

Abstract
Two aromatic residues of the muscle nicotinic receptor putative agonist binding site, a tyrosine in position α93 and a tryptophan in position α149, were mutated to phenylalanine and the effects of the mutations on receptor properties were investigated using single-channel patch clamp.
The αY93F mutation reduced the receptor affinity by ≈4-fold and the channel opening rate constant by 48-fold. The αW149F mutation reduced the receptor affinity by ≈12-fold and the channel opening rate constant by 93-fold.
The kinetic properties of hybrid receptors that contained one wild-type and one mutated α subunit were also examined. Only one type of hybrid receptor activity was detected. The hybrid receptors had a channel opening rate constant intermediate to those of the wild-type and mutant receptors. It was concluded that the ligand binding sites in the mutated muscle nicotinic receptor contributed equally to channel gating. In the case of the αW149F mutation, the presence of the mutation in one of the binding sites had no effect on the binding properties of the other, non-mutated, site.
The mutant channel opening and closing rate constants were also estimated in the presence of tetramethylammonium. The data suggested significant interaction between the acetyl group of acetylcholine and the αY93 residue.
The adult muscle nicotinic acetylcholine (ACh) receptor (AChR) is a pentameric protein consisting of two α subunits, and one each of β, δ and ε subunits. The receptor contains two agonist binding sites, which are formed by the α-δ and α-ε subunit pairs (Blount & Merlie, 1989). Affinity labelling and mutagenesis studies have identified three regions of the α subunit that contribute to the stabilization of the bound ACh (for review see Devillers-Thiery et al. 1993). Residues in region A include aromatic residues in positions 86 (tryptophan) and 93 (tyrosine). Region B contains Trp149 and Tyr151, and finally, region C contains Tyr190, Tyr198, Asp200 and vicinal cysteines in positions 192 and 193.
The high number of aromatic residues in the putative binding site is of interest. In another ACh binding protein, acetylcholinesterase, there is evidence that the quaternary ammonium portion of ACh interacts with an aromatic residue, tryptophan (Silman et al. 1994). For the nicotinic receptor, the data available are more controversial. Even though evidence exists to support the idea of a cation π-type interaction between a tryptophan residue (αW149) located within the putative binding site and the quaternary ammonium group of ACh (Zhong et al. 1998), other studies have proposed that different aromatic residues may contribute to stabilization of ACh in the binding pocket via interactions with the quaternary ammonium group (αY93, αY190 and αY198; Aylwin & White, 1994; Sine et al. 1994; Nowak et al. 1995). Furthermore, it has been proposed that in the AChR, the positively charged quaternary ammonium moiety of ACh does not interact with any of the aromatic residues, but rather with negatively charged residues on the δ and γ subunits (Asp180 in the δ and Asp174 in the γ subunit; Czajkowski & Karlin, 1995; Martin et al. 1996).
In the adult-type mouse receptor, the two agonist binding sites have equal affinities for ACh (Akk & Auerbach, 1996; Wang et al. 1997; Salamone et al. 1999), suggesting that the microenvironment surrounding the ACh molecule is similar within the two sites. Similarity in affinities also suggests the absence of any binding cooperativity or interactions between the two binding sites. It is not clear, however, whether the two binding sites contribute equally to channel gating. It has been argued previously that, when the binding sites have different affinities for the ligand, the contributions to channel gating are different (Jackson, 1989). Even though the ligand binding sites in the adult-type receptor have equivalent affinities, there is evidence that the binding site mutation αD200N has differing effects on gating depending on which of the two α subunits it is in (Akk et al. 1996).
In the present work, the kinetic properties of two putative binding site mutants, αY93F and αW149F, were investigated. Both mutations predominantly affect channel gating with smaller effect on the agonist binding properties of the receptor. In addition, the kinetic properties of hybrid receptors, i.e. receptors in which only one of the two a subunits contains the mutated residue, were examined. The results suggest that these residues at both binding sites contribute roughly equally to channel gating. For the αW149F site, the hybrid receptor activity can be descibed by one of the binding sites having wild-type-like affinity while the other has affinity close (a 1.1-fold difference) to what is observed in the pure mutant receptor. For the αY93F site, the KD of the mutated site in the hybrid receptor is similar (a 1.6-fold difference) to one in the pure mutant receptor, but the rate constants for agonist association and dissociation differ from the ones observed in the mutant receptor by 2- to 3-fold. Experiments on mutant receptors activated by tetramethylammonium (TMA) suggest that significant interaction takes place between the acetyl group of ACh and the αY93 site during channel gating.
METHODS
All chemicals, including acetylcholine chloride and tetramethylammonium iodide, were obtained from Sigma Chemical Co. (St Louis, MO, USA).
Expression systems and electrophysiology
Mouse muscle type nicotinic AChR (nAChR) subunit cDNAs (α, β, δ, ε) originally came from the laboratories of the late Dr John Merlie and Dr Norman Davidson, and were subcloned into a CMV promoter-based expression vector pcDNAIII (Invitrogen, San Diego, CA, USA). The α subunit differed from the sequence in the GenBank database (accession X03986) by having alanine rather than valine at position 433 (Salamone et al. 1999). The αY93F and αW149F mutant clones were provided by Dr Steven Sine.
The nAChR was expressed in HEK 293 cells (human embryonic kidney cell line) using transient transfection based on calcium phosphate precipitation (Ausubel et al. 1992). For muscle-type receptors, a total of 3.5 μg DNA per 35 mm culture dish in the ratio 2:1:1:1 (α:β:δ:ε) was used. In some experiments, mutant and wild-type α subunits were coexpressed with wild-type β, δ and ε subunits. In such cases, the total amount of DNA remained unchanged, but the ratio changed to 1:1:1:1:1 (αwild-type:αmutant:β:δ:ε). The DNA was added to the cells for 12-24 h, after which the medium was changed. Electrophysiological recordings were performed 24-48 h later.
All electrophysiological experiments were performed using the patch clamp technique in the cell-attached configuration (Hamill et al. 1981). The bath comprised Dulbecco's phosphate-buffered saline containing (mm): 137 NaCl; 0.9 CaCl2; 2.7 KCl; 1.5 KH2PO4; 0.5 MgCl2; and 6.6 Na2HPO4; pH 7.3. The pipette solution contained (mm): 142 KCl; 1.8 CaCl2; 1.7 MgCl2; 5.4 NaCl; and 10 Hepes; pH 7.4. In addition, the pipette solution contained the indicated concentration of ACh or tetramethylammonium (TMA). The pipette potential was held at +40-60 mV. Based on the reversal potential of ionic currents through the nAChR expressed in HEK cells (data not shown), the typical resting membrane potential is estimated to be -40 mV. Thus, the potential difference across the patch membrane was -80 to -100 mV. All experiments were performed at room temperature.
Kinetic analysis
Data collection and processing were as described previously (Akk et al. 1996; Salamone et al. 1999). Briefly, single-channel currents were amplified with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA, USA), digitized at 50 kHz, and saved on a PC hard disk using a Digidata 1200 Series interface (Axon Instruments). For event detection, the data were low-pass filtered at 3-5 kHz and idealized using the program SKM (www.qub.buffalo.edu). Lists of open and closed interval durations were generated via a half-amplitude threshold-crossing criterion. The analysis was restricted to clusters of channel openings that each reflected the activity of a single AChR (Sakmann et al. 1980). Clusters were defined as series of openings separated by closed intervals shorter than some critical duration (τcrit, 50-500 ms). The value of τcrit was chosen arbitrarily but depended on the type of agonist used and its concentration, and was always at least 10 times longer than the main component of closed intervals within clusters that scaled with the agonist concentration. The definition of clusters was usually not sensitive to the value of τcrit because the slowest component of intracluster closed times and the intercluster closed time component associated with desensitization typically were well separated. Clusters were typically 200-2000 ms in duration. When low concentrations of agonist were used, or in the case of the αY93F mutant receptor activated by TMA, the currents did not occur in identifiable clusters and a determination of all of the activation rate constants was not possible.
