay constants were assessed from ≥ 30 averaged events from control and GVG-treated cells and were not significantly different (control, 19.4 ± 1.5 ms; GVG, 17.3 ± 1.2 ms; n = 11 each, P = 0.37, Mann-Whitney U test). The increase in mean mIPSC amplitude (control, 45.5 ± 3.5 pA; GVG-teated, 61.6 ± 4.1 pA; n = 11, P < 0.02, Mann-Whitney U test) was confirmed by a significant shift of the amplitude histograms towards larger events (Fig. 1B and C). The functional changes in the presence of GVG were accompanied by an enhanced overall GABA content of hippocampal slice cultures (about 4-fold increase from 126 ± 12 to 507 ± 33 pmol (μg protein)−1; n = 3 separate cultures) and by an increased number of immunogold particles for GABA in inhibitory terminals on CA3 pyramidal cells (see below, Fig. 5).
Figure 1. Effects of GVG and MPA on mIPSCs in CA3 pyramidal layer neurons.
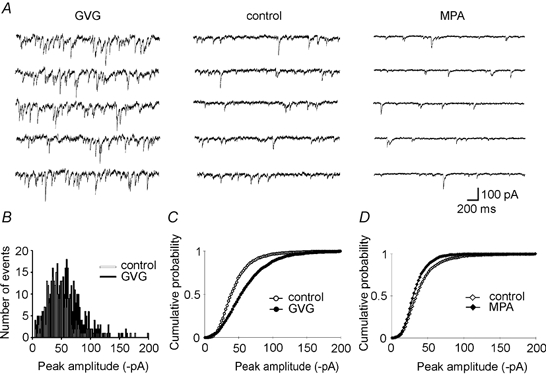
A, specimen recordings from CA3 cells of cultured rat hippocampal slices treated with GVG (left), kept in control conditions (middle) and treated with MPA (left). Note the increase in mIPSC amplitude and frequency in the presence of GVG. In the MPA-treated cell, mIPSC frequency reduction is obvious while the changes in amplitude are more subtle (see D). Baseline noise was enhanced (GVG) or reduced (MPA) compared with control. B, mIPSC amplitude histograms of the control (□) and GVG-treated (▪) cells shown in A. Note the broader distribution of amplitudes in the presence of GVG, which is reflected by an increased variance derived from Gaussian fits to the distributions (coefficient of variation (CV) = 0.39 ± 0.02 in control cells, n = 11 cells; CV = 0.47 ± 0.02 in GVG-treated cells, n = 11; P = 0.01, Mann-Whitney U test). C, cumulative probability of mIPSC amplitudes pooled from 11 control cells (○) and 11 GVG-treated cells (•), showing a significant shift towards larger amplitudes (P < 0.05, Kolmogorov-Smirnov test). D, cumulative probability of mIPSC amplitudes pooled from 6 control cells (⋄) and 7 MPA-treated cells (♦). Points are significantly shifted towards smaller amplitudes (P < 0.01) while the corresponding decrease in mean amplitude (17 %) was not significant.
Figure 2. Differences in baseline noise.
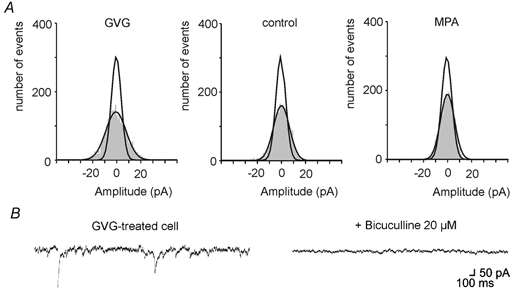
A, representative all-point histograms of 1 s of event-free recordings from a GVG-treated (left), control (middle) and MPA-treated (right) cell. Note the broader distribution of baseline noise (shaded area) in the cell with elevated GABA content (left) and the low noise after treatment with the GAD-blocker MPA. Mean (±s.d.) of Gaussians fitted to baseline noise distributions from 5 cells in each group was 5.2 ± 0.4 pA in control, 8.7 ± 0.4 pA in GVG-treated cells (P < 0.01 compared with control, Mann-Whitney U test) and 2.7 ± 0.2 pA in MPA-treated cells (P < 0.01 compared with control, Mann-Whitney U test). Additional Gaussian curves in the 3 graphs show the narrowing of baseline noise after addition of 20 μm bicuculline, indicating that a large proportion of the noise in control and GVG-treated cells is caused by activation of GABAA receptors. B, sections of current traces recorded in a GVG-treated cell before and after application of 20 μm bicuculline. Note the decrease of the noise.
