

Abstract
Effects of arachidonic acid (AA) on proton and electron currents in human eosinophils were studied using the permeabilized-patch voltage-clamp technique, using an applied NH4+ gradient to control pHi.
Superoxide anion (O2−) release was assessed by cytochrome c reduction in human eosinophils. Significant O2− release was stimulated by 5–10 μm AA.
AA activated diphenylene iodinium (DPI)-inhibitable inward current reflecting electron efflux through NADPH oxidase. These electron currents (Ie) were elicited in human eosinophils at AA concentrations (3–10 μm) similar to those that induced O2− release.
The voltage-gated proton conductance (gH) in eosinophils stimulated with AA was profoundly enhanced: H+ current amplitude (IH) increased 4.6 times, activation was 4 times faster, and the H+ conductance-voltage (gH−V) relationship was shifted to substantially more negative voltages. The electrophysiological effects of AA resembled those reported for PMA, except that AA did not consistently slow τtail (deactivation of H+ currents).
The stimulation of both proton and electron currents by AA was reversible upon washout. Repeated exposure elicited repeated responses. The activation of H+ currents by AA was dissociable from its activation of NADPH oxidase; H+ currents were enhanced at low concentrations of AA that did not elicit detectable Ie or when NADPH oxidase was inhibited by DPI.
Most of the effects of AA on H+ currents qualitatively resemble those reported in whole-cell studies, reflecting a more direct action than PMA. The results are compatible with AA being an immediate activator of both NADPH oxidase and proton channels in human eosinophils.
The importance of arachidonic acid (AA) during the respiratory burst - the rapid conversion of oxygen to superoxide anion (O2−) and other cytotoxic reactive oxygen species to kill bacteria and other invaders - is well established in neutrophils (Badwey et al. 1981; Curnutte et al. 1984; Henderson et al. 1989; Henderson & Chappell, 1992). Arachidonic acid is released from phagocytes during the respiratory burst, upon challenge with a wide variety of agonists including chemotactic peptides and phorbol myristate acetate (PMA) (Walsh et al. 1981; Bromberg & Pick, 1983; Galbraith, 1988; Tao et al. 1989). In addition, AA is itself a potent agonist that can trigger O2− release from human neutrophils (Badwey et al. 1981; Henderson et al. 1989, 1993), porcine macrophages (Bromberg & Pick, 1983), guinea-pig eosinophils (Lindsay et al. 1998) and, as demonstrated here, from human eosinophils. In fact, it has been proposed that AA is the immediate activator of NADPH oxidase (Bromberg & Pick, 1983; Henderson et al. 1989, 1993). Mechanisms of action proposed for AA include activation of a GTP binding protein (Abramson et al. 1991), increasing the activity of the assembled NADPH oxidase complex (Rubinek & Levy, 1993), and stimulation of the translocation of p47phox and its assembly with cytochrome b558 (Uhlinger et al. 1993; Cross et al. 1999). In a cell-free system, both AA and PKC-mediated phosphorylation of p47phox induce identical conformational changes in p47phox that result in NADPH oxidase activity (Swain et al. 1997; Park & Babior, 1997).
NADPH oxidase is electrogenic (Henderson et al. 1987, 1988a,b; Nanda & Grinstein, 1991); the flow of electrons along the transport chain and out of the cell comprises a measurable electrical current (Schrenzel et al. 1998; Bánfi et al. 1999; DeCoursey et al. 2000, 2001). During the respiratory burst, electrogenic H+ efflux, proposed to be mediated by proton channels, compensates the charge translocation by the oxidase complex (Henderson et al. 1987, 1988a,b; Nanda & Grinstein, 1991). Although AA-sensitive voltage-gated proton channels were observed in human neutrophils (DeCoursey & Cherny, 1993) and eosinophils (Gordienko et al. 1996; Schrenzel et al. 1996), their mediation of the H+ efflux that occurs during the AA-stimulated respiratory burst has not been demonstrated directly. AA enhances voltage-gated proton currents in whole-cell studies: the maximum H+ conductance (gH,max) is increased, the H+ chord conductance-voltage (gH-V) relationship is shifted ≈20 mV toward more negative potentials, and both activation (channel opening) and deactivation (channel closing) are roughly twice as fast (DeCoursey & Cherny, 1993; Kapus et al. 1994; Schrenzel et al. 1996; Gordienko et al. 1996; Suszták et al. 1997). Here we explore the effects of AA on voltage-gated proton currents in human eosinophils studied in the permeabilized-patch configuration, which allows the simultaneous monitoring of the activity of NADPH oxidase as electron current (DeCoursey et al. 2000, 2001). We demonstrate that AA activates NADPH oxidase in human eosinophils, measured both as O2− release and as electron current. We confirm that AA enhances voltage-gated proton currents. Although there is an intimate relationship between NADPH oxidase and voltage-gated proton channels, the enhancement of H+ current by AA was largely dissociable from the activation of NADPH oxidase.
A preliminary account of this work has been published (Cherny et al. 2001).
METHODS
Venous blood was drawn from healthy adult volunteers under informed consent according to procedures approved by our Institutional Review Board and in accordance with Federal regulations. Unless stated otherwise, the Methods are identical to those described in the accompanying paper (DeCoursey et al. 2001). Studies using AA are complicated by its tendencies to oxidize, to adhere to surfaces, to form micelles, and to damage cell membranes (Meves, 1994). The sodium salt of AA was purchased from Sigma Chemical Co. (St Louis, MO, USA). Immediately upon opening the container, AA was dissolved at 5 mm in 50 % ethanol, aliquotted into small glass vials, and gassed with nitrogen. Vials were stored at -20 °C for no more than 2-3 weeks before use. Once opened, a vial was used only for a few hours.
Eosinophils were positioned on glass coverslip chips precoated with bovine serum albumin, to reduce adherence-induced activation (DeCoursey et al. 2001). In light of the high affinity of AA for albumin (Ashbrook et al. 1975; Berde et al. 1979; Badwey et al. 1984), the local concentration of AA near the cell may have been reduced by binding to albumin. In some cells simply exchanging the bath with the same AA-containing solution produced additional effects. We attribute this result to the tendency of AA to adhere to surfaces, so that a bath exchange at the same nominal concentration transiently increased the local AA concentration.
Production of O2− was measured as described in the companion paper (DeCoursey et al. 2001) with three modifications. (1) Albumin was omitted from the incubation buffer and the 96-well plates were not pretreated with albumin. (2) Eosinophil concentration was increased to 106 cells ml−1. (3) The cells were pre-warmed in Hepes (10 mm)-buffered HBSS, pH 7.4, containing 50 μm cytochrome c for 3 min at 37 °C in a flat-bottom 96-well tissue culture plate (Costar, Acton, MA, USA) before addition of AA. The indicated concentrations of AA were added, and the contents were incubated for 14 min at 37 °C. Absorbance at 550 nm was recorded at 1 min intervals, and O2− production was calculated as described previously (Horie & Kita, 1994).