The following kinetic scheme (Model 1) was used to describe the current interval durations for the wild-type and mutant receptors. A closed, unoccupied receptor (C) binds two agonist molecules (A) to become a doubly liganded, open receptor (A2O):
Model 1.
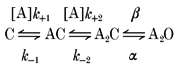
k+1 and k+2 are the agonist association rate constants, k-1 and k-2 are the agonist dissociation rate constants, β is the channel opening rate constant, and α is the channel closing rate constant. For mouse adult muscle nAChR activated by ACh, the two transmitter binding sites have essentially equivalent KD values (Akk & Auerbach, 1996; Wang et al. 1997; Salamone et al. 1999). Therefore, k+1= 2k+2, and k-2= 2k-1, and the microscopic dissociation equilibrium constant of each site (KD) is k-1/k+2. The analysis was also performed without such constraint. However, in most cases, no well-defined sets of rate constant estimates could be obtained. The gating equilibrium constant (Θ) is β/α. At the agonist concentrations used, monoliganded openings were rare and, consequently, were excluded from the analysis.
Hybrid receptors were expressed when both wild-type and mutant α subunits were coexpressed (along with wild-type β, δ and ε subunits). The activation rate constants for the hybrid receptors were estimated according to Model 2:
Model 2.
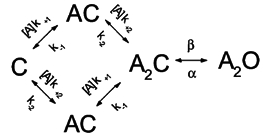
The symbols are as described above for Model 1. Model 2 takes into consideration that the two binding sites have different affinities for the agonist molecule. Both Models 1 and 2 assume a concerted gating reaction, i.e. a single conformational change occurs after both agonist binding sites are occupied. As an alternative, one could imagine a stepwise channel activation mechanism in which the two binding sites undergo independent conformational changes (Colquhoun & Ogden, 1988; Auerbach, 1993). According to such a mechanism, the channel is considered to open only after both binding sites have undergone the conformational changes. In the case of hybrid receptors, in light of only one of the binding sites containing the mutation, one could speculate that a stepwise scheme leads to a more correct description of experimental data. However, to reduce the complexity of models, Models 1 and 2 were used, both of which assume a single concerted gating event. Even though channel block was apparent at some high agonist concentrations, this step was not included in Models 1 and 2. The rate constant analysis was carried out at relatively low agonist concentrations (< 1 mm ACh), at which channel block is not prominent.
Two agonists were used, ACh and TMA. For receptors activated by ACh, two kinds of concentration-response curves were constructed. First, the probability of being open within a cluster (Po) was determined as a function of agonist concentration. This curve is similar to a whole-cell concentration-response curve, minus the effects of desensitization. Channel block by the agonists examined was rapid and was manifest at high concentrations mainly as a reduction in the current amplitude. At high agonist concentrations, block also prolonged the apparent open channel lifetime, thereby increasing the Po estimate. No correction for block was made in the analysis. At the single-channel level, desensitization of nicotinic receptors was manifest as the termination of the cluster. Only intracluster events were analysed; thus, desensitization did not influence the Po estimate.
For the second concentration-response profile, the component in the intracluster closed time histograms that scaled with agonist concentration was measured. The inverse of its duration was defined as the effective opening rate, β'. As the agonist concentration increased, the closed intervals within clusters became briefer, and β' increased. At very high agonist concentrations, Models 1 and 2 reduced to: A2C ⇋ A2O, and the effective opening rate approached the intrinsic opening rate constant (β) of the receptor. For most receptor-ligand combinations, this parameter is not affected by channel block at high agonist concentrations, because the two have widely differing time constants (channel block, 40 000 s−1, Maconochie & Steinbach, 1995; channel opening rate constant < 10 000 s−1 for most mutant receptors). For receptors activated by TMA, only the effective opening rate curve was constructed.
The Po and β' versus[agonist] curves were fitted by an empirical equation (the Hill equation):
| (1) |
where the response (Po or β‘) is at agonist concentration A, EC50 is the concentration that produces a half-maximal response and n is the Hill coefficient.
For single-channel kinetic analysis, the rate constants for agonist association, agonist dissociation and channel closing were determined from the analysis of idealized intracluster interval durations using Q-matrix methods. A maximum-likelihood method was employed that incorporated an approximate correction for missed events (program MIL, http://www.qub.buffalo.edu; Qin et al. 1997). Usually, the rate constants were optimized using interval durations combined from files obtained at several agonist concentrations. Error limits were estimated from the curvature of the likelihood surface at its maximum using the approximation of parabolic shape (Qin et al. 1996).
RESULTS
αy93f mutant receptors activated by ach
The properties of the αY93F mutant receptor activated by ACh have been published previously elsewhere (Auerbach et al. 1996; Akk & Steinbach, 2000). The following is a short review of the effect of the mutation. Figure 1C shows part of a sample cluster from a mutant receptor activated by 500 μm ACh. Compared with the wild-type receptor (Fig. 1A), clusters of activity from the mutant receptor had lower open probability. Figure 2 shows the concentration-response curves for the αY93F mutant receptor. The mutation in the putative binding site led to a rightward shift in the concentration- response curves. The mutation shifted the midpoint of the Po curve from 29 μm to 1123 μm. A shift of similar magnitude was observed in the effective opening rate curve (Fig. 2B), from 448 μm to 2738 μm. The opening rate constant of the mutant receptor was only 1239 s−1, compared with ≈60 000 s−1 for the wild-type receptor.
Figure 1. Sample single-channel clusters and open and closed time histograms for the wild-type, hybrid and mutant αY93F receptors.
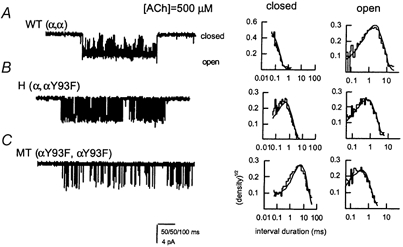
The receptors were activated by 500 μm ACh. Pipette potential was +50 mV. Inward current is shown downward. A, wild-type receptor (WT). B, hybrid receptor (H). C, mutant receptor (MT). For the mutant receptor (C), only a portion of a cluster is shown. Note also that a different time scale has been used for the mutant receptor cluster. Continuous lines in histograms were calculated according to the rate constants shown in Table 2.
Figure 2. Concentration-response properties of wild-type, hybrid and mutant αY93F receptors.
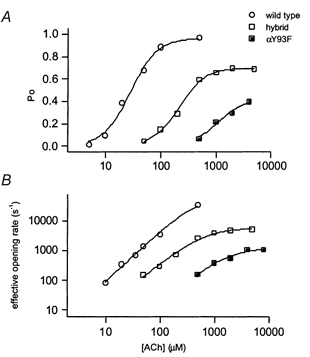
A, cluster open probability (Po) versus[ACh]. B, effective opening rate (β') versus[ACh]. Effective opening rate was measured as the inverse of the duration of the longest intracluster closed time component. The data for the wild-type receptor are from Akk & Auerbach (1996). Each point represents data from one patch. The number of events per patch was 739-40 198 (αY93F hybrid) and 2520-22 227 (αY93F mutant). Continuous lines are fits to eqn (1). The results from fits are shown in Table 1.