Figure 5. Morphology of inhibitory synapses in control- and GVG-treated slices.
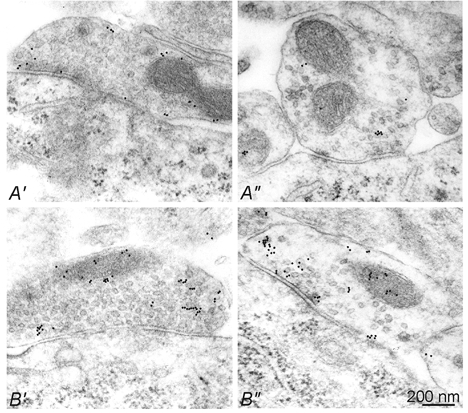
Hippocampal slices either untreated (A′, A′) or treated with 40 μm GVG (B′, B′) were subjected to post-embedding immunogold staining using a rabbit antiserum against GABA. The density of gold particles was increased in GVG-treated synapses. In the left and right panels, two representative types of GABAergic terminal contacting CA3 pyramidal cell somata are shown. One type is characterized by densely packed synaptic vesicles (A′, B′), while in the other type the vesicles are more loosely packed (A′, B′′), similar to the heterogeneity of synapses in normal rat neocortex (Peters et al. 1991).
As can be seen in Fig. 1A, suppression of the GABA-producing enzyme GAD by MPA (10 μm overnight) caused opposite effects to GVG: mIPSC amplitude distributions were significantly shifted towards smaller values (see Fig. 1D), baseline noise apparently decreased (Fig. 2) and the frequency of events appeared reduced (Fig. 1A). Similar to GVG, MPA did not change the event kinetics. The ∼17 % reduction in mean amplitude was, however, less dramatic than the opposite change in the presence of GVG and did not reach significance (MPA-treated cells versus control, P 0.5). We were also unable to demonstrate a reduction of global tissue GABA concentration or changes in immunogold reactivity for GABA in inhibitory terminals. Thus, functional changes in the presence of MPA are consistent with a lowered vesicular GABA content but are more subtle than the up-regulation of GABA in the presence of GVG. Nonetheless, the data show a bi-directional regulation of postsynaptic current amplitude following manipulation of GABA metabolism. The increased amplitude upon enhancing presynaptic GABA is consistent with sub-saturation of postsynaptic inhibitory GABAA receptors in CA3 pyramidal cells. This was confirmed by effects of the GABAA receptor modulator diazepam (10 μm), which significantly enhanced the amplitudes of mIPSCs in control and GVG-treated slices (data not shown).
In addition to changes in the amplitude of rapidly rising synaptic currents, we also found differences in baseline noise. Cells from GVG-treated slices showed a larger variation of the current baseline than control cells, while cells from MPA-treated slices were more quiescent (Fig. 2). The enhanced baseline noise in GVG-treated slices and the noise in control slices were partially sensitive to the GABAA receptor antagonist bicuculline. Thus, there is a tonic GABAergic inhibition in CA3 pyramidal cells from cultured hippocampal slices that depends on the cellular GABA content, at least in somatic recordings.