RESULTS
Superoxide anion release induced by AA in eosinophils
Although widely studied in neutrophils, AA has been evaluated only rarely as a stimulus for O2− production by eosinophils (Lindsay et al. 1998). We therefore examined AA-stimulated O2− release by eosinophils using some of the same eosinophil preparations used for patch-clamp studies. The mean dose-response relationship is plotted in Fig. 1 (○). We observed donor-to-donor variability, but 5 μm AA consistently stimulated O2− release as measured by cytochrome c reduction. The concentration-response relationship is steep, with no further increase in O2− generation at AA concentrations higher than 10 μm AA.
Figure 1. Comparison of the dose-response relationships for superoxide anion release and electron currents stimulated by AA in human eosinophils.

The background level of cytochrome c reduction has been subtracted to give the net O2− release which is plotted (○, left axis) as mean ±s.e.m. for n = 3 experiments. Measurements were made every 1 min, and the maximum increase in any 1 min time period was determined for each AA concentration. For low AA concentrations that induced a small response compared with background, the average over 2 min was determined. The peak Ie measured at -60 mV in single cells in the permeabilized patch configuration (▪, n = 2-12, right axis) is plotted for comparison. The values used for Ie are the maximum Ie measured in a given cell at each concentration; thus if a given concentration of AA was applied more than once, the largest response was selected. The ordinates have been scaled so that the O2− release rate corresponds with the Ie that would be measured in an average cell, assuming that each translocated electron results in the generation of one O2− molecule.
electron current (ie) elicited by AA
Because NADPH oxidase is electrogenic, its activity can be detected as a sustained inward current that presumably would be suppressed by extreme depolarization. Figure 1 shows the average peak Ie (▪) elicited by various AA concentrations. The concentration-response relationship is steep, with little Ie activated by 1 μm AA and a strong response at 3 μm AA. Determining the concentration dependence was complicated by the tendency of the cells to become non-specifically leaky within a few minutes of addition of high concentrations of AA (> 5 μm). The average amplitude of Ie elicited by 3-10 μm AA was -7.4 ± 3.8 pA (mean ±s.d., n = 22), slightly larger than the -6.0 pA elicited by PMA under identical conditions (DeCoursey et al. 2001). The concentration of AA required to stimulate Ie is similar to that required to stimulate O2− release.
Figure 2 illustrates Ie responses of three human eosinophils in continuous chart records of the holding current at -60 mV. The frequent interruptions are the result of depolarizing voltage pulses, which were applied to elicit H+ currents that were recorded separately. In the cell illustrated in Fig. 2A, soon after achieving a seal and establishing permeabilized patch configuration, the inward current increased spontaneously, reaching an amplitude of approximately -10 pA. The tail current decay during this time became slower, typical of the responses seen with PMA (DeCoursey et al. 2000, 2001), and suggesting that the cell was becoming activated. Spontaneous activation of putative Ie was reported previously in human eosinophils dialysed with a pipette solution containing NADPH (Schrenzel et al. 1998). At the time indicated by the first arrow, 6 μm DPI was added. The holding current decreased to approximately -2 pA and tail current decay became faster. DPI is an inhibitor of NADPH oxidase (Robertson et al. 1990); thus the DPI-sensitive fraction of inward current at the holding potential (Vh) is attributable to electron currents flowing through NADPH oxidase. Typical for non-activated eosinophils, the background holding current at -60 mV was approximately -2 pA, was not affected by DPI, and is presumably attributable to non-specific leakage. When 60 nm PMA was added several minutes later, it elicited an Ie that was inhibited by subsequent addition of DPI. As in this cell, the Ie elicited by AA or PMA was generally several times larger than the non-specific leak current at Vh. After washing DPI out of the bath, 5 μm AA was added, which elicited Ie that was inhibited by DPI. DPI was added together with AA as a control for possible non-specific leakage current that might be induced by AA. In the experiment shown in Fig. 2A, Ie attributable to NADPH oxidase was activated 3 times in the same eosinophil, first spontaneously, then by PMA, and finally by AA.
Figure 2. Examples of Ie in three different eosinophils.
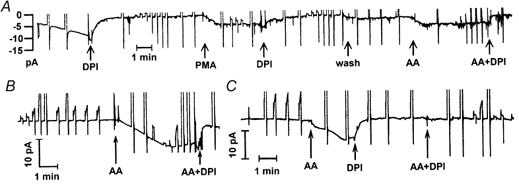
The holding current at -60 mV was recorded continuously on a chart recorder during each experiment. The frequent interruptions are the result of voltage pulses, which produced currents that were off-scale in most cases. All chart records were low-pass filtered at 4 Hz. A, this cell appeared to become activated spontaneously. At the times indicated by arrows, DPI (6 μm), PMA (60 nm), or AA (5 μm) was introduced. Additions were made by completely changing the bath solution. The bath changes took 10-20 s and the arrows indicate the middle of this time. B, activation of Ie by AA in another eosinophil. C, activation of Ie by AA. This is an example of a cell in which the Ie time course was biphasic. This pattern was seen with some cells stimulated with AA and others stimulated with PMA. Subsequent addition of AA together with DPI did not elicit detectable Ie.
The inhibition of Ie by DPI was slower when PMA was the agonist than when AA was the agonist. The average time required for DPI to reduce Ie to half its peak value was 1.8 ± 1.2 min (mean ±s.d.) in nine cells stimulated with PMA, but only 0.34 ± 0.51 min in eight cells stimulated by AA. The difference in time course is significant at P < 0.01 by Student's two-tailed t test.
The time course of the activation of Ie by AA in another cell is illustrated in Fig. 2B. The Ie began to increase within seconds after addition of AA to the bath, increased for 2-3 min, then reached a maximum and began to decline slowly. Spontaneous decline in Ie elicited by AA was often observed. DPI added together with AA rapidly eliminated most of the inward current. That the inward current did not return completely to the previous baseline might reflect a small component of AA-induced leak, or it may reflect that NADPH oxidase continues to function at a greatly reduced level in the presence of AA and DPI together.