The activation rate constants for the mutant receptor were estimated using Model 1. In the analysis, two constraints were used. First, the channel opening rate constant was fixed at the value obtained from the saturation limit of the effective opening rate curve. Second, equivalent binding site kinetics was assumed. The activation rate constant estimates are presented in Table 2. The αY93F mutation resulted in a 13-fold reduction in the agonist association rate constant and a ≈3-fold reduction in the dissociation rate constant, resulting in a KD of 628 μm (versus 164 μm in the wild-type receptor). The mutation also led to a ≈50-fold reduction in the channel opening rate constant and an ≈2-fold increase in the channel closing rate constant. The gating equilibrium constant was reduced by > 100-fold compared with the wild-type receptor (0.42 versus 45).
Table 2.
Activation rate constants of wild-type, hybrid and mutant αY93F and αW149F receptors in the presence of ACh
| Receptor | k+1 (μm−1 s−1) | k−1 (s−1) | Kd1 or Kd (μm) | k+2(μm−1 s−1) | k−2 (s−1) | Kd2 (μm) | β (s−1) | α (s−1) | Θ |
|---|---|---|---|---|---|---|---|---|---|
| Wild type | — | 18000 ± 422 | 164 | 110 ± 20 | — | — | (60 000) | 1321 ± 15 | 45 |
| αY93F hybrid | (110) | (18 000) | 164 | 27 ± 3 | 10 363 ± 2151 | 384 | 5657 | 2099 ± 35 | 2.7 |
| αY93F mutant | — | 5153 ± 763 | 628 | 8.2 ± 1.2 | — | — | 1200 | 2919 ± 65 | 0.42 |
| αW149F hybrid | (110) | (18 000) | 164 | 16.3 ± 0.2 | (27 320) | 1676 | 3337 | 1909 ± 17 | 1.75 |
| αW149F mutant | — | 27320 ± 15517 | 1910 | 14.3 ± 8.0 | — | — | 648 | 1651 ± 22 | 0.39 |
The rate constants and error limits were determined from single-channel kinetic analysis using Model 1 (wild-type and mutant receptors) or Model 2 (hybrid receptors). The opening rate constants for hybrid and mutant receptors were constrained to values obtained from fitting the β′ curve (Table 1). The rate constants for the unmutated site in the hybrid receptor were constrained to values for the wild-type receptor binding site. KD was calculated as k−/k+; Θ is β/α. The values for the wild-type receptor are from Akk & Auerbach (1996). The opening rate constant for the wild-type receptor is from Maconochie & Steinbach (1998). For the hybrid aY93F receptor, the rate constants were estimated from simultaneous fitting of data from two patches recorded with 200 μm (4366 events) or 500 μm ACh (4388 events). The dead time was 45 μs. For the hybrid αW149F receptor, the rate constants were estimated from simultaneous fitting of data from three patches recorded with 200 μm (7431 events), 500 μm (10 346 events) or 1000 μm ACh (7943 events). The dead time was 60 μs. For the mutant receptor, the rate constants were estimated from simultaneous fitting of data from three patches recorded at 1000 μm (3213 events), 2000 μm (7585 events) or 5000 μm ACh (1324 events). The dead time was 60 μs.
αw149f mutant receptors activated by ach
The tryptophan-to-phenylalanine mutation in position 149 led to a reduction in cluster open probability. Figure 3B shows an example cluster from αW149F mutant receptors activated by 1 mm ACh. The Po and β' curves are shown in Fig. 4. The EC50 of the Po curve was shifted towards higher agonist concentrations by almost two orders of magnitude (2309 versus 29 μm). In contrast to the wild-type receptor, the saturation of the effective opening rate for the mutant receptor was observed at high ACh concentrations, making it possible to estimate the channel opening rate constant directly from the effective opening rate curve. The β for the αW149F receptor was almost 100-fold lower than in the wild-type receptor (648 s−1), and the EC50 for the effective opening rate curve was 3521 μm. The concentration-response parameters are summarized in Table 1.
Figure 3. Sample single-channel clusters and open and closed time histograms for the hybrid and mutant αW149F receptors.
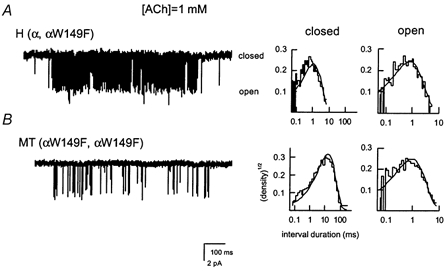
The receptors were activated by 1 mm ACh. Pipette potential was +50 mV. Inward current is shown downward. Continuous lines in histograms were calculated according to the rate constants shown in Table 2.
Figure 4. Concentration-response properties of the wild-type, hybrid and mutant αW149F receptors.
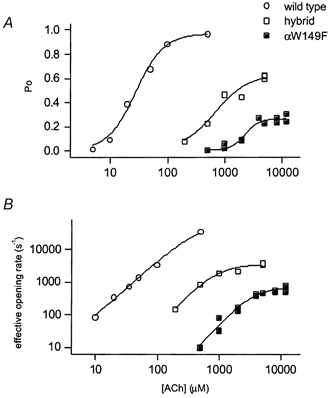
A, cluster open probability (Po) versus[ACh]. B, effective opening rate (β') versus[ACh]. Effective opening rate was measured as the inverse of the duration of the longest intracluster closed time component. The data for the wild-type receptor are from Akk & Auerbach (1996). Each point represents data from one patch. The number of events per patch was 1584-11 552 (αW149F hybrid) and 910-17 562 (αW149F mutant). Continuous lines are fits to eqn (1). The results from fits are shown in Table 1.
Table 1.
Concentration–response parameters of wild-type, hybrid and mutant αY93F and αW149F receptors activated by ACh
| Receptor | Po,max | EC50 (μm) | n | β (s−1) | EC50 (μm) | n |
|---|---|---|---|---|---|---|
| Wild-type | 0.96 ± 0.04 | 29 ± 3 | 1.8 ± 0.3 | 60 000 | 448 ± 52 | 1.7 ± 0.07 |
| αY93F hybrid | 0.69 ± 0.02 | 224 ± 13 | 1.9 ± 0.2 | 5657 ± 853 | 686 ± 180 | 1.4 ± 0.1 |
| αY93F mutant | 0.42 ± 0.08 | 1123 ± 357 | 1.8 ± 0.8 | 1239 ± 269 | 2738 ± 14 | 1.1 ± 0.03 |
| αW149F hybrid | 0.63 ± 0.06 | 698 ± 160 | 1.5 ± 0.5 | 3337 ± 337 | 958 ± 165 | 1.9 ± 0.2 |
| αW149F mutant | 0.26 ± 0.02 | 2309 ± 261 | 3.6 ± 1.4 | 648 ± 93 | 3521 ± 630 | 2.1 ± 0.2 |
Cluster open probability (Po) and the effective opening rate (β′) were fitted using eqn (1) on data from Figs 2 and 4. The values for the wild-type receptor are from Akk & Auerbach (1996). The values for the mutant αY93F receptor are from Akk & Steinbach (2000). The opening rate constant for the wild-type receptor is from Maconochie & Steinbach (1998). The values are best-fit parameters ± standard deviation estimated from the fit.
