

Abstract
Measurements of cell capacitance were used to investigate the molecular mechanisms by which somatostatin inhibits Ca2+-induced exocytosis in single rat glucagon-secreting pancreatic α-cells.
Somatostatin decreased the exocytotic responses elicited by voltage-clamp depolarisations by 80 % in the presence of cyclic AMP-elevating agents such as isoprenaline and forskolin. Inhibition was time dependent and half-maximal within 22 s.
The inhibitory action of somatostatin was concentration dependent with an IC50 of 68 nm and prevented by pretreatment of the cells with pertussis toxin. The latter effect was mimicked by intracellular dialysis with specific antibodies to Gi1/2 and by antisense oligonucleotides against G proteins of the subtype Gi2.
Somatostatin lacked inhibitory action when applied in the absence of forskolin or in the presence of the L-type Ca2+ channel blocker nifedipine. The size of the ω-conotoxin-sensitive and forskolin-independent component of exocytosis was limited to 60 fF. By contrast, somatostatin abolished L-type Ca2+ channel-dependent exocytosis in α-cells exposed to forskolin. The magnitude of the latter pool amounted to 230 fF.
The inhibitory effect of somatostatin on exocytosis was mediated by activation of the serine/threonine protein phosphatase calcineurin and was prevented by pretreatment with cyclosporin A and deltamethrin or intracellularly applied calcineurin autoinhibitory peptide. Experiments using the stable ATP analogue AMP-PCP indicate that somatostatin acts by depriming of granules.
We propose that somatostatin receptors associate with L-type Ca2+ channels and couple to Gi2 proteins leading to a localised activation of calcineurin and depriming of secretory granules situated close to the L-type Ca2+ channels.
The hormone glucagon is secreted from pancreatic α-cells during hypoglycaemia. Hormones and neurotransmitters such as adrenaline and somatostatin, which stimulate and inhibit glucagon release, respectively, modulate the release of the glucagon. Somatostatin is produced and secreted from the δ-cells, which are juxtaposed to the α-cells in the outer part of the islet of Langerhans (Göpel et al. 2000). This paracrine regulation of glucagon secretion is principally mediated by somatostatin receptors of the SSTR2-subtype (Kuman et al. 1999; Strowski et al. 2000) and is believed to involve several mechanisms. First, it suppresses α-cell electrical activity by activation of a sulphonylurea-insensitive low-conductance K+ channel (Yoshimoto et al. 1999; Gromada et al. 2001). Second, somatostatin inhibits cyclic AMP production and thus reduces protein kinase A-dependent secretion (Fehmann et al. 1995). Third, somatostatin suppresses Ca2+-dependent exocytosis by a mechanism exerted distally to the elevation of cytoplasmic Ca2+ concentration (Ding et al. 1997).
The molecular mechanisms by which somatostatin modulates exocytosis in the α-cells remain largely unestablished. There is evidence that the secretory granules in the α-cell, by analogy to what is the case in other neuroendocrine cells, can be functionally subdivided into a reserve pool and a readily releasable pool (RRP). Most of the granules belong to the reserve pool and only ∼100 are immediately available for release (Gromada et al. 1997). The process by which the granules go from the reserve pool to the RRP (mobilisation) is poorly characterised in α-cells but is accelerated by agents that activate protein kinase A (Gromada et al. 1997). In β-cells (Vallar et al. 1987; Eliasson et al. 1997) as well as pituitary melanotrophs (Parsons et al. 1995) and adrenal chromaffin cells (Holz et al. 1989) there is also evidence that ATP hydrolysis (priming) is required for the granules to attain release competence. Whether this is also the case in the α-cell is unclear. It is likewise unknown whether granules that have already been primed can lose their release competence by depriming.
Here we have used high-resolution capacitance measurements to explore the mechanisms by which somatostatin suppresses exocytosis in single rat pancreatic α-cells. We demonstrate that the secretory granules become release competent by ATP-dependent priming and that somatostatin inhibits exocytosis by Gi2-dependent activation of the protein phosphatase calcineurin. Evidence is also provided that somatostatin acts by selectively depriming granules associated with L-type Ca2+ channels whereas exocytosis of granules close to N-type Ca2+ channels is unaffected, suggesting that somatostatin receptors may exclusively locate to the L-type Ca2+ channels.
METHODS
Preparation of rat α-cells
Male Lewis rats (250-300 g; Møllegaard, Lille Skensved, Denmark) were anaesthetised with sodium pentobarbitone (100 mg kg−1i.p.) and killed by cervical dislocation. The animal procedures were approved by the local ethical committees in Copenhagen and in Lund. After removal of the pancreas, islets were isolated by collagenase digestion and dispersed into single cells using dispase as detailed elsewhere (Høy et al. 2000). Most of the experiments were performed on α-cells separated by fluorescence-activated cell sorting (FACS; Josefsen et al. 1996). Based on the hormone contents and their glucose sensitivities, we estimate that the preparations contain > 80 %α-cells and < 3 %β-cells (Josefsen et al. 1996; Gromada et al. 1997). A few experiments were performed on α-cells in a crude islet cell preparation. The α-cells were then identified by their small cell size (cell capacitance < 2.5 pF). Cells identified by this method had properties indistinguishable from those observed in α-cells obtained by FACS. For all experiments, the cells were plated on 35 mm diameter Petri dishes and incubated in a humidified atmosphere for up to 5 days in RPMI 1640 tissue culture medium (Gibco BRL, Life Technologies Ltd, Paisley, UK) supplemented with 10 % (v/v) heat-inactivated fetal calf serum, 100 i.u. ml−1 penicillin and 100 μg ml−1 streptomycin.
Electrophysiology
Patch pipettes were pulled from borosilicate glass (tip resistance 3-4 MΩ when filled with the pipette solution), coated with Sylgard and fire polished before use. The zero current potential was adjusted before establishment of the seal with the pipette in the bath. The holding potential in all experiments was -70 mV.
Exocytosis was monitored in single α-cells as changes in cell membrane capacitance using either the standard or the perforated-patch whole-cell configuration. An EPC-7 patch-clamp amplifier (List Elektronik, Darmstadt, Germany) was used and exocytosis was elicited by 500 ms voltage-clamp depolarisations from -70 to 0 mV. Changes in cell capacitance were detected using in-house software written in Axobasic (Axon Instruments, Foster City, CA, USA; Ämmäläet al. 1993). Briefly, a 28 mV peak-to-peak 800 Hz sine wave was added to the holding potential (-70 mV) and 10 cycles were averaged for each data point. The resulting current was analysed at two orthogonal phase angles with a resolution of 100 ms per point. The phase angle was determined before each depolarisation by varying the Gseries (series conductance) and Cslow (cell capacitance) settings of the patch-clamp amplifier until a change in Gseries did not influence the measured cell capacitance. During the experiments the cells were situated in an experimental chamber with a volume of 0.4 ml, which was continuously superfused at a rate of 1.5 ml min−1 to maintain the temperature at +33 °C.
Measurements of [Ca2+]i
The [Ca2+]i measurements were made using an Axiovert 135 inverted microscope equipped with a Plan-Neofluar × 100, 1.30 NA objective (Carl Zeiss, Oberkochen, Germany) and an Ionoptix (Milton, MA, USA) fluorescence imaging system as described elsewhere (Bokvist et al. 1995). The experiments were conducted using the perforated-patch whole-cell configuration with the pipette-filling solution specified below. Prior to the experiments, the cells were loaded with 0.2 μm fura-2 AM (Molecular Probes, Eugene, OR, USA) for 16-18 min. Calibration of the fluorescence ratios was performed by using the standard whole-cell configuration to infuse fura-2 with different mixtures of Ca2+ and EGTA to obtain a known [Ca2+]i.
Glucagon release measurements
Groups of 12 freshly isolated rat pancreatic islets were preincubated for 30 min at 37 °C in 1 ml Krebs-Ringer bicarbonate buffer consisting of (mm): 120 NaCl, 25 mm NaHCO3, 4.7 KCl, 1.2 MgSO4, 2.5 CaCl2, 1.2 KH2PO, 1 glucose and 10 Hepes (pH 7.4). The medium was gassed with 95 % O2-5 % CO2 to obtain constant pH and oxygenation. After preincubation, the buffer was changed to a medium supplemented with the test agents (from the sources stated below) and the islets were incubated for another 60 min at 37 °C. Immediately after incubation, a 25 μl aliquot of the medium was removed for assay of glucagon as described elsewhere (Panagiotidis et al. 1992).