Figure 2C illustrates a biphasic response to AA in another eosinophil. This type of response was seen occasionally in response to either AA or PMA. In this experiment, the AA-induced inward current was identified as NADPH-generated Ie by addition of DPI alone (i.e. without AA). Subsequent addition of AA together with DPI elicited little or no inward current. This result rules out the possibility that the AA-induced current was non-specific membrane leak. Later in this experiment AA was applied and DPI-inhibitable Ie was again stimulated (not shown), confirming that the failure of the cell to respond to AA when added together with DPI was due to DPI inhibition rather than to ‘desensitization’ or some other process.
effects of aa on h+ currents
Figure 3A illustrates the electrophysiological effects of AA on human eosinophils. From Vh = -60 mV, 8 s test pulses to +40 mV were applied at 30 s intervals. The H+ current elicited by the first pulse after 5 μm AA addition was already increased. IH continued to increase over the 4 min illustrated. The inward current at -60 mV also increased progressively over this time period. The activation time constant, τact, decreased from 4.4 to 1.5 s. In contrast, although the tail current amplitude increased substantially, τtail did not change (215 ms before and after AA). The main difference in the response to AA compared with PMA (DeCoursey et al. 2001) is that τtail was slowed markedly by PMA, but was minimally affected by AA.
Figure 3. Effects of AA on H+ currents.

A, from Vh = -60 mV, test pulses 8 s in duration to +40 mV were applied at 30 s intervals. The smallest current is the control. The first pulse after 5 μm AA addition elicited a larger H+ current, and IH continued to increase over the 4 min illustrated. The inward current at -60 mV also increased progressively. The larger tail currents were truncated. B, currents during pulses to +40 mV from Vh = -60 mV in an eosinophil in the presence of 0, 1 and 3 μm AA are superimposed. Note that in the presence of 3 μm AA there is distinct inward Ie not only at -60 mV, but also at +40 mV at the start of the test pulse. The peak tail current in 3 μm AA was off-scale. C, measurements were made in the same cell shown in B before exposure (•), and in the presence of 1 μm AA (□), and 3 μm AA (▵). Because the currents did not reach steady state during the pulses, we fitted them with a single exponential after a delay to obtain the steady-state amplitude. The chord conductance gH plotted here was calculated from the extrapolated currents using Vrev measured in each condition. The gH values plotted in A were normalized (arbitrarily) to the largest value in each condition and are replotted in D to illustrate the voltage dependence.
Figure 3B illustrates the concentration dependence of AA effects. A test pulse to +40 mV elicited a small H+ current (‘0′). After addition of 1 μm AA, IH increased and τact became faster but no Ie was detected. Similar effects were seen in four other cells treated with low AA concentrations. Addition of 3 μm AA not only produced a large additional increase in IH, but also activated Ie at -60 mV. Thus H+ current was enhanced by AA at lower concentrations than those required to elicit distinct Ie. This experiment further illustrates the complex effects of AA on tail current kinetics. Initially τtail was 230 ms and after addition of 1 μm AA decreased to 160-200 ms. In six of eight cells stimulated with AA at concentrations too low to activate Ie, τtail became faster. After addition of 3 μm AA τtail first decreased to 150 ms, but while Ie was activated maximally τtail slowed to 355 ms. It appears that AA has a tendency to speed up tail current decay, but also that activation of Ie by AA is associated with a slowing of τtail, as observed with PMA stimulation.
Plotted in Fig. 3C are gH-V relationships measured at 0 μm AA (•), 1 μm AA (□), and 3 μm AA (▵) in the same eosinophil shown in Fig. 3B. The gH calculated from the amplitude of a single exponential fitted to the currents was more than doubled at 1 μm AA at all voltages. When these gH data are normalized (Fig. 3D) it becomes clear that there was no change in the voltage dependence of gating in the presence of 1 μm AA. However, in the presence of 3 μm AA (▵) there was a 20-30 mV shift. Intriguingly, Ie was observed at 3 μm AA but was not detectable at 1 μm AA. In other cells, voltage shifts were quantified by estimating the threshold voltage for activating the gH. By this criterion, in five cells treated with low AA concentrations that did not activate Ie no voltage shift was detected, whereas in seven eosinophils in which 3-10 μm AA activated Ie voltage shifts in the range 20-50 mV were observed. In summary, at low concentrations AA increased gH and sped up τact and at higher concentrations AA had these effects but also activated Ie and shifted the gH-V relationship toward more negative voltages.
As observed with PMA stimulation (DeCoursey et al. 2001), Vrev measured after AA stimulation tended to be more negative than in the same cell before AA. In 13 eosinophils exposed to 3-5 μm AA, Vrev shifted -5.3 ± 1.2 mV (mean ±s.e.m.), corresponding to a drop in the apparent pHi of ≈0.1 unit. Thus, at most only a small part of the hyperpolarizing shift of the gH-V relationship can be ascribed to uncompensated cytoplasmic acidification.
The constellation of effects of AA is illustrated in more depth in Fig. 4. Test pulses to +40 mV were applied periodically. Figure 4A shows test currents before and after the first addition of 5 μm AA. The H+ current was greatly enhanced, with both increased amplitude (IH) and faster activation (smaller τact). These parameters are plotted in Fig. 4D as a function of time during the experiment. Soon after AA was added, the current at -60 mV (+ in Fig. 4D) increased from -1.6 to -9.4 pA, a net change of -7.8 pA that we attribute to Ie generated by NADPH oxidase. Addition of DPI and removal of AA (Fig. 4B) not only eliminated Ie, but also resulted in a slowing of H+ current activation (Fig. 4Bb and c). This result is in apparent contrast to the behaviour of PMA-stimulated neutrophils (DeCoursey et al. 2000) in which τact became faster after PMA, but this response was not reversed by DPI. However, we attribute the slowing of τact in Fig. 4Bc to removal of AA rather than to the effects of DPI, because addition of AA together with DPI accelerated τact (Fig. 4C and D, ▴). It appears that the faster activation is a relatively direct effect of AA on voltage-gated proton channels that occurs independently of NADPH oxidase activity.
Figure 4. Electrophysiological effects of AA and DPI on an eosinophil.
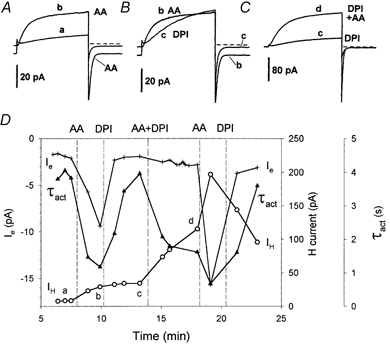
Current during 8 s pulses to +40 mV from Vh = -60 mV are superimposed. The raw current records plotted in A-C are labelled with lower case letters that also appear in D showing the time point of each illustrated record. A, currents before and ≈2 min after addition of 5 μm AA. The H+ current activated more rapidly - note the smaller activation time constant τact in D (▴) - and the H+ current amplitude IH (○) was several times larger, but τtail was not markedly changed (not plotted). The inward current at Vh = -60 mV (+ in D) increased by -7.8 pA after AA addition, attributed to Ie generated by NADPH oxidase. B, the same current record in the presence of AA (b) is superimposed on the current after replacement of AA with 6 μm DPI in the bath (c). Activation is slower and the putative Ie is eliminated. C, re-addition of 5 μm AA together with 6 μm DPI greatly enhanced IH (note different current calibration bar) but did not activate distinct Ie. D, the time course of the experiment is illustrated. After addition of AA and DPI together, 5 μm AA alone was added, and again Ie was activated, which was eliminated by further addition of 6 μm DPI in the absence of AA. A continuous record of Ie in this cell is also shown in Fig. 2C. Note that the total current at Vh is plotted (+) and labelled Ie, but we interpret only the AA-activated and DPI-inhibited fraction as genuine (net) Ie.
