

Abstract
Both GABAB and muscarinic acetylcholine receptors (mAChRs) influence hippocampal-dependent mnemonic processing. Here the possibility of a direct interaction between GABAB receptors and mAChR-mediated synaptic responses has been studied using intracellular recording in rat hippocampal slices.
The GABAB receptor agonist(−)-baclofen (5–10 μm) depressed an atropine-sensitive slow EPSP (EPSPM) and occluded the GABAB-receptor-mediated IPSP (IPSPB) which preceded it. These inhibitory effects were accompanied by postsynaptic hyperpolarization (9 ± 2 mV) and a reduction in cell input resistance (12 ± 3 %).
The selective GABAB receptor antagonist CGP 55845A (1 μm) fully reversed the depressant effects of (−)-baclofen (5–10 μm) such that in the combined presence of (−)-baclofen and CGP 55845A the EPSPM was 134 ± 21 % of control.
(−)-Baclofen (5–10 μm) caused a small (28 ± 11 %) inhibition of carbachol-induced (3.0 μm) postsynaptic depolarizations and increases in input resistance.
CGP 55845A (1 μm) alone caused an increase in the amplitude of the EPSPM (253 ± 74 % of control) and blocked the IPSPB that preceded it.
In contrast, the selective GABA uptake inhibitor NNC 05–0711 (10 μm) increased the amplitude of the IPSPB by 141 ± 38 % and depressed the amplitude of the EPSPM by 58 ± 10 %. This inhibition was abolished by CGP 55845A (1 μm).
Taken together these data provide good evidence that synaptically released GABA activates GABAB receptors that inhibit mAChR-mediated EPSPs in hippocampal CA1 pyramidal neurones. The mechanism of inhibition may involve both pre- and postsynaptic elements.
One of the most widely characterized extrinsic inputs to the CA1 region of the rat hippocampus is the septohippocampal input (Dutar et al. 1995). This input comprises a heterogeneous population of afferents that mediate their effects through the release of various neurotransmitters including acetylcholine (ACh), γ-aminobutyric acid (GABA), 5-hydroxytryptamine (5-HT) and a variety of neuropeptides (Decker & McGaugh, 1991; Dutar et al. 1995). Of these transmitters, both cholinergic and GABAergic inputs have received most attention because of their critical involvement in mnemonic processing (Cole & Nicoll, 1983; Decker & McGaugh, 1991; Dutar et al. 1995). However, whilst both sets of fibres have been shown to increase hippocampal excitability (through activation of a muscarinic acetylcholine receptor (mAChR)-mediated slow excitatory postsynaptic potential and a reduction in spike frequency adaptation (Cole & Nicoll, 1983, 1984; Madison et al. 1987; Morton & Davies, 1997) and by GABAA receptor-mediated disinhibition of CA3 circuits (Tóth et al. 1997)) the possibility of direct interactions between GABAergic and cholinergic inputs has not been extensively investigated.
In this respect, the classical inhibitory role of GABA synapses might be expected to be appropriate for providing negative regulatory control over the marked changes in excitability induced by mAChR activation. Certainly, the metabotropic nature of the GABAB receptor makes this receptor system a potential candidate for preventing the likely neurodegenerative and epileptogenic consequences of overactivation of mAChRs (Lothman et al. 1991; Wasterlain et al. 1993). Indeed, we have demonstrated recently that adenosine A1 receptors, which share many of the same cellular effectors as GABAB receptors (Dutar & Nicoll, 1988a; Thompson et al. 1992), provide a strong inhibitory influence over mAChR-mediated synaptic depolarization and loss of spike frequency adaptation (Morton & Davies, 1997). However, there are only a few reports in the peripheral and central nervous systems of interactions between GABAB and mACh receptors (Brown & Higgins, 1979; Worley et al. 1987; Wichmann et al. 1987; Libri et al. 1998; Scanziani, 2000). As such, the aim of the present study is to extend these investigations by examining how pharmacological, and synaptic, activation of GABAB receptors modifies mAChR-mediated synaptic transmission in the hippocampus. Some of these data have appeared previously in abstract form (Morton et al. 1997).