GABA metabolism influences mIPSC frequency
From the original recordings in Fig. 1A it is apparent that the frequency of mIPSCs increased after block of GABA degradation (with GVG) and decreased after suppression of GABA synthesis (with MPA). Indeed, interevent intervals were changed by both drugs (Fig. 3). In GVG-treated slices, the mean interevent interval decreased from 640 ms to 301 ms, increasing mIPSC frequency by 100 %. There is, however, a possible artefact when event frequencies are assessed by a threshold detection algorithm: the increased size of mIPSCs in the presence of GVG will facilitate the detection of currents that were below threshold in control slices. We therefore repeated event detection in recordings from GVG-treated slices with an enhanced detection threshold (+35 %, compensating for the mean amplitude increase). This did not alter the leftward shift of cumulative probability plots of interevent interval (Fig. 3B), indicating that the increased number of detected events per time interval was not secondary to the amplitude increase. Moreover, the pronounced baseline noise shown in Fig. 2 led to a deteriorated signal-to-noise ratio in recordings from GVG-treated cells, which counteracts the detection of events rather than causing artificially high frequencies. Again, MPA had the opposite effect: the mean frequency of mIPSCs was reduced by ∼40 %, as reflected by the rightward shift of cumulative interevent interval plots (Fig. 3C). This decrease, however, might be a consequence of the reduced amplitude, as has recently been reported for event frequencies after non-specific depletion of vesicular transmitter content by bafilomycin A1 (Zhou et al. 2000). Addition of the GABAB receptor antagonist CGP 55845A (1-2 μm) did not alter the frequency or amplitude of events in three control and one GVG-treated slice.
Figure 3. Effects of changes in GABA metabolism on mIPSC frequency.
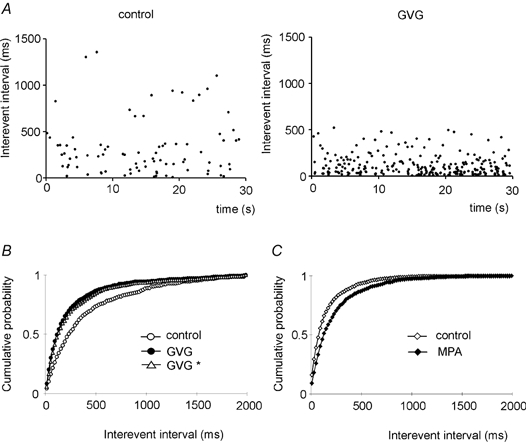
A, scatter plots of consecutive interevent intervals detected during 30 s of recordings from a control (left) and a GVG-treated (right) neuron. Note the high density of events with short interevent intervals in the cell with an enhanced GABA content. B, cumulative interevent intervals from control (○) and GVG-treated cells (•). Re-analysis of the GVG data with an increased detection threshold (+35 %, GVG *, ▵), which compensates for the mean increase in mIPSC amplitude, is also shown. Both curves from GVG-treated cells show significantly shorter interevent intervals (P < 0.01 for standard analysis and P < 0.05 for enhanced threshold, Kolmogorov-Smirnov test), corresponding to an increase in the frequency of spontaneous mIPSCs by 99 %. C, cumulative interevent interval plot for MPA-treated cells (n = 7, ♦) versus control (n = 6, ⋄). The rightward shift in the presence of MPA (P < 0.01, Kolmogorov-Smirnov test) indicates increased intervals between mIPSCs, corresponding to a decrease in mean frequency by 40 %.
Cells from tissue with an increased GABA content showed a peculiar distribution of interevent intervals, which appeared as bursts of mIPSCs (Fig. 4A, see also Fig. 1A). In interevent interval histograms, events following each other within ∼120 ms were much more frequent than expected from the mean interevent interval of 301 ms. Comparison with control cells revealed significant increases in the number of mIPSCs following the preceding events at such short intervals (Fig. 4B and C), most drastically within the first 40 ms. Thus, the general increase in frequency is partially due to the occurrence of brief bursts of mIPSCs in slices with an enhanced presynaptic GABA content.
Figure 4. Increased number of mIPSC bursts in GVG-treated cells.
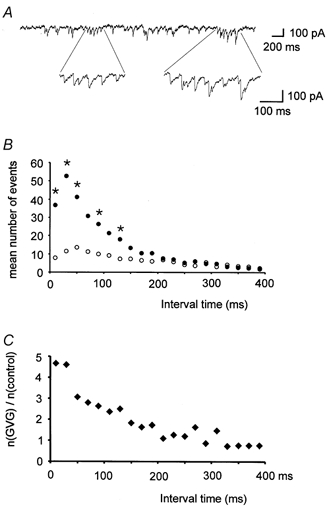
A, sample trace from a GVG-treated cell showing the occurrence of bursts of mIPSCs. B, mean number of events within 20 ms classes of interevent intervals (11 cells each for control (○) and GVG-treated cells (•)). Note the large increase in short interevent intervals in the presence of GVG. Significant differences between GVG-treated and control cells (P < 0.05, Mann-Whitney U test) are marked by asterisks. C, relative frequency of events in 20 ms classes of interevent intervals (mean number of events (n) in GVG-treated cells divided by mean number in control cells). GVG-treated cells show a 4- to 5-fold increase in the number of mIPSCs within 40 ms after the preceding mIPSC. At intervals beyond 150 ms there is virtually no difference in the number of detected events.