Addition of AA increased IH in this experiment (Fig. 4D, ○), and this increase was not reversed within a few minutes after the first addition of DPI and washout of AA. AA added together with DPI increased IH further even though Ie was not activated. Eventually, after AA was washed out during the final DPI addition, IH began to decrease. In other experiments, washout of AA usually resulted in a gradual decrease in H+ current. Our impression is that, similar to its acceleration of τact, AA increased IH amplitude independently of NADPH oxidase activity, and that this increase was at least partially reversible upon washout. In most experiments, IH and τact changed in parallel. When AA increased IH it usually also decreased τact, and when IH decreased after washout of AA, τact usually slowed with a similar time course.
Response of eosinophils to repeated stimulation
The ability of eosinophils to respond to repeated stimuli is illustrated in Fig. 2A and Fig. 5. As can be seen in Fig. 5A, distinct Ie (+) was activated by each of three consecutive additions of 3 μm AA. The amplitudes and time courses of the three responses were not identical, but in each there was a tendency toward spontaneous turn-off. The continuous records of holding current (Fig. 2) show that DPI turns off AA-induced Ie rapidly. There was a clear response to PMA when applied after washout of the third application of AA.
Figure 5. Time course of the response to multiple application of agonists.

Temporal changes in H+ current parameters and Ie during repeated application of 3 μm AA, 3 μm DPI, 60 nm PMA, and after washout, as indicated. Parameters from the same experiment are plotted separately in A and B for clarity. A, inward Ie (+) was activated during each of three applications of 3 μm AA. There were only minor changes in τtail (▪) after AA treatment, but PMA caused distinct slowing, which was reversed by DPI. B, the amplitude of the H+ current at +40 mV is plotted (○) along with τact (▴) during the same experiment as in A. Note that although DPI appears to reverse the AA-induced changes, similar reversal is seen with simple washout of AA. In contrast, the changes in these parameters induced by PMA were not reversed by DPI.
In this experiment, τtail became slower during the first application of AA. This slowing reversed in parallel with the shut-off of Ie, consistent with the parallel responses of these parameters during PMA application (DeCoursey et al. 2001). During subsequent applications of AA no further changes in τtail were observed. This cell remained responsive, however, because addition of PMA did result in a distinct slowing of τtail that occurred simultaneously with the induction of DPI-sensitive Ie. The slowing of τtail by PMA was reversed by DPI. The effects of DPI in this experiment taken together are consistent with the idea that DPI does not directly change τtail, but only reverses slowing that occurs in conjunction with NADPH oxidase activation. In most experiments in which Ie was elicited upon AA addition, slowing of τtail was observed. Subsequently, however, τtail often became progressively faster. Our impression is that AA has two competing effects on H+ current deactivation.
Like PMA, AA increased IH amplitude and made activation faster (smaller τact). Unlike PMA, these changes usually reversed at least partially when AA was washed out of the bath (Fig. 5B). Addition of DPI also appeared to reverse these changes, but it is likely that the simultaneous washout of AA was responsible.
The time course and amplitude of the effects of AA are summarized and compared with those of PMA in Table 1. Except for the lack of net effect on τtail, the magnitudes of the effects of AA and PMA were identical. The time course of the changes was consistently faster for AA than for PMA. The three parameters in Table 1 that were changed by AA all changed at least twice as fast as they did after PMA stimulation. As was the case with PMA, the most rapid response was the faster activation rate of H+ currents. Because τact was reduced before Ie had turned on significantly, it cannot be a consequence of NADPH oxidase activity.
Table 1.
Comparison of the magnitude and time course of effects of AA and PMA in human eosinophils
| τact | τtail | IH | Ie (pA) | |
|---|---|---|---|---|
| AA/control | 0.25 ± 0.12 (7) | 1.0 ± 0.4 (6)† | 4.6 ± 1.5 (7) | −7.4 ± 3.8 (22) |
| PMA/control | 0.24 ± 0.09 (12) | 5.4 ± 3.4 (14) | 4.7 ± 2.8 (12) | −6.0 ± 2.8 (23) |
| t1/2 AA (min) | 0.1 ± 0.2 (7)† | — | 0.7 ± 1.1 (7) | 1.2 ± 1.1 (17)* |
| t1/2 PMA (min) | 1.0 ± 0.6 (12) | 1.6 ± 0.7 (14) | 2.0 ± 1.5 (12) | 2.5 ± 2.1 (17) |
The average ratio of each parameter, except Ie, measured after treatment with 3–10 μm AA to its value before treatment. Mean ± S.D. is given for (n) observations. Usually IH and τact were measured during pulses to +40 or +60 mV, and τtail and Ie at −60 mV. Ie is taken as the average peak increase in inward current after PMA. The t1/2 value is the time required for the change to reach half-completion. The difference between AA and PMA is significant at
P < 0.05
P < 0.01 by Student's t test. The PMA values are for human eosinophils, taken from DeCoursey et al. (2001).
DISCUSSION
aa elicits o2− release and Ie in human eosinophils
Here we demonstrate that AA is a potent and effective activator of NADPH oxidase in human eosinophils. O2− release was observed at 5 μm AA and at 10 μm AA, the response was near maximal. Although the maximum rate of O2− generated by human eosinophils in response to AA (3.3 nmol (106 cells)−1 min−1) was lower than that elicited by PMA (DeCoursey et al. 2001), the maximal rate of AA-stimulated O2− release averaged 70 % of that elicited by an optimal concentration of PMA under identical conditions (data not shown). To our knowledge, the only other study of AA activation of NADPH oxidase in eosinophils is of guinea-pig eosinophils (Lindsay et al. 1998), in which the maximum AA-stimulated H2O2 generation rate was 6.15 nmol (106 cells)−1 min−1.