Solutions for electrophysiology
The pipette solution for standard whole-cell experiments (Figs 3, 4 and 11) contained (mm): 125 caesium glutamate, 10 CsCl, 10 NaCl, 1 MgCl2, 5 Hepes, 0.05 EGTA, 3 Mg-ATP or 3 AMP-PCP, 0.1 cAMP and 0.01 GTP (pH 7.15 with CsOH). In perforated-patch whole-cell experiments, the pipette solution consisted of (mm): 76 Cs2SO4, 10 NaCl, 10 KCl, 1 MgCl2 and 5 Hepes (pH 7.35 with CsOH). Electrical contact with the cell interior was established by adding 0.24 mg ml−1 amphotericin B to the pipette solution. Perforation required a few minutes and the voltage clamp was considered satisfactory when Gseries was constant and > 35-40 nS. The extracellular medium consisted of (mm): 118 NaCl, 20 tetraethylammonium (TEA)-Cl, 5.6 KCl, 1.2 MgCl2, 2.6 CaCl2, 5 Hepes (pH 7.40 using NaOH) and 5 d-glucose. TEA-Cl was included to block outward rectifying K+ currents, which persist even after replacement of intracellular K+ with Cs+. In some of the perforated-patch whole-cell recordings, forskolin was included in the extracellular solution to increase the exocytotic capacity. Somatostatin-14 (Sigma) was used throughout this study. ω-Conotoxin was obtained from Alomone Labs (Jerusalem, Israel). Calcineurin autoinhibitory peptide was supplied by Calbiochem (La Jolla, CA, USA). All other chemicals were purchased from Sigma. Somatostatin, forskolin, deltamethrin, permethrin and okadaic acid were dissolved in dimethylsulphoxide (DMSO; final concentration of DMSO: 0.01-0.1 %). All other compounds were dissolved in water.
Figure 3. Somatostatin produces G protein-dependent inhibition of exocytosis.
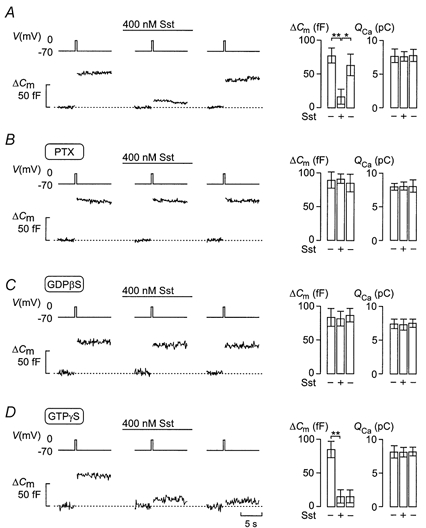
Effects of 400 nm somatostatin (Sst) on changes in cell capacitance (ΔCm) and whole-cell Ca2+ currents (ICa) elicited by 500 ms voltage-clamp depolarisations from -70 to 0 mV (Vm) using the standard whole-cell configuration in single rat α-cells. Changes in cell capacitance were measured before and 2 min after the addition of Sst, and 4 min after wash-out of Sst from the medium under control conditions (A), in cells treated for > 20 h with 100 ng ml−1 pertussis toxin (PTX; B), with 0.5 mm GDPβS included in the pipette solution (C) and in the presence of 0.1 mm intracellular GTPγS (D). Cyclic AMP (0.1 mm) was included in all pipette solutions. The histograms (right panels in A-D) show changes in cell capacitance (ΔCm) and integrated Ca2+ current (QCa) before (-, left), during (+) and after washout (-, right) of Sst. Data are means ±s.e.m. of 5-11 cells. *P < 0.05; **P < 0.01.
Figure 4. Anti-Gi1/2 prevents the inhibitory action of somatostatin on exocytosis.
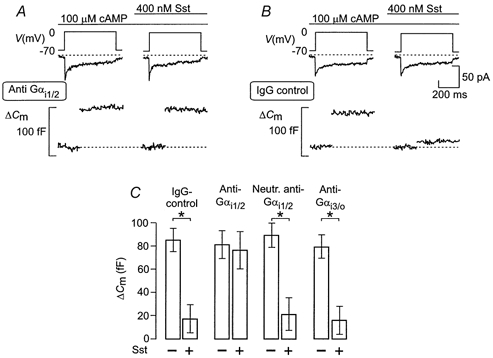
Effects of antibodies against the C-terminal end of the α-subunit of Gi1/2 (anti-Gi1/2; A) or non-immune IgG (IgG control; B) on changes in cell capacitance (ΔCm) and whole-cell Ca2+ currents (ICa) elicited by 500 ms voltage-clamp depolarisations from -70 to 0 mV (Vm) in single rat α-cells. The standard whole-cell configuration was used to record changes in cell capacitance before and 2 min after the addition of somatostatin (Sst). The antibodies were allowed to diffuse into the cells for 2 min before initiation of the experiments. Cyclic AMP (0.1 mm) was included in the pipette solution dialysing the cell interior. C, histogram summarising the effects of somatostatin under control conditions (IgG-control) or following application of anti-Gi1/2 (Anti-Gαi1/2), antibodies neutralised with the corresponding peptide of Gi1/2 (Neutr. anti-Gαi1/2) or antibodies against Gi3/o (Anti-Gαi3/o). Final concentration of the antibodies in the pipette solution was 17.5 μg ml−1. The data are means ±s.e.m. of 5-6 different cells in each group. *P < 0.01.
Figure 11. Somatostatin deprimes secretory granules in the readily releasable pool.
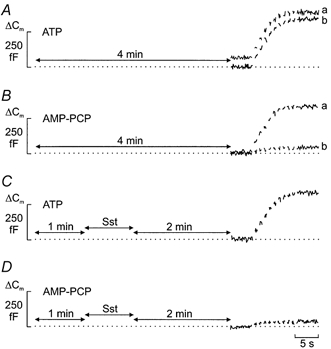
A, trains of ten 500 ms voltage-clamp depolarisations from -70 to 0 mV were applied at a frequency of 1 Hz (Vm) using the standard whole-cell configuration in single rat α-cells. The trains were applied 4 min (a) or and 7 min (b) after establishment of the whole-cell configuration. The pipette contained 3 mm Mg-ATP and 0.1 mm cAMP. The cells were exposed to 10 μm forskolin for ≥ 5 min before establishment of the standard whole-cell configuration to maximise the size of the RRP. B, as in A but ATP was replaced by the non-hydrolysable analogue AMP-PCP. C, as in A but somatostatin (Sst, 400 nm) was present for 1 min as indicated schematically and the train applied only once. D, as in C but ATP was replaced by AMP-PCP. The traces are typical for a total of 5-7 cells for each experimental condition. In C and D, the trains were applied 4 min after the establishment of the whole-cell configuration.
Antibodies
Affinity-purified antibodies (rabbit) against the common carboxyl terminal sequence (residues 345-354) of the α-subunit of either Gi1/2 or Gi3/o (both 3.5 mg ml−1 obtained from Calbiochem) were used in this study. No pre-immune antibodies were available and consequently a non-immune rabbit IgG (3.5 mg ml−1) was used as control. The carboxy-terminal (C-terminal) sequence peptide of Gαi1/2 (345-354, antigen for anti-Gαi1/2; Calbiochem) was used to neutralise anti-Gi1/2. In these experiments, 5 μl of the antigen peptide solution (50 mg ml−1) was added to 45 μl of the antibody solution and incubated for 1 h at 4 °C. These solutions were added to the recording media to produce final concentrations of Gi1/2, Gi3/o and IgG of 17.5 μg ml−1. After establishment of the whole-cell configuration, the antibodies were allowed to wash into the cells for 2 min before the experiment commenced.
Antisense and sense oligonucleotides
The following antisense and sense oligonucleotides were used.
Antisense-Gαi1: 5′-CATGGTGGCCGACGTCGCCCGCCCTCGGCGCCGGGGCCG-3′. This sequence is based on the 5′-non-coding sequence upstream of the initiation codon of the rat Gαi1 cDNA (Takano et al. 1997).
Antisense-Gαi2: 5′-CATCCTGCCGTCCGCCGGCCCGGCCTGGCCCCCACCACG-3′.
Sense-Gαi2: 5′-CGTGGTGGGGGCCAGGCCGGGCCGGCGGACGGCAGGATG-3′, from the leader sequence just before the initiation codon, based on the rat Gαi2 cDNA sequence (Itoh et al. 1988; Takano et al. 1997).
Antisense-Gαi3: 5′-CATGACGGCGGCCGGAGAGGGGACCGGGCCCTGGCTCCAC-3′, from the leader sequence just before the initiation codon, based on the rat Gαi3 cDNA sequence (Itoh et al. 1988; Takano et al. 1997).