The activation rate constants were estimated using Model 1. The results of the rate constant analysis are shown in Table 2. The αW149F mutation led to an 8-fold reduction in the agonist association rate constant. The agonist dissociation rate constant remained largely unaffected by the mutation. The channel closing rate constant, α, was also not greatly affected by the mutation. Therefore, the > 100-fold reduction in the gating equilibrium constant, Θ (β/α), was caused mainly by the reduction in the channel opening rate constant.
αy93f and αw149f hybrid receptors activated by ach
When wild-type and mutated α subunits are coexpressed, then AChRs having three types of α subunit compositions should result: (i) two wild-type, (ii) two mutant and (iii) hybrids having one wild-type and one mutant. The hybrid receptors can exhibit two types of activity depending on which of the two α subunits contains the mutation, because the two α subunits have different subunits as neighbours. The pure wild-type and pure mutant receptors can be characterized separately by examining receptors when only one type of α subunit is expressed. Then, by coexpressing wild-type and mutant α subunits, the hybrid receptor can be studied after isolating its currents from channel activity that belongs to wild-type or mutant receptors.
Figure 5 shows a stability plot of cluster Po in cells expressing both wild-type and mutant αY93F or αW149F subunits. For both mutations, only one population of intermediate activity was observed, suggesting that a mutation in either α subunit led to a similar change in receptor kinetics. Sample clusters of αY93F and αW149F hybrid receptors elicited by 0.5-1 mm ACh are presented in Fig. 1B and Fig. 3A.
Figure 5. The open probability (Po) of clusters.
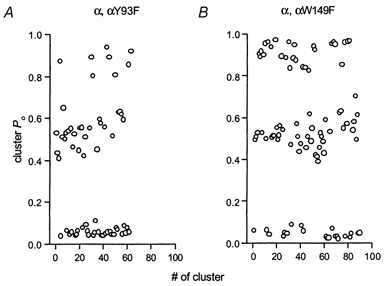
Intracluster Po is plotted for one patch expressing wild-type, hybrid and mutant αY93F (A) or αW149F (B) receptors. The receptors were activated by 500 μm (A) or 2 mm (B) ACh. The clusters with the highest Po belong to wild-type receptors, ones with the intermediate Po to hybrid receptors, and ones with the lowest Po to mutant receptors. Note that there is no overlap in the ranges of Po for the three classes of clusters. The total number of clusters was 63 in A, and 90 in B.
In a further effort to determine whether there was more than one type of hybrid receptor, the variability in Po in clusters classified as wild-type or hybrid was studied, by comparing the Po distributions for clusters from the wild-type receptor (50 μm ACh, Po= 0.59), αY93F hybrid receptor (500 μm ACh, Po= 0.53) and αW149F hybrid receptor (2 mm ACh, Po= 0.51). The results showed that the standard deviations were comparable (wild-type, 0.062; αY93F hybrid, 0.068; αW149F hybrid, 0.054). This observation supports the conclusion that clusters are not more heterogeneous in the postulated hybrid receptors than in the wild-type.
The concentration-response curves for the hybrid receptors are presented in Fig. 2 and Fig. 4. For the αY93F hybrid receptor (Table 1), the midpoint of the Po curve was shifted by almost 10-fold towards higher ACh concentrations (compared with the wild-type receptor), whereas the full mutant shifted the EC50 by ≈40-fold (see above, Table 2). The maximal Po of the hybrid receptor (0.69) was lower than that in the wild-type receptor (0.96) but higher than that in the mutant receptor (0.42). The channel opening rate constant, β, 5657 s−1, was > 10-fold lower than in the wild-type receptor, and was ≈5-fold greater than in the pure mutant AChR.
The αW149F hybrid receptor also had concentration- response parameters that were intermediate to the wild-type and full mutant receptors. The maximal Po of the hybrid was 0.63 and the EC50 of the Po curve was at 698 μm. The channel opening rate constant was 3337 s−1. The concentration-response parameters for the αW149F hybrid receptor are summarized in Table 1.
The activation rate constants for the hybrid receptors were estimated using Model 2. In this analysis, the channel opening rate constant was constrained to the value obtained from the fit of the effective opening rate curve. In addition, the agonist association and dissociation rate constants for one of the binding sites were fixed to the values estimated previously for the wild-type receptor binding sites (Akk & Auerbach, 1996). Thus, it was hypothesized that in the hybrid receptor, the mutation only altered the properties of the local binding site. It is reasonable to assume that if the binding sites are independent, then one of the sites should behave similarly to the wild-type receptor, and the other, similarly to the mutant receptor. By constraining the agonist association and dissociation rate constants in one of the binding sites, I aimed to estimate the same for the mutated site, in order to compare these values with the agonist association and dissociation rate constants in the pure mutant receptor. The results for the αY93F hybrid receptor are presented in Table 2. The agonist association rate constant for the mutant binding site was 27 μm−1 s−1. This was almost 3-fold higher than in the pure mutant receptor. The ACh dissociation rate constant was 10363 s−1, i.e. approximately 2-fold higher than in the mutant receptor. The equilibrium dissociation constant was 384 μm for the mutated site in the hybrid receptor. For comparison, the KD of the mutant receptor was 628 μm (a 1.6-fold difference).
A summary of the effects of the mutation on the free energy of channel gating is presented in Table 3. For the pure αY93F mutant receptor, the ΔΔG for the equilibrium gating constant Θ (β/α) demonstrated an increase of 2.77 kcal mol−1. For comparison, there was an increase of only 1.66 kcal mol−1 in the hybrid receptor. Thus, having a mutation in only one of the binding sites raised the free energy of the gating reaction by approximately one-half in comparison with having a mutation in both binding sites. It should be noted that the estimates of β and α were not affected by the constraint of equivalence of binding sites. The channel opening rate constant was estimated from the saturation of the effective opening rate curve. The channel closing rate constant was estimated from single-channel kinetic analysis, but the estimated value was, in general, not sensitive to the type of model used. Due to the relatively high k-2/β ratio, the estimated α was close to an inverse of the mean open duration. With the αY93F mutations in both binding sites (full mutant receptor), the increase in free energy for equilibrium binding was 0.8 kcal mol−1 per binding site. In the mutated binding site of the hybrid receptor, the increase in free energy for binding was 0.5 kcal mol−1.
Table 3.
Gating energetics of hybrid and mutant αY93F and αW149F receptors in the presence of ACh
| Mutation | Hybrid | Mutant | (2 ± hybrid) - mutant |
|---|---|---|---|
| αY93F | 1.66 | 2.77 | 0.55 |
| αW149F | 1.92 | 2.80 | 1.04 |
Values are given as ΔΔG in kcal mol−1 and are calculated as 0.59 ln(ΘWT/Θmutant), from the equilibrium gating constants given in Table 1.
The kinetic properties of the αW149F hybrid receptor were determined according to Model 2. As described above for the αY93F hybrid receptor, the agonist association and dissociation rate constants for the unmutated site were constrained to the values obtained earlier for the wild-type receptor (Akk & Auerbach, 1996). The channel opening rate constant was fixed at 3337 s−1, as estimated from the saturation of the effective opening rate. In addition, the agonist dissocation rate constant for the mutated site was constrained to 27 320 s−1. This value corresponds to the agonist dissocation rate constant in the mutant receptor. Without this constraint, the analysis program, MIL, did not converge to a well-defined set of rate constants. The analysis showed that the agonist association rate constant of the mutated site in the hybrid receptor was 16.3 μm−1 s−1. This value agrees well with the estimate for the agonist assocation rate constant in the mutant receptor (14.3 μm−1 s−1). Thus, the results suggest that a binding site with an αW149F mutation has almost identical affinity for ACh whether it is in a hybrid or pure mutant receptor, i.e. whether the other agonist binding site contains the αW149F mutation or not. The KD of the mutated site in the hybrid receptor was 1676 μm; for comparison, it was 1910 μm in the mutant receptor (a 1.1-fold difference).