In eosinophils studied with permeabilized-patch recording, AA at 3 μm or higher elicited distinct inward currents (Ie) that were largely inhibitable by DPI and were partially reversed by washout of AA. The dose-response relationship for Ie, like that for O2− release, was steep, with little or no activation by 1 μm AA and a maximal response at 5-10 μm AA. A similarly steep concentration-response relation has also been reported in human neutrophils (Henderson et al. 1993) and in guinea-pig eosinophils (Lindsay et al. 1998). Importantly, the concentration-response relationship for O2− release measured by cytochrome c reduction closely paralleled that observed for putative electron current (Ie). This similarity reinforces the identification of the observed inward currents as Ie that is generated by functioning NADPH oxidase. The correspondence between Ie and the electron flux rate calculated from O2− generation studies indicates that Ie as measured electrically under the conditions of this study is a good model system to study AA effects at the level of individual cells.
The Ie elicited by AA averaged -7.4 pA, and was similar in amplitude to that elicited by PMA, which averaged -6.0 pA (Table 1). This result is consistent with the finding that AA and PMA stimulate comparable maximal levels of O2− release in human neutrophils (Curnutte et al. 1984; Steinbeck et al. 1991). The maximum AA-stimulated H2O2 generation rate in guinea-pig eosinophils studied at 37 °C corresponds to -19.7 pA of Ie (Lindsay et al. 1998).
A surprising result was that DPI shut down Ie more rapidly when AA was the stimulus than when PMA was the stimulus or in spontaneously activated eosinophils. The most obvious interpretation of this result is that the NADPH oxidase complex activated by AA is functionally different from that activated by PMA or spontaneously. There is some evidence that AA and PMA might lead to two distinct forms of activated NADPH oxidase. At high concentrations (50-100 μm) AA alone (i.e. without phosphorylation of p47phox) is sufficient to induce the required interaction between cytosolic p47phox and p22phox of the membrane-bound cytochrome b, but at low concentrations (1-5 μm) AA acts synergistically with phosphorylation of p47phox to activate the NADPH oxidase (Shiose & Sumimoto, 2000). In this view, the more rapid inhibition of the Ie by DPI may indicate that the AA-activated NADPH oxidase complex is less stable than that assembled by the action of PMA.
AA causes membrane damage at high concentrations
High concentrations of AA induce non-specific membrane damage in many cell types (Meves, 1994). High concentrations of AA (> 5 μm), or long and repeated exposure to lower concentrations of AA, often resulted in non-specific increases in membrane permeability (leak) in individual eosinophils under voltage clamp. Similar sensitivity of the membrane to AA-induced damage was reported previously in murine macrophages studied in the whole-cell configuration (Kapus et al. 1994). AA partitions strongly into cell membranes, with a partition coefficient of ≈2 × 104; at 5 μm applied AA, 3.6 % of the membrane is composed of AA molecules intercalated between membrane phospholipids (Hutter, 1997). Micelle formation at high concentrations of AA has been suggested as a mechanism for non-specific damage (Meves, 1994).
As a control for non-specific membrane damage, we generally added AA together with DPI (Fig. 2 and Fig. 4). That little inward current appeared under these conditions eliminates the possibility that the inward current during the addition of AA alone was due to non-specific leakage. Further evidence that the inward current reflects NADPH oxidase activity (rather than leak) is provided by the finding that the AA-activated current is also inward at +40 mV, and is visible as net inward current at the start of the test pulses (Figs 3A, 3B, 4A and 4B). The zero-current potential of NADPH oxidase (viewed as an electron pump) is likely to be large and positive, and thus the Ie that it generates should be inward over a wide voltage range. In contrast, a non-specific leak current would be expected to reverse near 0 mV. In cells that became leaky, either spontaneously or after addition of high concentrations of AA, the inward current at -60 mV and outward current at +40 mV both increased. In summary, genuine Ie could be distinguished from non-specific membrane damage: Ie was reversible upon washout of AA or addition of DPI, and Ie was inward at the test potentials of +40 or + 60 mV, whereas leak was outward.
AA promotes opening of voltage-gated proton channels
AA profoundly enhanced voltage-gated proton currents in eosinophils studied in permeabilized-patch conditions: gH,max increased several-fold, activation was faster, and the gH-V relationship was shifted toward more negative voltages. Because these effects were concentration dependent and, as can be seen in Fig. 4D and Fig. 5, repeated applications of the same concentration of AA produced cumulative effects on IH, quantifying them is somewhat arbitrary. Nevertheless, the magnitude of the changes produced by 3-10 μm AA was identical to that seen after PMA stimulation (Table 1). Like the effects of PMA, all of the effects of AA on H+ channels lead to enhanced H+ current. Although the probability and rate of H+ channel opening at any voltage is increased by AA or PMA, the gH remains voltage gated. Thus, H+ channels in intact cells will open only with an appropriate combination of pH changes and depolarization.
The response of phagocytes to AA differs in four respects from the response to PMA. (1) PMA was completely ineffective in eosinophils (data not shown) or neutrophils (DeCoursey et al. 2000) studied in the whole-cell configuration, stimulating neither NADPH oxidase (Ie) nor voltage-gated proton channels. In contrast, AA enhances H+ currents even in whole-cell configuration (see next paragraph). (2) The increase in IH, speeding of τact, and activation of Ie all occurred more rapidly when AA was the stimulus. (3) Ie was inhibited by DPI much more rapidly when stimulated by AA than by PMA. (4) τtail was slowed profoundly by PMA but not by AA. AA appears to speed τtail in general, but in some experiments a slowing (< 2-fold) was observed (e.g. Fig. 5A), usually in conjunction with the appearance of Ie. These results can be explained if AA has two distinct effects on H+ channels, both slowing and speeding τtail. In spontaneously activated or PMA-treated eosinophils, τtail is profoundly slowed. If both effects occur in permeabilized patch conditions, they will counteract each other, resulting in little or no net change.
Whole-cell studies of AA effects on voltage-gated proton channels in human eosinophils (Gordienko et al. 1996; Schrenzel et al. 1996), human neutrophils (DeCoursey & Cherny, 1993), and murine macrophages (Kapus et al. 1994) described a hyperpolarizing shift of 15-20 mV in the gH-V relationship, an increase in the limiting gH (gH,max), and ≈2-fold faster activation and deactivation kinetics. It appears that AA increased gH,max more in permeabilized patch conditions. The hyperpolarizing shift in the gH-V relationship also appears to be greater than in whole-cell studies. Activation of H+ currents negative to Vrev after AA treatment was not reported in the whole-cell studies just cited. In the present study, AA shifted the voltage-activation curve sufficiently to activate the gH negative to Vrev in some cells and, consequently, produced inward currents over a small voltage range. Most of the effects of AA on H+ currents can be elicited in whole-cell configuration. Thus, although the PMA response of eosinophils requires diffusible second messengers, much of the AA enhancement of H+ currents does not. Hence, at least part of the enhancement of H+ currents by AA reflects a relatively direct action on the channel. The more rapid response to AA than to PMA is consistent with this conclusion.