Synaptic GABA content increases after treatment with GVG
In order to assess the effects of GVG on the GABA content of individual inhibitory synaptic terminals, we analysed slices at the electron microscopic level (Fig. 5). GABAergic synapses were identified by post-embedding immunogold staining for GABA. Incubation with GVG increased the densitiy of gold particles per unit area within inhibitory terminals approximately 2.5-fold (39 particles μm−2 in control versus 103 particles μm−2 in GVG, 11 versus 13 synapses, respectively; P = 0.025, Mann-Whitney U test). In tissue treated with MPA, however, we did not find any change in GABA immunoreactivity, consistent with the biochemical assay. We observed two morphologically distinct types of perisomatic synapses in the CA3 pyramidal cell layer (Fig. 5, compare A′ and B′ with A′ and B′). One type of GABAergic terminal showed a strikingly low density of vesicles close to the synaptic cleft (Fig. 5A′ and B′), reminiscent of recently published examples from chronically epileptic rats with a decreased frequency of mIPSCs (Hirsch et al. 1999). In our preparation this type of naturally occurring GABAergic synapse (Peters et al. 1991) was approximately doubled in number after GVG treatment (20/44 synapses versus 8/29 synapses in control slices). This went along, however, with increased, rather than decreased, mIPSC frequency, indicating that the changes in spontaneous vesicular release of GABA are unlikely to be caused by a structural reorganization of synaptic terminals.
DISCUSSION
GABA metabolism controls synaptic efficacy
Our data show a direct interdependence between GABAergic synaptic efficacy and GABA metabolism at mammalian central inhibitory synapses. Block of GABA degradation increased and block of GABA synthesis decreased the amplitude and frequency of mIPSCs. These findings have several implications for our understanding of synaptic function in the mammalian CNS and provide a rationale for many observations showing that GABA and GAD are regulated in an activity-dependent manner.
Vesicular GABA transport and postsynaptic GABAA receptors are not saturated
The increase in mIPSC amplitude after treatment of slice cultures with GVG is most easily explained by greater amounts of transmitter being released from individual vesicles. GABA is taken up into vesicles by the vesicular GABA transporter VGAT (McIntire et al. 1997), which is driven by an electrochemical gradient provided by the vesicular proton pump. This proton pump is present on all types of secretory vesicles and its block decreases vesicular content and thus depresses mIPSC and mEPSC amplitude without differentiating between vesicular subpopulations (Zhou et al. 2000). Conversely, over-expression of vesicular neurotransmitter transporters has been used to increase the quantal size of peripheral cholinergic Xenopus neurons (Song et al. 1997) and midbrain dopaminergic cells (Pothos et al. 2000). These findings indicate that vesicles can accept larger numbers of transmitter molecules, probably by increasing in volume (Frerking et al. 1995; Colliver et al. 2000; Hájos et al. 2000). At dopaminergic synapses, the intravesicular amount of transmitter can also be varied by enhancing dopamine production (Pothos et al. 1996, Pothos 2000). Here we show, for the first time, that similar mechanisms exist for a major central transmitter system, using small synaptic vesicles. Immunogold staining of GABA in sections prepared for electron micrographs revealed a clear increase in the presynaptic GABA content after treatment with GVG. The increased amplitudes of mIPSCs show that this increase in presynaptic transmitter concentration leads to an enhanced vesicular GABA content, indicating that the accumulation of GABA in synaptic vesicles does not normally reach saturation. Probably, cytosolic GABA levels are not far above the Km value of the vesicular GABA transporter VGAT (∼5 mm; McIntire et al. 1997), so the transport rate can easily be varied. This would leave room for an efficient up- and down-regulation of vesicular GABA content and for the fine-tuning of inhibitory strength.