Antisense-Gαo: 5′-CATGGTGGCCCCTTCCCTGCCACAGCCCGCACGACTCGG-3′, from the leader sequence just before the initiation codon, based on the rat Gαo cDNA sequence and common for Gαo1 and Gαo2 (Jones & Reed, 1987; Takano et al. 1997).
All oligonucleotides were obtained from TAG Copenhagen (Copenhagen, Denmark). Single α-cells were incubated for 24 h with 20 μm of the above antisense and sense oligonucleotides in tissue culture medium at 37 °C.
Data analysis
Results are presented as means ±s.e.m. for the indicated number of experiments. All current amplitudes are given without compensation for leak conductance. Significant differences were evaluated using Student's t test for paired data. Experiments commenced when two successive depolarisations or trains of pulses applied at a 1-2 min interval elicited exocytotic responses of the same amplitude (±10 %) to ascertain that the observed changes were not simply attributable to spontaneous long-term changes of the secretory capacity.
RESULTS
Somatostatin inhibits exocytosis in rat α-cells
Figure 1A shows Ca2+ currents and associated changes in cell membrane capacitance elicited by 500 ms voltage-clamp depolarisations going from -70 to 0 mV before and after addition of 400 nm somatostatin (Sst) in the presence of 10 μm forskolin. Under control conditions, the membrane depolarisation evoked a capacitance increase (ΔCm) of 88 fF. Two minutes after the application of somatostatin, the same membrane depolarisation evoked a capacitance increase of 17 fF. On average (Fig. 1B, left panel), somatostatin (400 nm) produced an 81 ± 9 % (P < 0.01; n = 9) inhibition of exocytosis. The latter effect was not accompanied by a change of the integrated Ca2+ current (Fig. 1B, right panel). The inhibitory action of somatostatin on exocytosis was reversible and 4 min following washout of the hormone from the bath solution, the depolarisation-evoked capacitance change amounted to 78 ± 16 fF (P < 0.01; n = 9). The depolarisations and increases in cell capacitance were not associated with any changes in cell conductance (Gm; Fig. 1A, bottom trace), suggesting that the observed increases in cell capacitance correspond to exocytosis.
Figure 1. Somatostatin inhibits exocytosis in rat pancreatic α-cells.
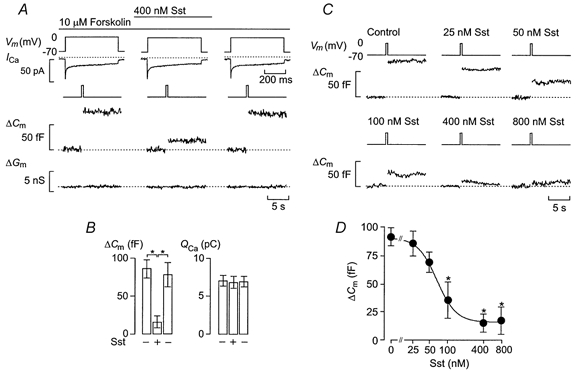
A, effects of somatostatin (Sst) (400 nm) on whole-cell Ca2+ currents (ICa), and changes in cell capacitance (ΔCm) and membrane conductance (ΔGm) elicited by 500 ms depolarisations from -70 to 0 mV as indicated (Vm) using the perforated-patch whole-cell configuration. The experiment was performed in the continuous presence of 10 μm of the adenylate cyclase activator forskolin. Note that the Ca2+ currents are displayed on an expanded time scale. The dotted lines indicate the zero current level as well as the prestimulatory capacitance and conductance levels. B, histograms summarising the increases in cell capacitance (ΔCm) and integrated Ca2+-current (QCa) elicited by 500 ms voltage-clamp depolarisation before (-, left) and 2 min after application of Sst (+), and 4 min following removal of the agonist from the bathing solution (-, right). C, increases in cell capacitance elicited as described in A in the absence and presence of increasing concentrations of Sst. D, concentration dependence of inhibitory action of Sst on exocytosis. The curve represents a least-squares fit of the mean data points to the Hill equation. Data are means ±s.e.m. of 5 (D) and 9 (B) experiments. * P < 0.01 vs. control (no somatostatin) in B and D.
The inhibitory effect of somatostatin on exocytosis was concentration dependent (Fig. 1C). No inhibition of exocytosis was observed at ≤ 25 nm somatostatin. At higher concentrations, somatostatin reduced exocytosis by 25-91 %. Approximating the mean data points to the Hill equation yielded a half-maximal inhibition of 68 nm and co-operativity factor of 2.7. Maximal inhibition of exocytosis was observed at concentrations of somatostatin ≥ 400 nm, which produced > 70 % inhibition (Fig. 1D).
Somatostatin-induced inhibition of exocytosis does not result from lowering of cytoplasmic Ca2+
Figure 2A shows simultaneous measurements of the voltage-clamp Ca2+ currents (ICa), changes in cytoplasmic Ca2+ levels ([Ca2+]i) and cell capacitance. Under control conditions, the depolarisation elicited an integrated Ca2+ current of 6.3 pC, increased [Ca2+]i to 0.8 μm and evoked a capacitance increase of 22 fF (Fig. 2A, left panel). Consistent with our previous observations (Gromada et al. 1997), the β-adrenergic agonist isoprenaline (1 μm) stimulated exocytosis by > 300 %. This effect was associated with 40 % enhancement of the whole-cell Ca2+ current and a corresponding increase in peak [Ca2+]i (+36 %). Somatostatin remained inhibitory in the presence of isoprenaline. Again, the effect on exocytosis occurred without any associated reduction of the whole-cell Ca2+ current or the [Ca2+]i transient and it was fully reversible (Fig. 2A). In a series of five experiments, isoprenaline increased exocytosis by 292 ± 27 % (P < 0.01; Fig. 2B, left), an effect that was associated with moderate increases in both the integrated Ca2+ current (40 ± 17 %; P < 0.01; Fig. 2B, right) and [Ca2+]i transient (37 ± 14 %; P < 0.01; Fig. 2B, middle). The somatostatin-induced inhibition of exocytosis amounted to 78 ± 10 % (P < 0.01; n = 5; Fig. 2B) whilst decreasing neither the integrated Ca2+ current nor the [Ca2+]i transient (Fig. 2B). We conclude that the inhibitory action of somatostatin on exocytosis is not secondary to reduced Ca2+ channel activity or changes in intracellular Ca2+ handling.
Figure 2. Inhibitory action of somatostatin does not involve reduction of cytoplasmic Ca2+.
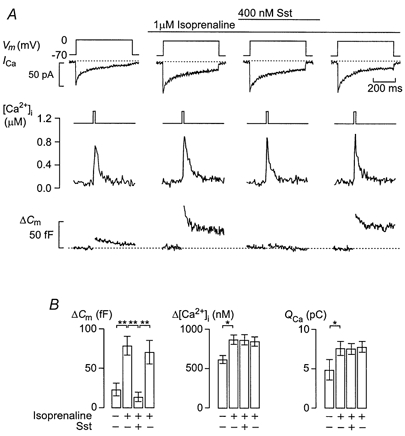
A, whole-cell Ca2+ currents (ICa), cytoplasmic Ca2+ levels ([Ca2+]i) and exocytosis (ΔCm) evoked by membrane depolarisations (500 ms; Vm) from -70 to 0 mV using the perforated-patch whole-cell configuration in single rat α-cells. Exocytosis was observed under control conditions, 2 min after addition of the β-adrenergic agonist isoprenaline (1 μm), in the simultaneous presence of both isoprenaline and Sst (400 nm applied for 2 min) and 4 min after wash-out of Sst from the medium. The dotted lines indicate the zero current level and the prestimulatory capacitance level. B, histograms summarising effects on changes of cell capacitance (ΔCm), cytoplasmic Ca2+ levels ([Ca2+]i) and integrated Ca2+ current (QCa). Data are means ±s.e.m. of 5 cells. *P < 0.05; **P < 0.01.