The properties of H+ currents during NADPH oxidase activity differ from those in resting cells. Bánfi et al. (1999) suggested that two types of voltage-gated proton channels exist, one that is active in resting eosinophils, and another that becomes active during the respiratory burst. We prefer the simpler interpretation of a single population of H+ channels that is modified extensively by NADPH oxidase activators like PMA (DeCoursey et al. 2000, 2001) and AA. With the exception of τtail, all the effects of AA on H+ currents were similar to those elicited by PMA, and are also similar to the properties of voltage-gated proton channels in eosinophils dialysed with NADPH in the whole-cell configuration (Bánfi et al. 1999).
The enhancement of voltage-gated proton currents by AA was dissociable from the activation of NADPH-oxidase-mediated Ie. Low concentrations of AA enhanced IH without activating detectable Ie (Fig. 3B). Also, addition of AA in the presence of DPI did not activate detectable Ie but enhanced IH substantially (Fig. 4). These results are consistent with the conclusion of Nanda et al. (1994) that the activation of H+ efflux by PMA does not require functional NADPH oxidase. Similarly, AA activates channel-mediated H+ efflux in PLB cells deficient in phospholipase A2, even in the presence of DPI (Lowenthal & Levy, 1999).
Response to repetitive stimulation
After the Ie elicited by AA was inhibited by DPI or by simple washout of AA, Ie could again be activated by subsequent application of AA. DPI is believed to inhibit NADPH oxidase by an irreversible chemical reaction (O'Donnell et al. 1994). However, DPI and the related compound iodinium biphenyl appear to interact with NADPH oxidase only under reducing conditions (i.e. when the oxidase is functioning) - no inhibition occurs if these drugs are applied when the oxidase in not active (Doussiere & Vignais, 1991; Doussiere et al. 1999). This mechanism of action of DPI can be reconciled with our observation of repetitive responses to AA if only a fraction of the total oxidase components in the cell are assembled during each AA application. Because Ie measured electrically reflects only the active NADPH oxidase complexes present in the plasma membrane, the observation of repetitive responses further suggests that not all of the available oxidase components are translocated to the plasma membrane (and inhibited) during the first exposure to AA. This conclusion is not surprising considering that sustained O2− generation requires continuous translocation of the cytosolic NADPH oxidase components to the membrane (Quinn et al. 1993), and that in intact cells O2− release persists for 20 min at 37 °C (DeCoursey et al. 2001).
In contrast to the repeated responses of eosinophils to AA described here, re-application of PMA generally elicited a weaker response or no response (DeCoursey et al. 2001). However, some eosinophils previously exposed to AA responded to PMA. Also, eosinophils that had recovered from spontaneous activation usually were capable of responding to either PMA or AA. Thus, it appears that PMA produces some changes that are not completely reversible either by DPI or by time, at least within the duration of these experiments. In contrast, AA is capable of activating Ie and increasing IH repeatedly. Even in cells in which previous exposure to AA had greatly enhanced IH, additional AA produced even greater responses. This pattern suggests that at least one step in the PMA signalling pathway is saturable. For example, once PKC has phosphorylated a target, additional PMA cannot produce a further response. In contrast, there appears to be no saturable intermediate or irreversible step in the AA response pathway.
Is AA the immediate activator of NADPH oxidase?
It has been proposed that in neutrophils AA is the immediate activator of NADPH oxidase (Henderson et al. 1989, 1993), and that AA is required for activation of H+ efflux during the respiratory burst (Henderson & Chappell, 1992; Suszták et al. 1997). Several of the present results impact on these hypotheses. We demonstrate that AA is a potent activator both of voltage-gated proton channels and also of the NADPH oxidase in human eosinophils. It is clear that AA alters the functional properties of the H+ channel, and that these effects are not simply the consequence of cytoplasmic acidification and/or membrane depolarization that tend to occur when NADPH oxidase is functioning. Activation occurs as a result of modification of the properties of the H+ channels themselves. Teleologically, this result is in keeping with the idea that voltage-gated proton channels provide a mechanism of H+ extrusion during the respiratory burst. The dual action of AA raises a question. Is it simply a coincidence that AA activates both NADPH oxidase and H+ channels? Two explanations are possible: NADPH oxidase and H+ channels might be activated by a common signalling pathway or they are activated by separate pathways that act concurrently and independently or perhaps in a coordinated fashion (Henderson & Chappell, 1992).
Overall, the most remarkable result of this study may be that AA, PMA, and spontaneous activation of eosinophils all produce the same constellation of effects. Both qualitatively and quantitatively, the speeding of τact, the increase in IH, the hyperpolarizing voltage shift in the gH-V relationship, and even the amplitude of Ie elicited by PMA or AA were quite similar (Table 1). This similarity in the responses to AA and PMA lends support to the common pathway idea.
Several distinct variants of common pathway mechanisms can be envisioned. The most extreme is that NADPH oxidase and voltage-gated proton channels are one and the same molecule. Although this idea has been discussed for some time (Henderson & Chappell, 1992, 1996; Nanda et al. 1994), our data do not resolve the question so we will not discuss it further. Another form of the common pathway idea is that AA is the immediate activator of NADPH oxidase (Henderson et al. 1993), whether it is stimulated by PMA or directly by AA. In this view, the effects of PMA on voltage-gated proton channels are mediated by AA that is released as a consequence of the action of PMA. That the action of AA is faster than PMA is consistent with this notion, as is the observation that much of the stimulation of H+ channels by AA occurs in whole-cell configuration, and thus does not require diffusible second messenger pathways. The failure of AA to change τtail consistently is more difficult to explain. AA might have two opposing effects on τtail, namely a slowing as seen with PMA, and an acceleration that is also seen in whole-cell studies. Even so, it is not clear why the acceleration would be seen when AA is applied as the sole stimulus, but not if AA is the final step in the PMA-induced cascade. Other evidence seems to argue against an absolute requirement for AA to activate NADPH oxidase in eosinophils (White et al. 1993). Phorbol 12,13-dibutyrate inhibits H2O2 generation by ≈80 % at concentrations that reduce AA release by leukotriene B4 by only ≈40 % (Lindsay et al. 1998). Evidently, PMA initiates responses via multiple pathways that overlap partially with the effects of AA.
The failure of AA to stimulate NADPH oxidase in whole-cell studies might indicate that diffusible second messengers are involved in the response. However, activation of NADPH oxidase requires assembly of at least six components, four of which in unstimulated cells are located in the cytoplasm. Diffusion of any required cytosolic component into the pipette would preclude NADPH oxidase assembly in whole-cell configuration. Overall, our data are most compatible with the idea that AA can directly enhance H+ channel function, but that the complete physiological response involves additional modulation by a pathway that requires a diffusible intermediary, and may be linked to NADPH oxidase function.