At the postsynaptic site, our data show that there are sufficient GABAA receptors to detect enhanced amounts of vesicular GABA. Non-saturating receptor occupancy has previously been concluded from experiments with benzodiazepines, which enhance the affinity of GABAA receptors to GABA (Frerking et al. 1995; Perrais & Ropert, 1999; Hájos et al. 2000). While this approach faces experimental complications like the temperature dependence of the effects (Perrais & Ropert, 1999) or possible changes in single channel current amplitude by the drugs (Eghbali et al. 1997), our experiments, using presynaptic changes in GABA metabolism as a tool, provide independent evidence favouring the concept of non-saturation.
The enhanced GABAergic background noise will add to the increase in tonic inhibition in the presence of GVG. This phenomenon may reflect a constantly enhanced extracellular GABA concentration, either due to the increased frequency and GABA content of vesicular release or due to non-vesicular release of GABA through GABA transporters, which can operate in the reversed direction (Schwartz, 1987). Alternatively, what appears as tonic GABAergic membrane noise at our somatic recording site may reflect summed and electrotonically filtered mIPSCs at remotely located synapses.
It is interesting to note that neither the increased mIPSC frequency nor the apparent tonic activation of GABAA receptors after GVG treatment decreased postsynaptic receptor sensitivity due to slow desensitization. This form of inactivation has recently been reported to decrease miniature and evoked IPSC amplitude in cultured hippocampal neurons after block of GABA uptake (Overstreet et al. 2000). In our system, if present, desensitization appears to be overrun by the increased number of synaptically released GABA molecules, activating additional postsynaptic GABAA receptors.
mIPSC bursts indicate release of multiple vesicles
The appearance of mIPSC bursts in slices with enhanced GABA content suggests that mIPSCs within such a sequence are not independent from each other. Enhanced GABA content may increase the speed of vesicle turnover or the number of vesicles in the readily releasable pool, so that spontaneous calcium fluctuations in single terminals cause release of multiple vesicles. Alternatively, exocytosis of one vesicle may facilitate the exocytosis of further vesicles. Such a positive feedback of GABA on its own release can result from depolarizing actions of GABA at presynaptic GABAA autoreceptors, as described for hippocampal interneurons (Vautrin et al. 1994). Presynaptic depolarization by GABA has been previously demonstrated in salamander retinal neurons (Kamermans & Werblin, 1992) and rat hippocampal CA3 pyramidal cells (Stasheff et al. 1993). In accordance with this idea, the reduction of GABA in crayfish synapses reduces the probability of release (Golan & Grossmann, 1994, Golan 1996).
Hippocampal neurons experience a constant bombardment with GABAergic vesicles at proximally located synapses (Soltesz et al. 1995). Thus, the observed increase in mIPSC frequency is likely to enhance somatic inhibition very efficiently. Moreover, an increased frequency of action potential-independent synaptic currents may go along with an increase in action potential-dependent release probability (Prange & Murphy, 1999), strengthening feedback and feedforward inhibition in hippocampal circuits.
Our electron micrographs did not reveal obvious structural alterations that could explain the facilitated and grouped spontaneous release of vesicles after treatment with GVG. Synapses with sparsely distributed vesicles were present in control and GVG-treated slices but were more frequent in preparations with an enhanced GABA content. This synaptic morphology is known from the intact brain (Peters et al. 1991), but has recently been related to a reduced mIPSC frequency in chronic epileptic tissue (Hirsch et al. 1999), contrary to our observations in GVG-treated slices. Thus, the functional characteristics of different morphological types of GABAergic synapse remains a complicated issue. In any case, we found no gross alterations at the ultrastructural level upon prolonged incubation with GVG and the observed changes in mIPSC frequency are likely to be caused by functional, rather than structural alterations.
mIPSCs are depressed by MPA
The depression of mIPSC amplitude and frequency in the presence of MPA, an inhibitor of GABA synthesis, shows that plasticity through GABA metabolism is bi-directional. Murphy et al. (1998) have reported decreased intracellular GABA levels in cultured hippocampal neurons that were exposed to MPA or to the gonadal steroid oestradiol. Treatment with the hormone depressed mIPSC amplitude and frequency, similar to our data, and caused a decreased GABA staining. Thus, our findings are in line with a decreased vesicular GABA content although we failed to directly demonstrate the altered GABA concentration with biochemical assays or electron microscopy. Possibly, MPA selectively depresses GABA synthesis at synaptic vesicles (where the GAD65 isoform is located; Hsu et al. 1999), while the cytosolic isoform GAD67 is up-regulated (Rimvall & Martin, 1992). Disruption of the gene coding for GAD65 in mice causes a rather mild phenotype without alteration of mIPSC amplitude (Tian et al. 1999), indicating that a radical depression of GAD65 can be almost completely compensated by increased activity of GAD 67. Interestingly, such animals have a reduced visual cortical plasticity (Hensch et al. 1998). Thus, the regulation of inhibitory strength by GAD65 contributes to the functional adaptation of cortical networks.