The inhibitory action of somatostatin on exocytosis involves G proteins and is preserved in standard whole-cell recordings
The ability of somatostatin to suppress exocytosis is maintained in standard whole-cell recordings, which have the advantage of permitting the cell interior to be dialysed by the pipette solution (Fig. 3A). Under these experimental conditions, the suppressor action of somatostatin averaged 78 ± 12 % (P < 0.01; n = 11), close to that observed during perforated-patch whole-cell recordings (compare with Fig. 1A and B). The effect of somatostatin on exocytosis is probably mediated by activation of an inhibitory G protein because pretreatment of α-cells with pertussis toxin for 20 h abolished the responsiveness to somatostatin (Fig. 3B). Somatostatin also failed to inhibit exocytosis in intact α-cells pretreated with pertussis toxin for 20 h using the perforated-patch whole-cell configuration (data not shown). Figure 3C shows that inclusion of 0.5 mm of the stable GDP analogue GDPβS in the intracellular solution likewise abolished the inhibitory action of somatostatin. On average, the exocytotic response in the presence of somatostatin (400 nm) amounted to 97 ± 9 % (n = 5) of that observed in the absence of the hormone when the cells were dialysed with the GDPβS-containing solution. By contrast, application of somatostatin resulted in 84 ± 12 % (P < 0.01; n = 5) inhibition of exocytosis after inclusion of 0.1 mm of the stable GTP analogue GTPγS in the pipette-filling solution (Fig. 3D). As expected if heterotrimeric G proteins are involved, the action of the agonist was irreversible in the presence of the non-hydrolysable GTP analogue.
Suppression of exocytosis is mediated by Gαi2
In order to investigate which type of G protein couples activation of somatostatin receptors to the inhibition of exocytosis, antibodies against Gαi1/2 or Gαi3/o were preloaded into single α-cells through the recording pipette for 2 min. Figure 4A shows that somatostatin failed to affect exocytosis in cells exposed to anti-Gi1/2. However, somatostatin retained its inhibitory action when the cell interior was dialysed with control IgG (Fig. 4B, see Methods). On average, the exocytotic response in the presence of somatostatin amounted to 93 ± 14 % (n = 6) of that observed under control conditions (absence of somatostatin) in cells loaded with anti-Gαi1/2 (Fig. 4C). The ability of the antibody to counteract the action of somatostatin was reversed by neutralising the antibody with the corresponding antigen (Fig. 4C). In cells loaded with anti-Gαi3/o, the somatostatin-induced inhibition of exocytosis was not different from control (cells loaded with a non-immune IgG; Fig. 4C).
Since Gαi1 and Gαi2 cannot be differentiated using antibodies, we used antisense oligonucleotides against sequences in the different Gi proteins. Figure 5A shows that in cells treated for 24 h with antisense oligonucleotides against Gαi2, somatostatin (400 nm) had no inhibitory effect on exocytosis. By contrast (Fig. 5B), exocytosis was completely inhibited in cells treated with the corresponding sense oligonucleotides against Gαi2. Figure 5C summarises the average effects of somatostatin on exocytosis in control cells as well as in cells treated with either sense or antisense oligonucleotides. It is clear that somatostatin decreased Ca2+-induced exocytosis by approximately 80 % under control conditions and in cells treated with antisense oligonucleotides against Gαi1, Gαi3 and Gαo. Sense oligonucleotides for Gαi1, Gαi2, Gαi3 and Gαo were likewise without effect (Fig. 5C). The only experimental manoeuvre that was effective was pretreatment with antisense oligonucleotides against Gαi2 (Fig. 5C). In all cases, the control response prior to addition of somatostatin averaged ∼80 fF (for clarity only one control group is shown). These data argue that somatostatin inhibits exocytosis via activation of Gαi2 proteins.
Figure 5. Gαi2 proteins mediate somatostatin-induced inhibition of exocytosis.
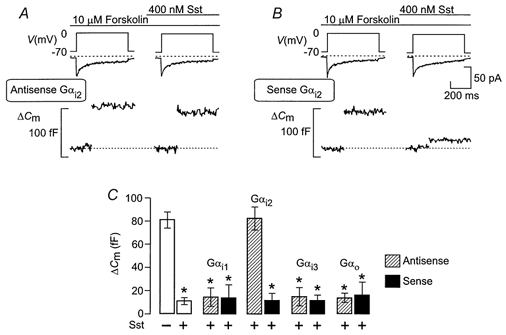
Changes in cell capacitance (ΔCm) and whole-cell Ca2+ currents (ICa) elicited by 500 ms voltage-clamp depolarisations from -70 to 0 mV (Vm) using the perforated-patch whole-cell configuration before and 2 min after the addition of 400 nm somatostatin (Sst) in cells exposed to 10 μm forskolin. The rat α-cells had been treated for 24 h with 20 μm antisense oligonucleotides against sequences in Gαi2 (antisense Gαi2, A) or the corresponding sense oligonucleotides (sense Gαi2, B). C, histogram summarising average increases in cell capacitance in response to 500 ms voltage-clamp depolarisations from -70 to 0 mV under control conditions (-) and following application of somatostatin (+) in untreated cells (open bar, 2nd from left) or in cells treated with either antisense (hatched bars) or the corresponding sense (filled bars) oligonucleotides against Gαi1, Gαi2, Gαi3, or Gαo, The data are means ±s.e.m. of 5 different cells in each group. *P < 0.01 (based on comparison with the responses prior to the addition of somatostatin).
Influence of phosphatase inhibitors on somatostatin-induced inhibition of exocytosis
In pancreatic β-cells the inhibitory action of somatostatin on exocytosis has been suggested to involve activation of the serine/threonine protein phosphatase calcineurin (Renström et al. 1996). We explored whether this also applies to somatostatin-induced inhibition of exocytosis in pancreatic α-cells. Indeed, somatostatin failed to suppress exocytosis following inhibition of calcineurin with cyclosporin A (1 μm for > 20 min; Fig. 6A). A similar abolition was observed with the calcineurin inhibitor deltamethrin (20 nm for > 1 h; Fig. 6B) but not in the presence of its inactive analogue permethrin (Fig. 6C). Intracellular application (through the patch electrode during whole-cell recordings) of calcineurin autoinhibitory peptide (100 μm), a highly selective inhibitor of calcineurin (Perrino et al. 1995), likewise removed the inhibitory action of somatostatin. The average capacitance increase 2 min after addition of 400 nm somatostatin averaged 81 ± 16 fF (n = 5), which is not different from the 86 ± 14 fF (n = 5) observed under control conditions (not shown). In contrast, okadaic acid (100 nm for > 10 min; Fig. 6D), an inhibitor of type 1, 2A and 3 serine/threonine protein phosphatases, failed to counteract the inhibitory action of somatostatin on exocytosis. Under these conditions, somatostatin reduced exocytosis by 75 ± 14 % (P < 0.01; n = 5), close to that observed in the absence of the phosphatase inhibitor (compare with Fig. 3A).
Figure 6. Somatostatin-induced inhibition of exocytosis involves activation of the protein phosphatase calcineurin.
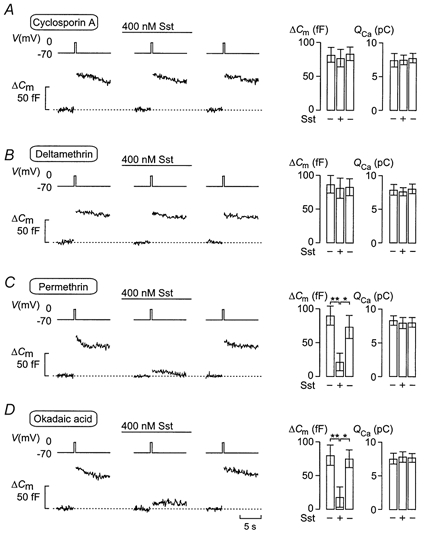
Effects of 400 nm somatostatin (Sst) on changes in cell capacitance (ΔCm) and whole-cell Ca2+ currents (ICa) elicited by 500 ms voltage-clamp depolarisations from -70 to 0 mV (Vm) using the perforated-patch whole-cell configuration before and 2 min after the addition of Sst, and 4 min after wash-out of Sst from the medium. The rat α-cells were pretreated with cyclosporin A (1 μm for > 20 min; A), deltamethrin (20 nm for 1 h, B), permethrin (20 nm for > 1 h, an inactive analogue of deltamethrin; C) or okadaic acid (100 nm for > 10 min; D). The histograms (right panels) show mean changes in cell capacitance (ΔCm) and integrated Ca2+ current (QCa) before (-), during (+) and after washout (-) of Sst. Data are means ±s.e.m. of 5 cells for each experimental condition. *P < 0.05; **P < 0.01.