The gating energetics for the hybrid and mutant αW149F receptors are shown in Table 3. Similarly to the αY93F receptor, having an αW149F mutation in both binding sites increased the free energy for the gating reaction by approximately two times more than when only one binding site contained the mutated α subunit. The increase in the free energy for the binding reaction in a mutated binding site was similar in the mutant (1.45 kcal mol−1) and hybrid receptors (1.37 kcal mol−1).
Activation of mutant and hybrid receptors by TMA
TMA opens the mouse nicotinic receptor with a rate constant that is > 7-fold lower than that for ACh (Zhang et al. 1995; Akk & Auerbach, 1996). In the adult mouse receptor, the equilibrium dissociation constant of the nicotinic receptor for TMA is > 8-fold greater than that for ACh (Akk & Auerbach, 1996). Here, the effects of the αY93F and αW149F mutations on the gating properties of TMA-activated nicotinic receptors were examined.
Figure 6 shows single-channel currents from pure and hybrid αY93F receptors activated by 10 mm TMA. In the pure mutant AChR, the currents consisted of isolated openings with no clusters apparent. Thus, in these experiments it was not possible to establish the number of simultaneously active receptors and, therefore, the kinetic properties of a single receptor. From this record, only the upper limit for the channel opening rate constant β (30 s−1) could be set. This value equals the inverse of the main closed time component in the patch. Therefore, a different approach was employed, examining the effect of a single mutation (hybrid receptors) and ‘extrapolating’ the effect to the full mutant receptor. Figure 6B shows a single-channel cluster from the hybrid αY93F receptor activated by 10 mm TMA. The effective opening rate curve for the αY93F hybrid receptors is shown in Fig. 6E, and the results are summarized in Table 4. The effective opening rate curve saturates at 228 s−1, i.e. with a mutation in only one binding site, the channel opening rate constant was reduced by ≈36-fold. The channel closing rate constant was less affected (3068 s−1 in the hybrid, determined at 2 mm TMA). Thus, the gating equilibrium constant for TMA was 73-fold lower in the hybrid than in the wild-type receptor. This corresponds to a 2.53 kcal mol−1 difference in free energies. If we assume that the effect of the Y93F mutation is equal and independent at each binding site, the gating equilibrium constant for TMA in the pure mutant receptor can be estimated by doubling the increase in free energy in the hybrid receptor. Therefore, we can estimate a 5.06 kcal mol−1 increase in the gating free energy corresponding to a Θ of 0.001 for the αY93F mutant receptor. With the channel closing rate constant α at 4540 s−1 (estimated from the mutant receptor activity in the presence of 2 mm TMA, pipette potential =+60 mV), the αY93F mutant receptor opening rate constant in the presence of TMA was 4.5 s−1.
Figure 6. Mutant and hybrid αY93F and αW149F receptors activated by 10 mm TMA.
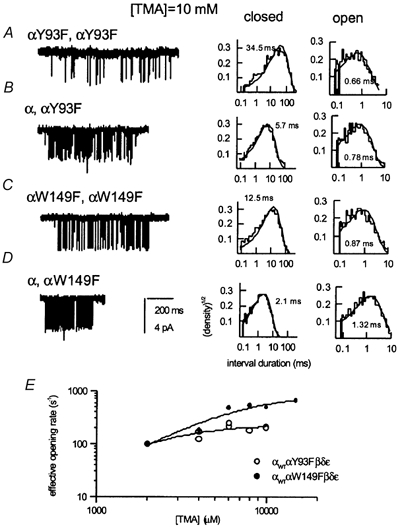
Single-channel activity and open and closed time histograms from the αY93F mutant (A) and hybrid (B) receptors, and αW149F mutant (C) and hybrid (D) receptors. For the αY93F mutant receptor, no clusters were detected at 10 mm TMA; an episode of 1200 ms in duration is shown. For the rest, a representative single-channel cluster is shown. The values within open and closed time histograms are results of fits to a single exponential. E, an effective opening rate (β') versus[TMA] for the αY93F and αW149F hybrid receptors. Each point represents data from one patch. The number of events per patch was 459-7378 (αY93F) and 598-4518 (αW149F). The continuous lines are fits to eqn (1). The concentration-response parameters are shown in Table 4.
Table 4.
Gating properties of wild-type and hybrid αY93F and αW149F receptors in the presence of TMA
| Receptor | β (s−1) | EC50 (μm) | n | α (s−1) | Θ |
|---|---|---|---|---|---|
| wild-type | 8156 ± 382 | 2375 ± 265 | 1.2 ± 0.1 | 1508 ± 205 | 5.4 |
| αY93F hybrid | 228 ± 67 | 2474 ± 1037 | 1.7 ± 1.2 | 3068 ± 68 | 0.074 |
| αW149F hybrid | 844 ± 430 | 7041 ± 4862 | 1.7 ± 0.6 | 2001 ± 57 | 0.42 |
The values for the wild-type receptor are from Akk & Auerbach (1996). β was determined from the saturation of the effective opening rate by fitting to eqn (1). α was determined in the presence of 2 mM TMA (pipette potential = 50–60 mV). Θ is β/α.
The kinetic properties of the αW149F receptor activated by TMA were also investigated. Single-channel clusters could be obtained in the presence of high concentrations (8-10 mm) of TMA. The effective opening rate for the patch shown in Fig. 6C was 80 s−1. In another patch, recorded in the presence of 8 mm TMA, the effective opening rate was 66 s−1. However, due to the lack of clear saturation of the effective opening rate curve, direct determination of the opening rate constant was not possible. Therefore, the approach described above for the αY93F mutant receptor was used. After determining the effect of a single αW149F mutation per receptor, the value was doubled to estimate the effect two mutations in the receptor would have. Figure 6D shows a sample cluster of the αW149F hybrid receptor, and the results of kinetic analysis are summarized in Table 4. The equilibrium gating constant (Θ) was 0.42 for hybrid receptors. Again, assuming that the effect of the mutant on gating was equal and independent in each binding site, it was estimated that Θ for the pure αW149F mutant receptor was 0.033. The closing rate constant of the αW149F receptor was 2298 s−1 (estimated in the presence of 2 mm TMA, pipette potential =+50 mV). Thus, the opening rate constant in the TMA-activated αW149F mutant receptor was 76 s−1.
DISCUSSION
The effects of two putative binding site mutations on the kinetic properties of the muscle nAChR were studied. The results showed that both mutations, αY93F and αW149F, led to a rightward shift in the concentration- response curves. The αY93F mutation mostly affected channel gating (the gating equilibrium constant was reduced by 107-fold) with a relatively small effect on ACh binding. The αW149F mutation affected both binding and gating: the equilibrium dissociation constant was reduced ≈12-fold, and the gating equilibrium constant by 115-fold. Experiments on hybrid receptors (containing one mutated and one wild-type α subunit) showed that when the receptor contained only one mutation, the effect on channel gating was one-half of the effect when the receptor contained mutations in both binding sites. In the αW149F hybrid receptor, a mutation in one of the binding sites had little effect on the affinity of the other, unmutated, site. On the other hand, with only one αY93F mutation per receptor, the mutated binding site had an affinity that was 1.6 times higher than in the pure mutant receptor. The individual binding rate constants were affected by 2- to 3-fold.