Variations of GABA metabolism as a mechanism of synaptic plasticity
Our data demonstrate that the regulation of GABA metabolism provides a versatile tool for synaptic plasticity in hippocampal interneurons. Indeed, GABA is up- and down-regulated in the intact and diseased brain. Besides regulation by gonadal steroids (Grattan et al. 1996; Murphy et al. 1998), there is evidence for an activity-dependent tuning of GABA metabolism: hyper-activity of local networks, as induced in experimental models of epilepsy, up-regulates GAD in interneurons (Feldblum et al. 1990; Houser & Esclapez, 1996). Conversely, depriving cortical circuits of their natural input results in the down-regulation of GABA, possibly maintaining a higher level of activity in the deafferented cells (Garraghty et al. 1991). Moreover, it has been reported that plastic changes at GABAergic striatal synapses occur in conjunction with altered elementary IPSC amplitude, possibly by varying vesicular GABA content (Behrends & ten Bruggencate, 1998).
Our data reveal the microphysiological changes upon alteration of presynaptic GABA content and thus provide a rationale for the previously observed plasticity of GABA and GAD expression in the mammalian hippocampus, neocortex and hypothalamus. The machinery for vesicle filling, release and postsynaptic response does not work at maximal load and is therefore sensitive to variations of presynaptic GABA concentration. Thus, GABA metabolism is a variable parameter for the adaptation of synaptic strength to altered requirements.
Acknowledgments
We thank Drs Rosemarie Grantyn, Sergej Kirischuk, Kai Kaila and Uwe Heinemann for helpful discussions, and Evelyn Heuckendorf and Oliver Kann for excellent technical help. γ-Vinyl-GABA (Vigabatrin) was a kind gift from Hoechst Marion Roussel. This work was supported by the Deutsche Forschungsgemeinschaft, SFB 515 A4 and B1.
References
- Behrends JC, Ten Bruggencate G. Changes in quantal size distributions upon experimental variations in the probability of release at striatal inhibitory synapses. Journal of Neurophysiology. 1998;79:2999–3011. doi: 10.1152/jn.1998.79.6.2999. [DOI] [PubMed] [Google Scholar]
- Blanton MG, Loturco JJ, Kriegstein AR. Whole cell recording from neurons in slices of reptilian and mammalian cerebral cortex. Journal of Neuroscience Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- Colliver TL, Pyott SJ, Achalabun M, Ewing AG. VMAT-mediated changes in quantal size and vesicular volume. Journal of Neuroscience. 2000;20:5276–5282. doi: 10.1523/JNEUROSCI.20-14-05276.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eghbali M, Curmi JP, Birnir B, Gage PW. Hippocampal GABAA channel conductance increased by diazepam. Nature. 1997;388:71–75. doi: 10.1038/40404. [DOI] [PubMed] [Google Scholar]
- Feldblum S, Ackermann RF, Tobin AJ. Long-term increase of glutamate decarboxylase mRNA in a rat model of temporal lobe epilepsy. Neuron. 1990;5:91–100. doi: 10.1016/0896-6273(90)90172-c. [DOI] [PubMed] [Google Scholar]
- Frerking M, Borges S, Wilson M. Variation in GABA mini amplitude is the consequence of variation in transmitter concentration. Neuron. 1995;15:885–895. doi: 10.1016/0896-6273(95)90179-5. [DOI] [PubMed] [Google Scholar]
- Garraghty PE, Lachica EA, Kaas JH. Injury-induced reorganization of somatosensory cortex is accompanied by reductions in GABA staining. Somatosensory and Motor Research. 1991;8:347–354. doi: 10.3109/08990229109144757. [DOI] [PubMed] [Google Scholar]
- Golan H, Grossmann Y. Block of GABA-transaminase modifies GABAergic transmission at the crayfish synapses. Journal of Neurophysiology. 1994;71:48–58. doi: 10.1152/jn.1994.71.1.48. [DOI] [PubMed] [Google Scholar]