Effects of somatostatin during repetitive stimulation of exocytosis
We have demonstrated previously that both N- and L-type Ca2+ channels mediate the influx of Ca2+ initiating exocytosis in rat α-cells (Gromada et al. 1997). Under basal conditions, exocytosis is tightly linked to Ca2+ influx through N-type Ca2+ channels, whereas secretion in the presence of cAMP-elevating agents (such as forskolin) is principally due to Ca2+ influx through L-type Ca2+ channels. We next investigated the effects of somatostatin in response to a train consisting of ten 500 ms depolarisations (1 Hz stimulation) from -70 to 0 mV. The experiments were conducted in the continuous presence of 10 μm forskolin to maximally fill the RRP. Two trains were first applied at an interval of 1 min apart under control conditions. It is clear that the total increase in cell capacitance as well as the time course of exocytosis were identical during the two trains (Fig. 7A and B). The total increases in cell capacitance averaged 289 ± 24 and 291 ± 22 fF for the first and second train, respectively. Clearly, a 1 min interval is sufficient for complete refilling of the RRP. The exhaustion of the exocytotic capacity during the trains is likely to reflect depletion of the RRP rather than inactivation of the Ca2+ current with resultant suppression of Ca2+-induced exocytosis. This is suggested by the observation that the integrated Ca2+ current measured at the end of the train, when secretion had ceased, was only reduced by 24 ± 12 % (1st train; n = 5) and by 22 ± 13 % (2nd train; n = 5) with respect to the first depolarisation. The cell was then allowed to rest for 4 min before application of the next series of depolarisations. However, the cell was exposed to 400 nm somatostatin during the last 2 min of this period. As expected, somatostatin strongly inhibited exocytosis elicited by the train (Fig. 7C) and the total increase in cell capacitance evoked by the train fell to 66 ± 21 fF (n = 5). Again, the cessation of exocytosis cannot be accounted for by inactivation of the Ca2+ current as the integrated Ca2+ current was only reduced by 19 ± 11 % (n = 5) during the train. The grey trace, for comparison, shows the exocytotic response 4 min after the preceding train in the absence of somatostatin. The inhibitory action of somatostatin was reversible and 4 min after removal of somatostatin from the bathing solution, exocytosis had returned to that observed before exposure to the agonist (Fig. 7D). On average, the total increase in cell capacitance amounted to 263 ± 34 fF (n = 5).
Figure 7. Somatostatin decreases the size of the RRP of granules.
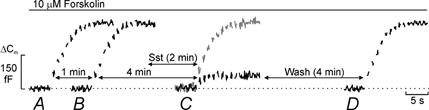
A, a rat α-cell was stimulated by a train consisting of ten 500 ms voltage-clamp depolarisations from -70 to 0 mV applied at 1 Hz frequency (Vm). B, the capacitance increase elicited in the same cell by the train of stimulating pulses applied 1 min after that displayed in A. C, same as in B except that the interval was 4 min and somatostatin (Sst) had been present for the last 2 min. The grey trace shows the responses in a different cell, which was stimulated 4 min after the preceding train but in the absence of Sst. D, the increase in cell capacitance evoked in the same cell as in A-C but 4 min after the washout of Sst. Note that the time scale used for the display of capacitance is not the same as the times indicated for the intervals between the different trains (horizontal arrows). The perforated-patch whole-cell method was used. Forskolin (10 μm) was included in the extracellular medium to maximise the filling of the RRP. Data are representative for a series of 5 separate cells.
We repeated the same protocol in the absence of forskolin (Fig. 8). Unexpectedly, somatostatin was without inhibitory action on exocytosis under these experimental conditions even when applied at a maximally inhibitory concentration (400 nm; compare with Fig. 1D). In a series of eight experiments, the maximum capacitance increase amounted to 59 ± 5 fF under control conditions and 54 ± 4 fF 2 min after the application of somatostatin. These values are close to the somatostatin-resistant component of exocytosis in the presence of forskolin (compare with Fig. 7C).
Figure 8. Somatostatin fails to inhibit basal exocytosis.
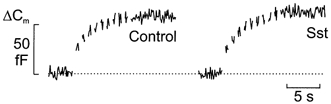
Trains of ten 500 ms voltage-clamp depolarisations from -70 to 0 mV were applied at a frequency of 1 Hz (Vm) using the perforated-patch whole-cell configuration in single rat α-cells. The trains of depolarisations were applied in the absence (left) and presence (right) of 400 nm somatostatin (Sst). Forskolin was not present in these experiments. The interval between the trains of depolarisations was 2 min. Data are representative of 8 different experiments.
Differential effects of ω-conotoxin and nifedipine on basal and forskolin-stimulated exocytosis
As remarked above, forskolin stimulates exocytosis by promoting mobilisation of granules to L-type Ca2+ channels (Gromada et al. 1997). We next investigated whether somatostatin acts by reversal of this effect. Somatostatin was therefore applied in the presence of forskolin and either 50 μm nifedipine (Fig. 9A) or 1 μmω-conotoxin (Fig. 9B). Exocytosis was stimulated by a train of ten 500 ms depolarisations from -70 to 0 mV. In the presence of the L-type Ca2+ channel blocker nifedipine, the train elicited an average capacitance increase of 55 ± 12 fF (n = 5). This is significantly (P < 0.01) lower than that observed under control conditions (Fig. 7A). Application of somatostatin under these conditions failed to reduce exocytosis and the average increase amounted to 49 ± 11 fF (n = 5; not significantly different from that in the absence of somatostatin).
Figure 9. Differential effects of ω-conotoxin and nifedipine on somatostatin-induced inhibition of exocytosis.
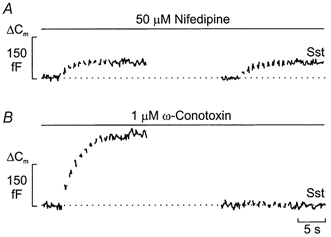
Trains of ten 500 ms voltage-clamp depolarisations from -70 to 0 mV were applied at a frequency of 1 Hz (Vm) using the perforated-patch whole-cell configuration in single rat α-cells. The experiments were performed in the presence of 10 μm forskolin and the trains of depolarisations were applied before (left) or after (right) the addition of 400 nm somatostatin (Sst) in the continuous presence of either 50 μm nifedipine (A) or 1 μmω-conotoxin (B). Note differential effects of Sst when applied in the presence of nifedipine and ω-conotoxin. The interval between the trains of depolarisations was 2 min. Data are representative of 5 separate experiments in both A and B.
When the same experiments were repeated in the presence of the N-type Ca2+ channel blocker ω-conotoxin, very different results were obtained. Under these conditions, the capacitance increase elicited by the train in the absence of somatostatin was 231 ± 29 fF (n = 5; Fig. 9B, left panel). This value is ∼50 fF less than that observed in the absence of the Ca2+ channel blocker (Fig. 7A). Unlike the situation in the absence of ω-conotoxin (e.g. Fig. 7C), addition of somatostatin under these conditions resulted in nearly complete inhibition of exocytosis and the total increase in cell capacitance amounted to 15 ± 6 fF.
We correlated the effects of nifedipine and ω-conotoxin on exocytosis seen in the capacitance measurements to changes in glucagon release in the presence of 1 mm glucose (Table 1). We point out that hypoglycaemia is a stimulus of glucagon release. In the presence of 1 mm glucose alone, application of ω-conotoxin (1 μm) reduced glucagon release by 45 % whereas nifedipine (50 μm) lacked inhibitory action (+10 %). Addition of forskolin (10 μm) resulted in a 3-fold stimulation of glucagon release. Under these conditions, ω-conotoxin inhibited glucagon release by only 8 % whereas addition of the L-type Ca2+ channel blocker nifedipine resulted in 45 % inhibition of glucagon release. These results corroborate the notion that glucagon release depends variably on Ca2+-influx through N- and L-type Ca2+ channels under basal conditions and following stimulation with forskolin or other agents leading to activation of protein kinase A (PKA).
Table 1.
Effects of Ca2+ channel blockers on glucagon release
| Condition | 1 mM glucose | 1mM glucose + 10 μM forskolin |
|---|---|---|
| Control | 34.0 ± 2.6 (12) | 100.5 ± 6.6 (12) * |
| 1μM ω-conotoxin | 18.8 ± 1.9 (10) * | 92.1 ± 7.5 (10) |
| 50 μM nifedipine | 36.9 ± 4.1 (10) | 55.5 ± 4.9 (10)† |
Glucagon release (pg islet−1 h−1) was measured following 1 h incubations in the presence of 1 mM glucose alone (left column) or following activation of adenylate cyclase with 10 μM forskolin (right column) in the presence of the L- and N-type Ca2+ channel blockers nifedipine and ω-conotoxin. Data are means ± S.E.M. of the indicated number of experiments (in parentheses).