This is the first detailed kinetic study on the αW149Fβδε receptor. It has been shown previously that the αW149 residue can be labelled by [3H]p-(dimethylamino) benzenediazonium fluoroborate, a photoaffinity ligand for the AChR binding site (Dennis et al. 1988). The electrophysiological effects of a natural amino acid mutation to the αW149 site have been studied only in α7 neuronal AChR. Galzi et al. (1991) examined the effect of the W-to-F mutation in position 148 (homologous to 149 in mouse muscle α subunit) and showed that the mutation led to a reduction in the apparent affinity for ACh and nicotine. However, a dissection of the concentration- response data into binding and gating components was not performed. The results presented in the current study showed that a W-to-F mutation in the α149 site affected both processes but with greater impact on channel gating. Zhong et al. (1998) investigated the effect of the presence of several unnatural amino acids in the α149 site. Their results suggested that an interaction existed between the aromatic residue in the α149 site and the quaternary ammonium moiety of the ACh molecule.
It appears that the αY93F and αW149F mutations have similar effects on channel kinetics when the mutated α subunit is in either of the subunit pairs (α-δversusα-ε). When hybrid receptors were expressed, only one type of activity was detected in addition to wild-type and full mutant activity. As expected, the additional component appeared intermediate, in terms of Po, to the wild-type and pure mutant clusters. Both wild-type and mutant receptors demonstrated high levels of expression when expressed as homologous α subunits (data not quantified or shown). Hybrid receptors were expressed with a similarly high efficiency when wild-type and mutant α subunits were used. Thus, it is improbable that misassembly of one but not another type of hybrid receptor could result in the absence of one type of hybrid activity. Similarly, it is unlikely that a mutation in one of the binding sites, but not in another (or when mutations are present in both binding sites), could lead to a non-functional receptor. Finally, a possibility exists that one of the hybrids is undistinguishable from either the pure mutant or the wild-type receptor. The methods used here cannot rule out this possibility.
The present results contrast with those of previous studies, in which we examined the effect of mutations of another residue located in the vicinity of the ligand binding site, αD200N, on gating by ACh (Akk et al. 1996). Receptors containing only one αD200N mutation exhibited two types of hybrid activity, with the channel opening rate constants differing by 5- to 10-fold depending on whether the receptors were expressed in fetal or adult configurations. By comparing the hybrid receptor activity in fetal and adult receptors, we proposed that an αD200N mutation present in the α-δ pair reduced the channel opening rate constant to a lesser degree than a mutation present in the α-γ or α-ε site.
For the αW149 site, the hybrid receptor activity can be adequately described by assuming that the ligand binding sites behave independently of each other. A model with one binding site possessing wild-type-like affinity, and the other, mutant-like affinity, well characterizes single-channel data obtained for the hybrid receptors. For the αY93 site, the agonist association rate constant was ≈3-fold higher in the mutated binding site of the hybrid receptor than in the mutant receptor. The agonist dissociation rate constant was ≈2-fold higher in the hybrid receptor than in the mutant receptor. This led to a KD estimate of 384 μm in the hybrid receptor mutant site, while the KD of the pure mutant receptor was 628 μm. Therefore, the results appear to suggest the presence of interaction between the two agonist binding sites in the αY93F receptor.
The effect on gating by a single mutation per receptor was approximately half of the effect seen in the full mutant receptor (two mutations per receptor). For the αY93F receptor, the ΔΔG for gating was somewhat smaller (≈0.55 kcal mol−1) in the mutant receptor than the value obtained by doubling the effect on hybrid receptor gating. So the Θ for the mutant receptor was ≈3-fold higher than would be expected by assuming complete equality and independence of contributions. Also, in the αW149F receptor, the ΔΔG for gating was smaller (by 1.04 kcal mol−1) in the mutant receptor than would have been predicted from doubling the effect seen in the hybrid receptor. The additivity of the contributions of the mutated α subunits to receptor function can be estimated by computing a coupling coefficient (Ω) (see Hidalgo & MacKinnon, 1995):
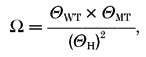 |
where Θ is the gating equilibrium constant, and WT, MT and H refer to wild-type, mutant and hybrid receptors respectively. For the αY93F mutation, Ω= 2.6 and for the αW149F mutation, Ω= 5.7. The coupling energy, calculated as ΔG =RTlnΩ, is 0.6 kcal mol−1 for the αY93F mutation, and 1.0 kcal mol−1 for the αW149F mutation. According to LiCata & Ackers (1995), only coupling energies greater than (±)1.5 kcal mol−1 (corresponding to Ω of less than 0.077 or more than 13) are considered non-additive and significant in terms of interaction between the two sites. Therefore, the difference between the effect in the pure mutant receptor and the doubled effect observed in the hybrid receptor is relatively small, and it is concluded that the mutation-caused effects on gating by ACh can be considered as additive.
The orientation of the ACh molecule in the binding site has been a topic of dispute. In another ACh-binding protein, acetylcholinesterase, the positively charged quaternary ammonium group interacts with the indole group of a tryptophan located on the acetylcholinesterase molecule (Silman et al. 1994). Detection of several aromatic residues near or at the putative ligand binding site led to a proposal of a similar cation π interaction between ACh and protein in the case of the AChR (Dougherty & Stauffer, 1990). Sine et al. (1994) demonstrated that mutations to αY93, αY190 and αY198 shifted the equilibrium binding of carbamylcholine and TMA similarly, suggesting that all tyrosine residues contribute to the stabilization of the quaternary ammonium moiety. Aylwin & White (1994) and Nowak et al. (1995) studied mouse AChR expressed in Xenopus oocytes and concluded that, while all three tyrosine residues participated in agonist binding, they appeared to have different roles in the interaction between the receptor and the ACh molecule. Kearney et al. (1996) found that shifts in the EC50 of whole-cell concentration- response curves were similar for ACh and TMA when various mutations were made to the αY93 site. Together, these results suggest that one or more aromatic residues of the binding site interacts directly with the positively charged quaternary ammonium group present on nicotinic ligands. After examining the properties of several tryptophan residues in the vicinity of the receptor binding site, it was also proposed that the quaternary ammonium group of ACh interacts with the indole side chain of the αW149 residue via cation π interaction (Zhong et al. 1998). In agreement with such a proposal, it was found that when the αW149 residue was substituted with an unnatural amino acid Tyr-O-(CH2)3-N(CH3)+, which placed the quaternary ammonium group in a position roughly similar to that occurring in the case of ACh binding to the wild-type receptor (i.e. in close vicinity of the α149 site), the receptors become constitutively active (ibid.). Recently, it was found that treatment of αY198C mutant receptors with [2-(trimethylammonium)ethyl]-methanethiosulfonate (MTSET) rendered the receptors constitutively active (Sullivan & Cohen, 2000). MTSET treatment resulted in an attachment of a thiocholine molecule to the α198 site. Comparison of sulfhydryl modifying agents of different length led to the conclusion that the site interacting with the quaternary ammonium group was at ≈6.9 Å from the α198 residue.