Acknowledgments
This work was supported by the National Heart, Lung and Blood Institute of the National Institutes of Health (grants HL52671 and HL61437 to T.D.), by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (grant AI48160 to L.T.), and Arthritis Research Council (grant H0604 to L.H.). The authors appreciate the able technical assistance of Tatiana Iastrebova.
References
- Abramson SB, Leszczynska-Piziak J, Weissmann G. Arachidonic acid as a second messenger: interactions with a GTP-binding protein of human neutrophils. Journal of Immunology. 1991;147:231–236. [PubMed] [Google Scholar]
- Ashbrook JD, Spector AA, Santos EC, Fletcher JE. Long chain fatty acid binding to human plasma albumin. Journal of Biological Chemistry. 1975;250:2333–2338. [PubMed] [Google Scholar]
- Badwey JA, Curnutte JT, Karnovsky ML. cis-Polyunsaturated fatty acids induce high levels of superoxide production by human neutrophils. Journal of Biological Chemistry. 1981;256:12640–12643. [PubMed] [Google Scholar]
- Badwey JA, Curnutte JT, Robinson JM, Berde CB, Karnovsky MJ, Karnovsky ML. Effects of free fatty acids on release of superoxide and on change of shape by human neutrophils. Journal of Biological Chemistry. 1984;259:7870–7877. [PubMed] [Google Scholar]
- Bánfi B, Schrenzel J, Nüsse O, Lew DP, Ligeti E, Krause K-H, Demaurex N. A novel H+ conductance in eosinophils: unique characteristics and absence in chronic granulomatous disease. Journal of Experimental Medicine. 1999;190:183–194. doi: 10.1084/jem.190.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berde CB, Hudson BS, Simoni RD, Sklar LA. Human serum albumin: spectroscopic studies of binding and proximity relationships for fatty acids and bilirubin. Journal of Biological Chemistry. 1979;254:391–400. [PubMed] [Google Scholar]
- Bromberg Y, Pick E. Unsaturated fatty acids as second messengers of superoxide generation by macrophages. Cellular Immunology. 1983;79:240–252. doi: 10.1016/0008-8749(83)90067-9. [DOI] [PubMed] [Google Scholar]
- Cherny VV, DeCoursey AG, Henderson LM, Thomas LL, DeCoursey TE. Proton and electron currents during the respiratory burst in human eosinophils. Biophysical Journal. 2001;80:506a–507a. [Google Scholar]
- Cross AR, Erickson RW, Curnutte JT. Simultaneous presence of p47phox and flavocytochrome b-245 are required for the activation of NADPH oxidase by anionic amphiphiles: evidence for an intermediate state of oxidase activation. Journal of Biological Chemistry. 1999;274:15519–15525. doi: 10.1074/jbc.274.22.15519. [DOI] [PubMed] [Google Scholar]
- Curnutte JT, Badwey JA, Robinson JM, Karnovsky MJ, Karnovsky ML. Studies on the mechanism of superoxide release from human neutrophils stimulated with arachidonate. Journal of Biological Chemistry. 1984;259:11851–11857. [PubMed] [Google Scholar]
- DeCoursey TE, Cherny VV. Potential, pH and arachidonate gate hydrogen ion currents in human neutrophils. Biophysical Journal. 1993;65:1590–1598. doi: 10.1016/S0006-3495(93)81198-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE, Cherny VV, DeCoursey AG, Xu W, Thomas LL. Interactions between NADPH oxidase-related proton and electron currents in human eosinophils. Journal of Physiology. 2001;535:767–781. doi: 10.1111/j.1469-7793.2001.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE, Cherny VV, Zhou W, Thomas LL. Simultaneous activation of NADPH oxidase-related proton and electron currents in human neutrophils. Proceedings of the National Academy of Sciences of the USA. 2000;97:6885–6889. doi: 10.1073/pnas.100047297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doussiere J, Gaillard J, Vignais PV. The heme component of the neutrophil NADPH oxidase complex is a target for aryliodonium compounds. Biochemistry. 1999;38:3694–3703. doi: 10.1021/bi9823481. [DOI] [PubMed] [Google Scholar]
- Doussiere J, Vignais PV. Inhibition of O2− generating oxidase of neutrophils by iodinium biphenyl in a cell free system: effect of the redox state of the oxidase complex. Biochemical and Biophysical Research Communications. 1991;175:143–151. doi: 10.1016/s0006-291x(05)81212-4. [DOI] [PubMed] [Google Scholar]
- Galbraith GMP. Chemotactic peptide-induced arachidonic acid mobilization in human polymorphonuclear leukocytes. American Journal of Pathology. 1988;133:347–354. [PMC free article] [PubMed] [Google Scholar]
- Gordienko DV, Tare M, Parveen S, Fenech CJ, Robinson C, Bolton TB. Voltage-activated proton current in eosinophils from human blood. Journal of Physiology. 1996;496:299–316. doi: 10.1113/jphysiol.1996.sp021686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson LM, Chappell JB. The NADPH-oxidase-associated H+ channel is opened by arachidonate. Biochemical Journal. 1992;283:171–175. doi: 10.1042/bj2830171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson LM, Chappell JB. NADPH oxidase of neutrophils. Biochimica et Biophysica Acta. 1996;1273:87–107. doi: 10.1016/0005-2728(95)00140-9. [DOI] [PubMed] [Google Scholar]
- Henderson LM, Chappell JB, Jones OTG. The superoxide-generating NADPH oxidase of human neutrophils is electrogenic and associated with an H+ channel. Biochemical Journal. 1987;246:325–329. doi: 10.1042/bj2460325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson LM, Chappell JB, Jones OTG. Internal pH changes associated with the activity of NADPH oxidase of human neutrophils, further evidence for the presence of an H+ conducting channel. Biochemical Journal. 1988a;251:563–567. doi: 10.1042/bj2510563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson LM, Chappell JB, Jones OTG. Superoxide generation by the electrogenic NADPH oxidase of human neutrophils is limited by the movement of a compensating charge. Biochemical Journal. 1988b;255:285–290. [PMC free article] [PubMed] [Google Scholar]
- Henderson LM, Chappell JB, Jones OTG. Superoxide generation is inhibited by phospholipase A2 inhibitors: role for phospholipase A2 in the activation of the NADPH oxidase. Biochemical Journal. 1989;264:249–255. doi: 10.1042/bj2640249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson LM, Moule SK, Chappell JB. The immediate activator of the NADPH oxidase is arachidonate not phosphorylation. European Journal of Biochemistry. 1993;211:157–162. doi: 10.1111/j.1432-1033.1993.tb19882.x. [DOI] [PubMed] [Google Scholar]