P < 0.001vs. Control (1 mM glucose alone)
P < 0.001vs. glucagon release measured in 1 mM glucose + 10 μM forskolin.
Time course of somatostatin-dependent inhibition of exocytosis
Next we determined the time course of somatostatin-induced inhibition of exocytosis. The cells were first subjected to two trains (each consisting of ten 500 ms depolarisations from -70 to 0 mV at 1 Hz) of depolarisations with an interval of 60 s to ensure reproducibility. Following the second train, the cells were exposed to a maximally inhibitory concentration of somatostatin (400 nm, Fig. 1D) for variable time periods (15-180 s) before initiation of a third series of depolarisations (Fig. 10A). The interval between the 2nd and 3rd train was always 240 s and somatostatin was present during the last part of this period. In the absence of somatostatin, the total increases in cell capacitance elicited by the 2nd and 3rd trains were 289 ± 33 and 281 ± 41 fF (n = 5), respectively. Exposure of the α-cells to somatostatin for increasing time periods resulted in a progressive decrease in the total increase in cell capacitance elicited by the train. The relationship between the exocytotic response to the train and the duration of somatostatin exposure is shown in Fig. 10B (filled circles). We estimated that a 22 s exposure to somatostatin is required to produce half-maximal inhibition. We also examined the effect of somatostatin on the response to the first depolarisation in the train. These data are summarised in Fig. 10B (open circles). The difference between the effects of somatostatin on exocytosis elicited by the first pulse and the total increase during the train is illustrated in Fig. 10C. It is clear that exocytosis evoked by single depolarisations (open circles) is less affected by somatostatin than that evoked by the train (filled circles) and requires longer exposures, half-maximal inhibition being observed at 68 s. It also becomes evident that whereas exocytosis elicited by the trains is reduced by > 80 %, the corresponding value for the first depolarisation is < 60 %. Irrespective of the length of somatostatin application, the amplitude of the integrated Ca2+ current remained almost constant and the observed decreases in the exocytotic responses can accordingly not be explained as inhibition of the Ca2+ currents (not shown).
Figure 10. Time-dependent decrease in the readily releasable pool of granules by somatostatin.
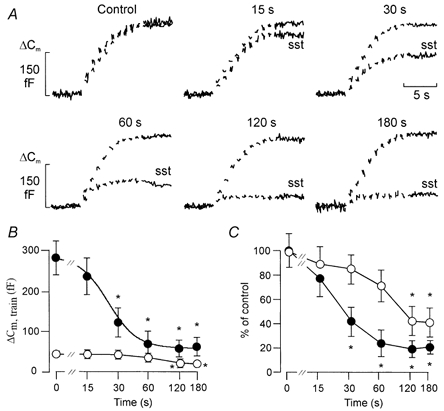
A, trains of 500 ms voltage-clamp depolarisations from -70 to 0 mV were applied at a frequency of 1 Hz (Vm) using the perforated-patch whole-cell configuration in single rat α-cells. The trains of depolarisations were applied in the absence (Control) and 15-180 s after addition of 400 nm somatostatin (sst). Forskolin (10 μm) was present in the extracellular medium throughout the recordings. B, time dependence of the inhibitory action of somatostatin on exocytosis elicited by the first depolarisation (○) or the entire train (•). The curves represent a least-squares fit of the mean values to the Hill equation. C, same as in B but the responses have been normalised to the capacitance increase evoked in the absence of somatostatin. A new cell was used for each exposure. Data in B and C represent means ±s.e.m. of 5-7 cells. *P < 0.01vs. control (0 s exposure to somatostatin).
Effects of somatostatin in the presence of intracellular AMP-PCP
The data of Fig. 10 indicate that the inhibitory action of somatostatin is time rather than use dependent and that somatostatin acts by reducing the releasability of the granules belonging to the RRP (depriming). In pancreatic β-cells it has been demonstrated that priming requires ATP-hydrolysis (Eliasson et al. 1997). We investigated whether this applies also to the α-cell. Figure 11A shows a whole-cell experiment in which the α-cell was dialysed with a medium containing 3 mm Mg-ATP and supplemented with 0.1 mm cAMP. The extracellular medium contained 10 μm forskolin to maximise the size of the RRP. Following establishment of the whole-cell configuration, the cell was allowed a 4 min equilibration period. A train consisting of ten 500 ms depolarisations from -70 to 0 mV was then applied to evoke exocytosis. In a series of five experiments, the total increase in cell capacitance amounted to 288 ± 35 fF (Fig. 11A, trace a). A second train applied to the same cell after an interval of 3 min evoked a capacitance increase of 271 ± 31 fF (Fig. 11A, trace b). When the same experiment was repeated after substitution of AMP-PCP for ATP, the first train was similar to that observed in the presence of standard ATP and averaged 267 ± 43 fF (n = 5; Fig. 11B, trace a). However, exocytosis during the second train (Fig. 11B, trace b) was almost abolished and the total increase averaged 21 ± 12 fF (P < 0.001; n = 5). This clearly demonstrates that ATP hydrolysis is required for the recruitment of new granules into the RRP. Application of somatostatin (400 nm) for 1 min during the 4 min wash-in period, did not affect the magnitude of the increase in exocytosis subsequently (2 min after exposure to somatostatin) evoked by a train of depolarisations when Mg-ATP was present in the pipette solution (Fig. 11C). However, exocytosis was dramatically reduced in the presence of AMP-PCP (Fig. 11D). In series of five experiments, the increases in cell capacitance amounted to 265 ± 28 and 52 ± 15 fF in the presence of ATP and AMP-PCP, respectively. The latter effect was not due to an action of AMP-PCP itself since a train applied in the presence of the stable analogue in the absence of the agonist evoked a capacitance increase close to that observed in the presence of standard ATP (see Fig. 11B). Collectively, the above data suggest that somatostatin inhibits exocytosis by depriming of granules in the RRP.
DISCUSSION
Somatostatin inhibits exocytosis by activation of calcineurin by a Gαi2-dependent mechanism
Our results demonstrate that somatostatin produces a fast and potent inhibition of Ca2+- and cAMP-induced exocytosis in rat pancreatic α-cells. This effect was mediated by pertussis toxin-sensitive G proteins. Using both antibodies and antisense oligonucleotide techniques we provide evidence for the involvement of Gαi2. This is consistent with the previous identification of Gαi2 in rat pancreatic islets (Berrow et al. 1992).
Evidence exists that SSTR2 is expressed in α-cells and mediates the inhibitory action of somatostatin on glucagon release (Rossowski & Coy, 1994; Kuman et al. 1999; Strowski et al. 2000). Interestingly, SSTR2 couples to Gαi2 in the presence of somatostatin (Law & Reisine, 1992; Murray-Whelan & Schlegel, 1992; Luthin et al. 1993) whereas it couples to Gαi1 and Gαi3 but not Gαi2 in the absence of the agonist (Law et al. 1991). The combination of these previous biochemical data and the present electrophysiological results suggests that the somatostatin receptor subtype that modulates exocytosis in rat α-cells couples to Gαi2 and that coupling only occurs when the receptor is occupied by the agonist. However, it remains to be established whether calcineurin activity is controlled by direct interaction of either granular or plasma membrane-associated Gαi2 proteins or whether intermediate proteins are responsible for signal transduction.
Our data demonstrate that the ability of somatostatin to inhibit Ca2+-dependent exocytosis is rapid and readily reversible and probably involves activation of the protein phosphatase calcineurin, which is expressed at high levels in rat α-cells (Gagliardino et al. 1991; Redecker & Cetin, 1997), as suggested by the effects of deltamethrin and cyclosporin. Although pharmacological tools are never perfectly selective, the fact that intracellular administration of calcineurin autoinhibitory peptide shared the capacity of deltamethrin and cyclosporin to antagonise the action of somatostatin makes it reasonable to conclude that the hormone mediates its inhibitory effect via activation of this protein phosphatase. These results are consistent with the results previously obtained in mouse pancreatic β-cells (Renström et al. 1996; but see Kampermann et al. 2000). However, little is known about the regulation of calcineurin by G proteins.
Recent studies have revealed the presence of Gi and Go proteins in secretory granules from βTC3 insulinoma cells, chromaffin cells and melanotrophs as well as synaptic vesicles from rodent and bovine brain (Aronin & DiFiglia, 1992; Ahnert-Hilger et al. 1994, 1998; Konrad et al. 1995; Kreft et al. 1999), but it remains unknown whether they are also present in glucagon-containing granules. We acknowledge that the direct demonstration that the granular G proteins participate in the secretory process is still lacking but they are clearly in an ideal position for controlling granule priming and exocytosis.