In the present work, analysis of the kinetics of single-channel records was used to evaluate the interaction between two aromatic residues in the putative ligand binding site and the agonist molecule. Using the approach of thermodynamic mutant cycles, it is possible to calculate the coupling coefficient and energy for the receptor and the ligand molecule (Carter et al. 1984; Hidalgo & MacKinnon, 1995). This method has usually been applied to evaluate additivity and pair-wise interactions of specific amino acid residues in protein-protein or protein-toxin interactions (Horovitz & Fersht, 1990; Serrano et al. 1990; Ackermann et al. 1998). Here, a binding site mutation (αY93F or αW149F) served as one of the mutants, while the elimination of the tail group on the ACh molecule (change of agonist: ACh → TMA) served as the other. So, the interaction between the αY93 or αW149 site and the acetyl group of ACh could be probed. The interaction during channel gating was assessed according to the following scheme:
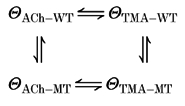 |
Θ is the equlibrium gating constant (β/α), WT is a wild-type receptor, MT is a mutant receptor, and ACh and TMA stand for acetylcholine and tetramethylammonium, respectively. The values for Θ for the ACh-activated wild-type and mutant receptors are those shown in Table 2. The Θ for the TMA-activated wild-type receptor has been established previously (5.4; Akk & Auerbach, 1996). The Θ for the TMA-activated αY93F receptor was 0.001, and for the αW149F receptor, 0.033 (see above). The coupling coefficient, Ω, was then calculated as:
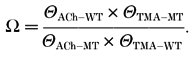 |
For the αY93F receptor, Ω was 0.02 and the coupling energy ΔG = -2.31 kcal mol−1. For the αW149F receptor, Ω was 0.71 and ΔG = -0.20 kcal mol−1. Coupling energies greater than (±)1.5 kcal mol−1 are considered non-additive and significant in terms of interaction between the two sites (LiCata & Ackers, 1995; Ackermann et al. 1998). Therefore, the results suggest that significant interaction takes place between the acetyl group and the αY93 site, but not the αW149 site, during channel gating. The Θ of the TMA-activated αY93F mutant receptor was ≈50-fold less than that which would be expected assuming independent contributions by the mutation and loss of the tail group of ACh. It should be noted, however, that ‘interaction’ in the present case does not necessarily refer to chemical bonding between the α93 site and the tail group of ACh, but is rather an indicator of non-additivity in more general terms. Also, the double mutant cycle approach was used to examine only channel gating. No evidence is available on whether the αY93F mutant receptor similarly discriminates in the binding of ACh and TMA. In light of previous electrophysiological data on receptors with mutations at the α93 site (Kearney et al. 1996), this appears less likely.
The standard ways to determine the channel opening rate constant are to measure β from the saturation of the effective opening rate curve (Auerbach, 1993), from the analysis of intraburst closed times (Colquhoun & Sakmann, 1981), or from the rate of development of current upon fast perfusion to outside-out patches (Maconochie & Steinbach, 1998). All these methods have limitations when certain receptor-ligand combinations result in very low apparent affinity. In such cases, the concentration-response curves are shifted towards higher agonist concentrations where channel block becomes dominant. A low value for β may lead to the absence of single-channel clusters, making it impossible to measure the saturation of the effective opening rate. The usual intraburst glitch analysis at low agonist concentrations is hindered by the low β values, leading to low β/k-2 ratios, and hence to little or no reopenings within the burst. To overcome the obstacles of small responses, an approach of using an additional mutation has been employed (Kearney et al. 1996; Zhong et al. 1998). In these studies, in addition to the mutation to be studied, the receptors contained another mutation, βL262S, which led to a leftward shift in the concentration-response curve. The authors then operated under the assumption that the effects of the mutation to be investigated and the βL262S mutation were independent. In the present work, a novel approach was used to estimate the channel opening rate constant. First, the gating properties (Θ) of the hybrid receptor were estimated. This receptor had a channel opening rate constant intermediate to that of the wild-type and the pure mutant receptor. Since only one hybrid population was detected, it was concluded that mutations in either binding site resulted in similar changes. Therefore, to estimate the gating properties of the mutant receptor, the effect observed in the hybrid receptor was doubled. Experiments in the presence of a high-efficacy agonist, ACh, were used as a control. In the presence of ACh, the gating properties could be determined independently for both hybrid and mutant receptors, revealing the relationship between the number of mutated binding sites and the shift in the channel gating equilibrium constant. Using this method, it was possible to estimate that the β was only 5 s−1 in the TMA-activated αY93F mutant receptor!
In conclusion, this study demonstrates that mutations to the aromatic residues αY93 and αW149 greatly affect ligand binding and channel gating albeit to a different degree. The data also suggest that agonist specific effects on gating take place in the αY93F mutant receptor. These results coupled to previous electrophysiological and biochemical data demonstrate the significance of binding site aromatic residues in the conformational change involved in the activation of the nicotinic receptor.
Acknowledgments
I thank Drs J. H. Steinbach and A. Auerbach for advice and comments during the course of the work. G.A. is a McDonnell Center for Cellular and Molecular Neurobiology fellow. This work was supported in part by NS-22356 to J. H. Steinbach and NS-23513 to A. Auerbach.
References
- Ackermann EJ, Ang ET-H, Kanter JR, Tsigelny I, Taylor P. Identification of pairwise interactions in the α-neurotoxin-nicotinic acetylcholine receptor complex through double mutant cycles. Journal of Biological Chemistry. 1998;273:10958–10964. doi: 10.1074/jbc.273.18.10958. [DOI] [PubMed] [Google Scholar]
- Akk G, Auerbach A. Inorganic, monovalent cations compete with agonists for the transmitter binding site of nicotinic acetylcholine receptors. Biophysical Journal. 1996;70:2652–2658. doi: 10.1016/S0006-3495(96)79834-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Sine S, Auerbach A. Binding sites contribute unequally to the gating of mouse nicotinic αD200N acetylcholine receptors. Journal of Physiology. 1996;496:185–196. doi: 10.1113/jphysiol.1996.sp021676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Steinbach JH. Structural elements near the C-terminus are responsible for changes in nicotinic receptor gating kinetics following patch excision. Journal of Physiology. 2000;527:405–417. doi: 10.1111/j.1469-7793.2000.t01-2-00405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach A. A statistical analysis of acetylcholine receptor activation in Xenopus myocytes: stepwise versus concerted models of gating. Journal of Physiology. 1993;461:339–378. doi: 10.1113/jphysiol.1993.sp019517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach A, Sigurdson W, Chen J, Akk G. Voltage dependence of mouse acetylcholine receptor gating: different charge movements in di-, mono- and unliganded receptors. Journal of Physiology. 1996;494:155–170. doi: 10.1113/jphysiol.1996.sp021482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Short Protocols in Molecular Biology. New York: John Wiley & Sons, Inc.; 1992. [Google Scholar]