- Horie S, Kita H. CD11b/CD18 (Mac-1) is required for degranulation of human eosinophils induced by human recombinant granulocyte-macrophage colony-stimulating factor and platelet-activating factor. Journal of Immunology. 1994;152:5457–467. [PubMed] [Google Scholar]
- Hutter OF. Partition coefficient of arachidonic acid estimated from pipette aspiration experiments on sarcolemmal vesicles from rabbit skeletal muscle. Journal of Physiology. 1997;499.P:133P. [Google Scholar]
- Kapus A, Romanek R, Grinstein S. Arachidonic acid stimulates the plasma membrane H+ conductance of macrophages. Journal of Biological Chemistry. 1994;269:4736–4745. [PubMed] [Google Scholar]
- Lindsay MA, Perkins RS, Barnes PJ, Giembycz MA. Leukotriene B4 activates the NADPH oxidase in eosinophils by a pertussis toxin-sensitive mechanism that is largely independent of arachidonic acid mobilization. Journal of Immunology. 1998;160:4526–4534. [PubMed] [Google Scholar]
- Lowenthal A, Levy R. Essential requirement of cytosolic phospholipase A2 for activation of the H+ channel in phagocyte-like cells. Journal of Biological Chemistry. 1999;274:21603–21608. doi: 10.1074/jbc.274.31.21603. [DOI] [PubMed] [Google Scholar]
- Meves H. Modulation if ion channels by arachidonic acid. Progress in Neurobiology. 1994;43:175–186. doi: 10.1016/0301-0082(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Nanda A, Curnutte JT, Grinstein S. Activation of H+ conductance in neutrophils requires assembly of components of the respiratory burst oxidase but not its redox function. Journal of Clinical Investigation. 1994;93:1770–1775. doi: 10.1172/JCI117162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda A, Grinstein S. Protein kinase C activates an H+ (equivalent) conductance in the plasma membrane of human neutrophils. Proceedings of the National Academy of Sciences of the USA. 1991;88:10816–10820. doi: 10.1073/pnas.88.23.10816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell VB, Smith GCM, Jones OTG. Involvement of phenyl radicals in iodinium compound inhibition of flavoenzymes. Molecular Pharmacology. 1994;46:778–785. [PubMed] [Google Scholar]
- Park JW, Babior BM. Activation of the leukocyte NADPH oxidase subunit p47phox by protein kinase C: a phosphorylation-dependent change in the conformation of the C-terminal end of p47phox. Biochemistry. 1997;36:7474–7480. doi: 10.1021/bi9700936. [DOI] [PubMed] [Google Scholar]
- Quinn MT, Evans T, Loetterle LR, Jesaitis AJ, Bokoch GM. Translocation of Rac correlates with NADPH oxidase activation: evidence for equimolar translocation of oxidase components. Journal of Biological Chemistry. 1993;268:20983–20987. [PubMed] [Google Scholar]
- Robertson AK, Cross AR, Jones OTG, Andrew PW. The use of diphenylene iodinium, an inhibitor of NADPH oxidase, to investigate the antimicrobial action of human monocyte derived macrophages. Journal of Immunological Methods. 1990;133:175–179. doi: 10.1016/0022-1759(90)90357-2. [DOI] [PubMed] [Google Scholar]
- Rubinek T, Levy R. Arachidonic acid increases the activity of the assembled NADPH oxidase in cytoplasmic membranes and endosomes. Biochimica et Biophysica Acta. 1993;1176:51–58. doi: 10.1016/0167-4889(93)90176-p. [DOI] [PubMed] [Google Scholar]
- Schrenzel J, Lew DP, Krause K-H. Proton currents in human eosinophils. American Journal of Physiology. 1996;271:C1861–1871. doi: 10.1152/ajpcell.1996.271.6.C1861. [DOI] [PubMed] [Google Scholar]
- Schrenzel J, Serrander L, Bánfi B, Nüsse O, Fouyouzi R, Lew DP, Demaurex N, Krause K-H. Electron currents generated by the human phagocyte NADPH oxidase. Nature. 1998;392:734–737. doi: 10.1038/33725. [DOI] [PubMed] [Google Scholar]
- Shiose A, Sumimoto H. Arachidonic acid and phosphorylation synergistically induce a conformational change of p47phox to activate the phagocyte NADPH oxidase. Journal of Biological Chemistry. 2000;275:13793–13801. doi: 10.1074/jbc.275.18.13793. [DOI] [PubMed] [Google Scholar]
- Steinbeck MJ, Hegg GG, Karnovsky MJ. Arachidonate activation of the neutrophil NADPH-oxidase: synergistic effects of protein phosphatase inhibitors compared with protein kinase activators. Journal of Biological Chemistry. 1991;266:16336–16342. [PubMed] [Google Scholar]
- Suszták K, Mócsai A, Ligeti E, Kapus A. Electrogenic H+ pathway contributes to stimulus-induced changes of internal pH and membrane potential in intact neutrophils: role of cytoplasmic phospholipase A2. Biochemical Journal. 1997;325:501–510. doi: 10.1042/bj3250501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain SD, Helgerson SL, Davis AR, Nelson LK, Quinn MT. Analysis of activation-induced conformational changes in p47phox using tryptophan fluorescence spectroscopy. Journal of Biological Chemistry. 1997;272:29502–29510. doi: 10.1074/jbc.272.47.29502. [DOI] [PubMed] [Google Scholar]
- Tao W, Molski TFP, Sha'afi RI. Arachidonic acid release in rabbit neutrophils. Biochemical Journal. 1989;257:633–637. doi: 10.1042/bj2570633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlinger DJ, Tyagi SR, Inge KL, Lambeth JD. The respiratory burst oxidase of human neutrophils: guanine nucleotides and arachidonate regulate the assembly of a multicomponent complex in a semirecombinant cell-free system. Journal of Biological Chemistry. 1993;268:8624–8631. [PubMed] [Google Scholar]
- Walsh CE, Waite BM, Thomas MJ, Dechatelet LR. Release and metabolism of arachidonic acid in human neutrophils. Journal of Biological Chemistry. 1981;256:7228–7234. [PubMed] [Google Scholar]
- White SR, Strek ME, Kulp GVP, Spaethe SM, Burch RA, Neeley SP, Leff AR. Regulation of human eosinophil degranulation and activation by endogenous phospholipase A2. Journal of Clinical Investigation. 1993;91:2118–2125. doi: 10.1172/JCI116436. [DOI] [PMC free article] [PubMed] [Google Scholar]