Since activation of PKA leads to enhancement of Ca2+-dependent exocytosis in rat α-cells (Gromada et al. 1997), it could be argued that suppression of exocytosis by somatostatin is the result of reduced cAMP levels and inhibition of PKA-mediated exocytosis. However, this possibility can be discarded since the ability of somatostatin to suppress exocytosis was maintained in standard whole-cell experiments in which the cytoplasmic cAMP concentration was clamped by inclusion of the nucleotide in the pipette solution.
Somatostatin-dependent suppression of exocytosis by depriming of RRP granules?
We demonstrate that the ability of somatostatin to inhibit exocytosis varies depending on whether PKA is activated or not. As discussed above, this is not a consequence of somatostatin affecting cytoplasmic cAMP levels. It is also of interest that the somatostatin-resistant component of exocytosis in the presence of the PKA activator forskolin (20 % or 65 fF) is close to the magnitude of the exocytosis that can maximally be elicited by a train of depolarisations under control conditions (absence of forskolin). These considerations argue that somatostatin acts by reversal of the process catalysed by PKA activation.
The priming of secretory granules in endocrine cells including pancreatic β-cells requires ATP hydrolysis. We demonstrate here that this also applies to the glucagon-releasing α-cell and that the secretory capacity rapidly diminishes when priming is prevented by replacement of Mg-ATP with its non-hydrolysable analogue AMP-PCP. Exposure of the α-cells to somatostatin leads to depriming of granules already belonging to the RRP and does not result from suppression of refilling of the RRP. This scenario is suggested by the experiments of Fig. 11 where somatostatin was found to irreversibly inhibit exocytosis in α-cells infused with the non-hydrolysable ATP analogue AMP-PCP, a condition suppressing replenishment of the RRP but not release of granules that have already proceeded into this pool (compare Fig. 11B and D). Regulation of exocytosis by reversible priming and depriming of granules by phosphorylation and dephosphorylation of (exocytotic?) proteins clearly provides the α-cells with the means to rapidly adjust their secretory capacity in response to circulating hormones as well as locally released neurotransmitters and hormones without any physical translocation of granules within the cell.
Somatostatin deprimes granules at the L-type Ca2+ channel
Why is basal (PKA-independent) exocytosis not affected by somatostatin? Here we demonstrate that the effects of the Ca2+ channel blockers ω-conotoxin and nifedipine vary depending on whether the cells are stimulated with forskolin or not (Table 1). Accordingly, glucagon secretion under basal conditions was almost resistant to the L-type channel blocker but strongly (45 %) inhibited by ω-conotoxin. Following stimulation with forskolin, glucagon release increased 3-fold. The latter effect correlated with an increased efficacy of nifedipine (45 % reduction) and a reduced efficacy of ω-conotoxin (8 % inhibition). These results compare favourably with measurements of cell capacitance (Gromada et al. 1997) where ω-conotoxin blocks 62 % of exocytosis under control conditions but only 12 % of that observed in the presence of forskolin. In the same type of experiments, nifedipine reduced exocytosis by 30 % under control conditions but 81 % of that elicited in the presence of the adenylate cyclase activator. The observation that both glucagon release and the capacitance changes observed in the absence of forskolin are highly sensitive to ω-conotoxin makes it reasonable to conclude that the ω-conotoxin-sensitive component of the capacitance increase reflects release of glucagon-containing secretory granules and that it cannot be accounted for by exocytosis of other types of vesicles.
These results reinforce previous observations (Gromada et al. 1997) that activation of PKA principally increases the number of RRP granules in the vicinity of the L-type Ca2+ channels. We further speculate that the somatostatin receptors selectively associate with L-type Ca2+ channels and thus lead to a localised activation of Gαi2 and calcineurin. This concept would account for the failure of somatostatin to affect basal glucagon secretion, which is due to Ca2+ influx through N-type Ca2+ channels. We postulate that the somatostatin receptors do not associate with the N-type Ca2+ channels and that granules in the vicinity of these channels are not deprimed, thus accounting for the lack of agonist-induced suppression of this component. The finding that somatostatin abolishes exocytosis in the presence of ω-conotoxin provides further support for the idea that somatostatin exclusively deprimes granules associated with L-type Ca2+ channels.
Concluding remarks
In conclusion, our data suggest that secretory granules in rat α-cells exist in three functional pools: the reserve pool, granules associated with N-type Ca2+ channels and granules in the vicinity of L-type Ca2+ channels. Whereas the size of the pool close to the N-type Ca2+ channels (60 fF or 30 granules) is not modulated, the number of RRP granules residing at the L-type Ca2+ channels is regulated by phosphorylation. Activation of PKA (by β-adrenergic stimulation or forskolin) increases the number of release-competent granules at the L-type Ca2+ channels from no granules under control conditions to ∼120 granules (240 fF) in the presence of the activator. Inhibitory agonists such as somatostatin do not affect exocytosis of granules close to the N-type Ca2+ channels but selectively deplete the pool of granules at the L-type Ca2+ channels. This effect is Gαi2 protein dependent and involves activation of the protein phosphatase calcineurin. Accordingly, agents that suppress the activity of this phosphatase (including cyclosporin A, deltamethrin and calcineurin autoinhibitory peptide) antagonise the inhibitory action of somatostatin. The latter effect is use independent, develops rapidly (inhibition being half-maximal within ∼20 s) and is readily reversible. It will be interesting to determine whether the changes in releasability are due to the physical translocation of the granules (i.e. out of the active zones) or whether a chemical modification is sufficient. Finally, the fact that calcineurin mediates the inhibitory action of somatostatin in both mouse β-cells (Renström et al. 1996) and rat α-cells suggests that this might represent a general mechanism for agonist regulation of exocytosis.
Acknowledgments
This study was supported in part by the Swedish Medical Research Council (grants 8647 and 13147), the Swedish Diabetes Association, the Crafoord Foundation, the Knut and Alice Wallenberg Foundation, the Juvenile Diabetes Research Foundation, the Novo Nordisk Foundation and the Swedish Council for Planning and Coordination of Research.