- Aylwin ML, White MM. Ligand-receptor interactions in the nicotinic acetylcholine receptor probed using multiple substitutions at conserved tyrosines on the α subunit. FEBS Letters. 1994;349:99–103. doi: 10.1016/0014-5793(94)00649-0. [DOI] [PubMed] [Google Scholar]
- Blount P, Merlie JP. Molecular basis of the two nonequivalent ligand binding sites of the muscle nicotinic acetylcholine receptor. Neuron. 1989;3:349–357. doi: 10.1016/0896-6273(89)90259-6. [DOI] [PubMed] [Google Scholar]
- Carter PJ, Winter G, Wilkinson AJ, Fersht AR. The use of double mutants to detect structural changes in the active site of the tyrosyl-tRNA synthetase (Bacillus stearothermophilus) Cell. 1984;38:835–840. doi: 10.1016/0092-8674(84)90278-2. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Ogden DC. Activation of ion channels in the frog end-plate by high concentrations of acetylcholine. Journal of Physiology. 1988;395:131–159. doi: 10.1113/jphysiol.1988.sp016912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Sakmann B. Fluctuations in the microsecond time range of the current through single acetylcholine receptor ion channels. Nature. 1981;294:464–466. doi: 10.1038/294464a0. [DOI] [PubMed] [Google Scholar]
- Czajkowski C, Karlin A. Structure of the nicotinic receptor acetylcholine-binding site. Identification of acidic residues in the δ subunit within 0.9 nm of the 5 α subunit-binding. Journal of Biological Chemistry. 1995;270:3160–3164. doi: 10.1074/jbc.270.7.3160. [DOI] [PubMed] [Google Scholar]
- Dennis M, Giraudat J, Kotzyba-Hibert F, Goeldner M, Hirth C, Chang JY, Lazure C, Chretien M, Changeux JP. Amino acids of the Torpedo marmorata acetylcholine receptor α subunit labeled by a photoaffinity ligand for the acetylcholine binding site. Biochemistry. 1988;27:2346–2357. doi: 10.1021/bi00407a016. [DOI] [PubMed] [Google Scholar]
- Devillers-Thiery A, Galzi JL, Eisele JL, Bertrand S, Bertrand D, Changeux JP. Functional architecture of the nicotinic acetylcholine receptor: a prototype of ligand-gated ion channels. Journal of Membrane Biology. 1993;136:97–112. doi: 10.1007/BF02505755. [DOI] [PubMed] [Google Scholar]
- Dougherty DA, Stauffer DA. Acetylcholine binding by a synthetic receptor: implications for biological recognition. Science. 1990;250:1558–1560. doi: 10.1126/science.2274786. [DOI] [PubMed] [Google Scholar]
- Galzi JL, Bertrand D, Devillers-Thiery A, Revah F, Bertrand S, Changeux JP. Functional significance of aromatic amino acids from three peptide loops of the α7 neuronal nicotinic receptor site investigated by site-directed mutagenesis. FEBS Letters. 1991;294:198–202. doi: 10.1016/0014-5793(91)80668-s. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hidalgo P, MacKinnon R. Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science. 1995;268:307–310. doi: 10.1126/science.7716527. [DOI] [PubMed] [Google Scholar]
- Horovitz A, Fersht AR. Strategy for analysing the co-operativity of intramolecular interactions in peptides and proteins. Journal of Molecular Biology. 1990;214:613–617. doi: 10.1016/0022-2836(90)90275-Q. [DOI] [PubMed] [Google Scholar]
- Jackson MB. Perfection of a synaptic receptor: kinetics and energetics of the acetylcholine receptor. Proceedings of the National Academy of Sciences of the USA. 1989;86:2199–2203. doi: 10.1073/pnas.86.7.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney PC, Nowak MW, Zhong W, Silverman SK, Lester HA, Dougherty D A. Dose-response relations for unnatural amino acids at the agonist binding site of the nicotinic acetylcholine receptor: tests with novel side chains and with several agonists. Molecular Pharmacology. 1996;50:1401–1412. [PubMed] [Google Scholar]
- LiCata VJ, Ackers GK. Long-range, small magnitude nonadditivity of mutational effects in proteins. Biochemistry. 1995;34:3133–3139. doi: 10.1021/bi00010a001. [DOI] [PubMed] [Google Scholar]
- Maconochie DJ, Steinbach JH. Block by acetylcholine of mouse muscle nicotinic receptors, stably expressed in fibroblasts. Journal of General Physiology. 1995;106:113–147. doi: 10.1085/jgp.106.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maconochie DJ, Steinbach JH. The channel opening rate of adult- and fetal-type mouse muscle nicotinic receptors activated by acetylcholine. Journal of Physiology. 1998;506:53–72. doi: 10.1111/j.1469-7793.1998.053bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Czajkowski C, Karlin A. The contributions of aspartyl residues in the acetylcholine receptor γ and δ subunits to the binding of agonists and competitive antagonists. Journal of Biological Chemistry. 1996;271:13497–13503. doi: 10.1074/jbc.271.23.13497. [DOI] [PubMed] [Google Scholar]
- Nowak MW, Kearney PC, Sampson JR, Saks ME, Labarca CG, Silverman S K, Zhong W, Thorson J, Abelson JN, Davidson N, Schultz PG, Dougherty DA, Lester HA. Nicotinic receptor binding site probed with unnatural amino acid incorporation in intact cells. Science. 1995;268:439–442. doi: 10.1126/science.7716551. [DOI] [PubMed] [Google Scholar]
- Qin F, Auerbach A, Sachs F. Estimating single-channel kinetic parameters from idealized patch-clamp data containing missed events. Biophysical Journal. 1996;70:264–280. doi: 10.1016/S0006-3495(96)79568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F, Auerbach A, Sachs F. Maximum likelihood estimation of aggregated Markov processes. Proceedings of the Royal Society. 1997;B 264:375–383. doi: 10.1098/rspb.1997.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakmann B, Patlak J, Neher E. Single acetylcholine-activated channels show burst-kinetics in presence of desensitizing concentrations of agonist. Nature. 1980;286:71–73. doi: 10.1038/286071a0. [DOI] [PubMed] [Google Scholar]
- Salamone FN, Zhou M, Auerbach A. A re-examination of adult mouse nicotinic acetylcholine receptor channel activation kinetics. Journal of Physiology. 1999;516:315–330. doi: 10.1111/j.1469-7793.1999.0315v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano L, Horovitz A, Avron B, Bycroft M, Fersht AR. Estimating the contribution of engineered surface electrostatic interactions to protein stability by using double-mutant cycles. Biochemistry. 1990;29:9343–9352. doi: 10.1021/bi00492a006. [DOI] [PubMed] [Google Scholar]
- Silman I, Harel M, Axelsen P, Raves M, Sussman JL. Three-dimensional structures of acetylcholinesterase and of its complexes with anticholinesterase agents. Biochemical Society Transactions. 1994;22:745–749. doi: 10.1042/bst0220745. [DOI] [PubMed] [Google Scholar]
- Sine SM, Quiram P, Papanikolaou F, Kreienkamp HJ, Taylor P. Conserved tyrosines in the α subunit of the nicotinic acetylcholine receptor stabilize quaternary ammonium groups of agonists and curariform antagonists. Journal of Biological Chemistry. 1994;269:8808–8816. [PubMed] [Google Scholar]
- Sullivan DA, Cohen JB. Mapping the agonist binding site of the nicotinic acetylcholine receptor: orientation requirements for activation by covalent agonist. Journal of Biological Chemistry. 2000;275:12651–12660. doi: 10.1074/jbc.275.17.12651. [DOI] [PubMed] [Google Scholar]
- Wang HL, Auerbach A, Bren N, Ohno K, Engel AG, Sine SM. Mutation in the M1 domain of the acetylcholine receptor α subunit decreases the rate of agonist dissociation. Journal of General Physiology. 1997;109:757–766. doi: 10.1085/jgp.109.6.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen J, Auerbach A. Activation of recombinant mouse acetylcholine receptors by acetylcholine, carbamylcholine and tetramethylammonium. Journal of Physiology. 1995;486:189–206. doi: 10.1113/jphysiol.1995.sp020802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W, Gallivan JP, Zhang Y, Li L, Lester HA, Dougherty DA. From ab initio quantum mechanics to molecular neurobiology: a cation-π binding site in the nicotinic receptor. Proceedings of the National Academy of Sciences of the USA. 1998;95:12088–12093. doi: 10.1073/pnas.95.21.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]