- Golan H, Grossmann Y. Block of glutamate decarboxylase decreases GABAergic inhibition at the crayfish synapses: possible role of presynaptic metabotropic mechanisms. Journal of Neurophysiology. 1996;75:2089–2098. doi: 10.1152/jn.1996.75.5.2089. [DOI] [PubMed] [Google Scholar]
- Grattan DR, Rocca MS, Strauss KI, Sagrillo CA, Selmanoff M, McCarthy MM. GABAergic neuronal activity and mRNA levels for both forms of glutamic acid decarboxylase (GAD65 and GAD67) are reduced in the diagonal band of Broca during the afternoon of proestrus. Brain Research. 1996;733:46–55. doi: 10.1016/0006-8993(96)00532-x. [DOI] [PubMed] [Google Scholar]
- Hájos N, Nusser Z, Rancz EA, Freund TF, Mody Y. Cell-type and synapse-specific variability in synaptic GABAA receptor occupancy. European Journal of Neuroscience. 2000;12:810–818. doi: 10.1046/j.1460-9568.2000.00964.x. [DOI] [PubMed] [Google Scholar]
- Hensch TK, Fagiolini M, Mataga N, Stryker MP, Baekkeskov S, Kash SF. Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science. 1998;282:1504–1508. doi: 10.1126/science.282.5393.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JC, Agassandian C, Merchan-Perez A, Ben-Ari Y, Defelipe J, Esclapez M, Bernard C. Deficit of quantal release of GABA in experimental models of temporal lobe epilepsy. Nature Neuroscience. 1999;2:499–500. doi: 10.1038/9142. [DOI] [PubMed] [Google Scholar]
- Houser CM, Esclapez M. Vulnerability and plasticity of the GABA system in the pilocarpine model of spontaneous recurrent seizures. Epilepsy Research. 1996;26:207–218. doi: 10.1016/s0920-1211(96)00054-x. [DOI] [PubMed] [Google Scholar]
- Hsu CC, Thomas C, Chen W, Davis KM, Foos T, Chen JL, Wu E, Floor E, Schloss JV, Wu J-Y. Role of synaptic vesicle proton gradient and protein phosphorylation on ATP-mediated activation of membrane-associated brain glutamate decarboxylase. Journal of Biological Chemistry. 1999;274:24366–24371. doi: 10.1074/jbc.274.34.24366. [DOI] [PubMed] [Google Scholar]
- Jung MJ, Lippert B, Metcalf BW, Bohlen P, Schechter PJ. gamma-Vinyl GABA (4-amino-hex-5-enoic acid), a new selective irreversible inhibitor of GABA-T: effects on brain GABA metabolism in mice. Journal of Neurochemistry. 1977;29:797–802. doi: 10.1111/j.1471-4159.1977.tb10721.x. [DOI] [PubMed] [Google Scholar]
- Kamermans M, Werblin F. GABA-mediated positive autofeedback loop controls horizontal cell kinetics in tiger salamander retina. Journal of Neuroscience. 1992;12:2451–2463. doi: 10.1523/JNEUROSCI.12-07-02451.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W, Jäckel R, Müller F. Anticonvulsant and proconvulsant effects of inhibitors of GABA degradation in the amygdala-kindling model. European Journal of Pharmacology. 1989;163:1–14. doi: 10.1016/0014-2999(89)90389-0. [DOI] [PubMed] [Google Scholar]
- McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM. Identification and charaterization of the vesicular GABA transporter. Nature. 1997;389:870–876. doi: 10.1038/39908. [DOI] [PubMed] [Google Scholar]
- Murphy DD, Cole NB, Greenberger V, Segal M. Estradiol increases dendritic spine density by reducing GABA neurotransmission in hippocampal neurons. Journal of Neuroscience. 1998;18:2550–2559. doi: 10.1523/JNEUROSCI.18-07-02550.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet LS, Jones MV, Westbrook GL. Slow desensitization regulates the availability of synaptic GABAA receptors. Journal of Neuroscience. 2000;20:7914–7921. doi: 10.1523/JNEUROSCI.20-21-07914.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrais D, Ropert N. Effect of zolpidem on miniature IPSCs and occupancy of postsynaptic GABAA receptors in central synapses. Journal of Neuroscience. 1999;19:578–588. doi: 10.1523/JNEUROSCI.19-02-00578.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Paley SL, Webster DH. Neurons and their Supporting Cells. 3. New York Oxford: Oxford University Press; 1991. The Fine Structure of the Nervous System; p. p.164. [Google Scholar]