References
- Ahnert-Hilger G, Nürnberg B, Exner T, Schäfer T, Jahn R. The heterotrimeric G protein Go2 regulates catecholamine uptake by secretory vesicles. EMBO Journal. 1998;17:406–413. doi: 10.1093/emboj/17.2.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahnert-Hilger G, Schäfer T, Spicher K, Grund C, Schultz G, Wiedenmann B. Detection of G-protein heterotrimers on large dense core and small synaptic vesicles of neuroendocrine and neuronal cells. European Journal of Cell Biology. 1994;65:26–38. [PubMed] [Google Scholar]
- Ämmälä C, Eliasson L, Bokvist K, Larsson O, Ashcroft FM, Rorsman P. Exocytosis elicited by action potentials and voltage-clamp calcium currents in individual mouse pancreatic B-cells. Journal of Physiology. 1993;472:665–688. doi: 10.1113/jphysiol.1993.sp019966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronin N, Difiglia M. The subcellular localization of the G-protein Giα in the basal ganglia reveals its potential role in both signal transduction and vesicle trafficking. Journal of Neuroscience. 1992;12:3435–3444. doi: 10.1523/JNEUROSCI.12-09-03435.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrow NS, Milligan G, Morgan NG. Immunological characterization of the guanine-nucleotide binding proteins Gi and Go in rat islets of Langerhans. Journal of Molecular Endocrinology. 1992;8:103–108. doi: 10.1677/jme.0.0080103. [DOI] [PubMed] [Google Scholar]
- Bokvist K, Eliasson L, Ämmälä C, Renström E, Rorsman P. Co-localization of L-type Ca2+ channels and insulin-containing secretory granules and its significance for the initiation of exocytosis. EMBO Journal. 1995;14:50–57. doi: 10.1002/j.1460-2075.1995.tb06974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W-G, Renström E, Rorsman P, Buschard K, Gromada J. Glucagon-like peptide I and glucose-dependent insulinotropic polypeptide stimulate Ca2+-induced secretion in rat α-cells by a protein kinase A-mediated mechanism. Diabetes. 1997;46:792–800. doi: 10.2337/diab.46.5.792. [DOI] [PubMed] [Google Scholar]
- Eliasson L, Renström E, Ding W-G, Proks P, Rorsman P. Rapid ATP-dependent priming of secretory granules precedes Ca2+-induced exocytosis in mouse pancreatic B-cells. Journal of Physiology. 1997;503:399–412. doi: 10.1111/j.1469-7793.1997.399bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehmann HC, Strowski M, Göke B. Functional characterisation of somatostatin receptors expressed on hamster glucagonoma cells. Americal Journal of Physiology. 1995;268:E40–47. doi: 10.1152/ajpendo.1995.268.1.E40. [DOI] [PubMed] [Google Scholar]
- Gagliardino JJ, Krinks MH, Gagliardino EE. Identification of the calmodulin-regulated protein phosphatase, calcineurin, in rat pancreatic islets. Biochemica et Biophysica Acta. 1991;1091:370–373. doi: 10.1016/0167-4889(91)90202-9. [DOI] [PubMed] [Google Scholar]
- Göpel SO, Kanno T, Barg S, Rorsman P. Patch-clamp characterization of somatostatin-secreting δ-cells in intact mouse pancreatic islets. Journal of Physiology. 2000;528:497–507. doi: 10.1111/j.1469-7793.2000.00497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromada J, Bokvist K, Ding W-G, Barg S, Buschard K, Renström E, Rorsman P. Adrenaline stimulates glucagon secretion in pancreatic A-cells by increasing the Ca2+ current and the number of granules close to the L-type Ca2+ channels. Journal of General Physiology. 1997;110:217–228. doi: 10.1085/jgp.110.3.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromada J, Høy M, Olsen HL, Gotfredsen CF, Buschard K, Rorsman P, Bokvist K. Gi2 proteins couple somatostatin receptors to low-conductance K+ channels in rat pancreatic α-cells. Pflügers Archiv. 2001;442:19–26. doi: 10.1007/s004240000474. [DOI] [PubMed] [Google Scholar]
- Holz RW, Bitter MA, Peppers SC, Senter RA, Eberhard DA. MgATP-independent and MgATP-dependent exocytosis. Journal of Biological Chemistry. 1989;264:5412–5419. [PubMed] [Google Scholar]
- Høy M, Olsen HL, Bokvist K, Buschard K, Barg S, Rorsman P, Gromada J. Tolbutamide stimulates exocytosis of glucagon by inhibition of a mitochondrial-like ATP-sensitive K+ (KATP) conductance in rat pancreatic A-cells. Journal of Physiology. 2000;527:109–120. doi: 10.1111/j.1469-7793.2000.00109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh H, Toyama R, Kozasa T, Tsukamoto T, Matsuoka M, Kaziro Y. Presence of three distinct molecular species of Gi protein α subunit. Structure of rat cDNAs and human genomic DNAs. Journal of Biological Chemistry. 1988;263:6656–6664. [PubMed] [Google Scholar]
- Josefsen K, Stenvang JP, Kindmark H, Berggren P-O, Horn T, Kjær T, Buschard K. Fluorescence-activated cell sorted rat islet cells and studies of the insulin secretory process. Journal of Endocrinology. 1996;149:145–154. doi: 10.1677/joe.0.1490145. [DOI] [PubMed] [Google Scholar]
- Jones DT, Reed RR. Molecular cloning of five GTP-binding protein cDNA species from rat olfactory neuroepithelium. Journal of Biological Chemistry. 1987;262:14241–14249. [PubMed] [Google Scholar]
- Kampermann J, Herbst M, Ullrich S. Effects of adrenaline and tolbutamide on insulin secretion in INS-1 cells under voltage control. Cellular Physiology and Biochemistry. 2000;10:81–90. doi: 10.1159/000016337. [DOI] [PubMed] [Google Scholar]
- Konrad RJ, Young RA, Record RD, Smith RM, Butkerait P, Manning D, Jarett L, Wolf BA. The heterotrimeric G-protein Gi is localized to the insulin secretory granules of β-cells and is involved in insulin exocytosis. Journal of Biological Chemistry. 1995;270:12869–12876. doi: 10.1074/jbc.270.21.12869. [DOI] [PubMed] [Google Scholar]
- Kreft M, Gasman S, Chasserot-Golaz S, Kuster V, Rupnik M, Sikdar SK, Bader M-F, Zorec R. The heterotrimeric Gi3 protein acts in slow but not in fast exocytosis of rat melanotrophs. Journal of Cell Science. 1999;112:4143–4150. doi: 10.1242/jcs.112.22.4143. [DOI] [PubMed] [Google Scholar]
- Kuman U, Sasi R, Suresh S, Patel A, Thangaraju M, Metrakos P, Patel SC, Patel YC. Subtype-selective expression of the five somatostatin receptors (hSSTR1–5) in human pancreatic islet cells. Diabetes. 1999;48:77–85. doi: 10.2337/diabetes.48.1.77. [DOI] [PubMed] [Google Scholar]
- Law SF, Manning D, Reisine T. Identification of the subunits of GTP-binding proteins coupled to somatostatin receptors. Journal of Biological Chemistry. 1991;266:17885–17897. [PubMed] [Google Scholar]
- Law SF, Reisine T. Agonist binding to rat brain somatostatin receptors alters the interaction of the receptor with guanine nucleotide-binding regulatory proteins. Molecular Pharmacology. 1992;42:398–402. [PubMed] [Google Scholar]
- Luthin DR, Eppler CM, Linden J. Identification and quantification of Gi-type GTP-binding proteins that copurify with a pituitary somatostatin receptor. Journal of Biological Chemistry. 1993;268:5990–5996. [PubMed] [Google Scholar]
- Murray-Whelan R, Schlegel W. Brain somatostatin receptor-G protein interaction. Journal of Biological Chemistry. 1992;267:2960–2965. [PubMed] [Google Scholar]
- Panagiotidis G, Salehi A, Westermark P, Lundquist I. Homologous islet amyloid polypeptides: effects on plasma levels of glucagon, insulin and glucose in the mouse. Diabetes Research in Clinical Practice. 1992;18:167–171. doi: 10.1016/0168-8227(92)90142-e. [DOI] [PubMed] [Google Scholar]
- Parsons TD, Coorseen JR, Horstmann H, Almers W. Docked granules, the exocytotic burst and the need for ATP hydrolysis in endocrine cells. Neuron. 1995;15:1085–1096. doi: 10.1016/0896-6273(95)90097-7. [DOI] [PubMed] [Google Scholar]
- Perrino BA, Ng LY, Soderling TR. Calcium regulation of calcineurin phosphatase activity by its B subunit and calmodulin: role of the autoinhibitory domain. Journal of Biological Chemistry. 1995;270:340–346. doi: 10.1074/jbc.270.1.340. [DOI] [PubMed] [Google Scholar]
- Redecker P, Cetin Y. Rodent pancreatic islet cells contain the calcium-binding proteins calcineurin and calretinin. Histochemical and Cell Biology. 1997;108:133–139. doi: 10.1007/s004180050154. [DOI] [PubMed] [Google Scholar]
- Renström E, Ding W-G, Bokvist K, Rorsman P. Neurotransmitter-induced inhibition of exocytosis in insulin-secreting β cells by activation of calcineurin. Neuron. 1996;17:513–522. doi: 10.1016/s0896-6273(00)80183-x. [DOI] [PubMed] [Google Scholar]
- Rossowski WJ, Coy DH. Specific inhibition of rat pancreatic insulin or glucagon release by receptor-selective somatostatin analogs. Biochemcal and Biophysical Research Communications. 1994;205:341–346. doi: 10.1006/bbrc.1994.2670. [DOI] [PubMed] [Google Scholar]
- Strowski MZ, Parmar RM, Blake AD, Schaeffer JM. Somatostatin inhibits insulin and glucagon secretion via two receptor subtypes: An in vitro study of pancreatic islets from somatostatin receptor 2 knockout mice. Endocrinology. 2000;141:111–117. doi: 10.1210/endo.141.1.7263. [DOI] [PubMed] [Google Scholar]
- Takano K, Yasufuku-Takano J, Kozasa T, Nakajima S, Nakijima Y. Different G-proteins mediate somatostatin-induced inward rectifier K+ currents in murine brain and endocrine cells. Journal of Physiology. 1997;502:559–567. doi: 10.1111/j.1469-7793.1997.559bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallar L, Biden TJ, Wollheim CB. Guanine nucleotides induced Ca2+-independent insulin secretion from permeabilized RINm5F-cells. Journal of Biological Chemistry. 1987;262:5049–5056. [PubMed] [Google Scholar]
- Yoshimoto Y, Fukuyama Y, Horio Y, Inanobe A, Gotoh M, Kurachi Y. Somatostatin induces hyperpolarization in pancreatic islet α cells by activating a G protein-gated K+ channel. FEBS Letters. 1999;444:265–269. doi: 10.1016/s0014-5793(99)00076-9. [DOI] [PubMed] [Google Scholar]