- Pothos EN, Desmond M, Sulzer D. l-3,4-Dihydroxyphenylalanine increases the quantal size of exocytotic dopamine release in vitro. Journal of Neurochemistry. 1996;66:629–636. doi: 10.1046/j.1471-4159.1996.66020629.x. [DOI] [PubMed] [Google Scholar]
- Pothos EN, Larsen KE, Krantz DE, Liu YJ, Haycock JW, Setlik W, Gershon MD, Edwards RH, Sulzer D. Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size. Journal of Neuroscience. 2000;20:7297–7306. doi: 10.1523/JNEUROSCI.20-19-07297.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prange O, Murphy TH. Correlation of miniature synaptic activity and evoked release probability in cultures of cortical neurons. Journal of Neuroscience. 1999;19:6427–6438. doi: 10.1523/JNEUROSCI.19-15-06427.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimvall K, Martin DL. Increased intracellular γ-aminobutyric acid selectively lowers the level of the larger of two glutamate decarboxylase proteins in cultured GABAergic neurons from rat cerebral cortex. Journal of Neurochemistry. 1992;58:158–166. doi: 10.1111/j.1471-4159.1992.tb09291.x. [DOI] [PubMed] [Google Scholar]
- Schwartz EA. Depolarization without calcium can release γ-aminobutyric acid from a retinal neuron. Science. 1987;238:350–355. doi: 10.1126/science.2443977. [DOI] [PubMed] [Google Scholar]
- Soltesz I, Smetters DK, Mody I. Tonic inhibition originates from synapses close to the soma. Neuron. 1995;14:1273–1283. doi: 10.1016/0896-6273(95)90274-0. [DOI] [PubMed] [Google Scholar]
- Song H-J, Ming G, Fon E, Bellocchio E, Edwards RH, Poo M. Expression of a putative vesicular acetylcholine transporter facilitates quantal transmitter packaging. Neuron. 1997;18:815–826. doi: 10.1016/s0896-6273(00)80320-7. [DOI] [PubMed] [Google Scholar]
- Stasheff SF, Mott DD, Wilson WA. Axon terminal hyperexcitability associated with epileptogenesis in vitro. II. Pharmacological regulation by NMDA and GABAA receptors. Journal of Neurophysiology. 1993;70:976–984. doi: 10.1152/jn.1993.70.3.976. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs P-A, Muller D. A simple method for organotypic cultures of nervous tissue. Journal of Neuroscience Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Taylor CP, Vartanian MG, Andruszkiewicz R, Silverman RB. 3-Alkyl GABA and 3-alkylglutamic acid analogues: two new classes of anticonvulsant agents. Epilepsy Research. 1992;11:103–110. doi: 10.1016/0920-1211(92)90044-t. [DOI] [PubMed] [Google Scholar]
- Tian N, Petersen C, Kash S, Baekkeskov S, Copenhagen D, Nicoll R. The role of the synthetic enzyme GAD65 in the control of neuronal γ-aminobutyric acid release. Proceedings of the National Academy of Sciences of the USA. 1999;96:12911–12916. doi: 10.1073/pnas.96.22.12911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vautrin J, Schaffner AE, Barker JL. Fast presynaptic GABAA receptor-mediated Cl− conductance in cultured rat hippocampal neurones. Journal of Physiology. 1994;479:53–63. doi: 10.1113/jphysiol.1994.sp020277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Petersen CCH, Nicoll RA. Effects of reduced vesicular filling on synaptic transmission in rat hippocampal neurones. Journal of Physiology. 2000;525:195–206. doi: 10.1111/j.1469-7793.2000.t01-1-00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]