METHODS
Female Wistar rats (2-4 weeks old) were killed by cervical dislocation and exsanguination followed by decapitation in accordance with UK Home Office guidelines. The brain was removed rapidly and transverse hippocampal slices prepared by hemisecting the whole brain minus the cerebellum and cutting 400 μm thick transverse slices containing hippocampal slices using a vibroslicer (Campden Instruments, Loughborough, UK). The CA3 region of each slice was then cut away to eliminate changes in network function that can occur due to epileptiform bursting in area CA3 when picrotoxin is applied to the slice. The resultant CA3-ectomized slices were placed on a nylon mesh at the interface of a warmed (32-34 °C), perfusing (1-2 ml min−1) artificial cerebrospinal fluid and an oxygen-enriched (95 % O2-5 % CO2), humidified atmosphere. The standard perfusion medium comprised (mm): NaCl, 124; KCl, 3; NaHCO3, 26; NaH2PO4, 1.25; CaCl2, 2; MgSO4, 1; d-glucose, 10; and was bubbled with 95 % O2-5 % CO2.
Following a 1 h equilibration period intracellular recordings were obtained from the CA1 pyramidal cell body region using 2 m potassium methylsulphate-filled microelectrodes (60-110 MΩ). (This recording configuration was chosen to limit run-down of G-protein-coupled receptor-mediated responses.) An Axoclamp-2B amplifier (Axon Instruments, Foster City, CA, USA) was used in discontinuous (3-5 kHz switching frequency) current-clamp mode. All impalements were made in control medium. Once stable recordings had been made for at least 10 min, all fast ionotropic glutamate receptor-mediated synaptic transmission was blocked using a combination of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate receptor antagonist 6-nitro-7-sulphamoylbenzo(f)quinoxaline-2,3-dione (NBQX; Tocris Cookson Ltd, Bristol, UK; 2-4 μm), and the N-methyl-d-aspartate (NMDA) receptor antagonists d-(E)-2-amino-4-methyl-5-phosphono-3-pentanoic acid (CGP 40116; Ciba-Geigy Ltd, Basle, Swizerland; 50 μm) or d-2-amino-5-phosphonopentanoate (AP5; Tocris Cookson Ltd, Bristol, UK; 50 μm). GABAA receptor-mediated synaptic transmission was abolished using the GABAA receptor antagonist picrotoxin (Sigma, St Louis, MO, USA; 50 μm).
Bipolar stimulating electrodes, made from 55 μm diameter insulated nickel-chromium wire (Advent Research Materials Ltd, Eynsham, UK), were positioned in the stratum oriens close to the recording electrode in the stratum pyramidale, to provide extracellular orthodromic activation of CA1 neurones. In every series of experiments stimuli comprised square-wave pulses (20-200 μs; 5-30 V) delivered at a fixed intensity every 5-10 min. Data were captured using pCLAMP6 software (Axon Instruments Inc.) and digitized records were stored on the hard disk of a PC for off-line analysis using Clampfit software.
During the period between stimuli the input resistance and extent of spike frequency adaptation of each neurone were measured routinely every 2 min using 300-600 ms long negative and positive current steps (± 0.15-0.40 nA), respectively. Input resistance was calculated from the voltage deflection in response to brief negative current injections. In all experiments in which mAChR-mediated EPSPs (EPSPMs) were evoked, baseline recordings consisted of three or more successive EPSPMs which had peak amplitudes that differed by no more than 15 %. In responses in which action potentials were evoked peak amplitudes were measured following low-pass filtering of the response at frequencies that excluded action potentials but which maintained similar kinetic profiles of the rising and decay phases of the unfiltered EPSPM. In experiments examining spike frequency adaptation, stimulation intensity was set to a level sub-threshold for evoking an EPSPM. The extent of spike frequency adaptation was monitored 1 s prior to delivery of this stimulus and 2 s after by injecting identical 600 ms depolarizing current pulses through the recording electrode. Baseline responses were recorded in this manner for 15-20 min prior to drug applications to ensure stationarity of responses.
All drugs were applied by addition to the perfusion medium. To compare the EPSPMs evoked in the presence and absence of a drug, DC was injected through the electrode to compensate for any drug-induced changes in membrane potential. Atropine and picrotoxin were purchased from Sigma. [1-(S)-3,4-dichlorophenyl)ethyl]amino-2-(S)-hydroxypropyl-p-benzyl-phosphonic acid (CGP 55845A) was obtained from Ciba-Geigy Ltd, Basle, Switzerland. Data are presented as means ± standard error of the mean (s.e.m.) and statistical significance was assessed using Student's paired or unpaired t tests performed on raw data with P < 0.05 being taken as indicating statistical significance. The n values refer to the number of times a particular experiment was performed, each in a different slice taken from a different rat.
RESULTS
Data were obtained from 42 stable intracellular recordings (1-6 h) from CA1 pyramidal neurones with overshooting action potentials, resting membrane potentials more negative than -55 mV and input resistance values of 30 MΩ or greater.
Characterization of cholinergic synaptic responses
Single shock stimulation in the stratum oriens evoked an EPSP that was followed by a biphasic IPSP (n = 42, data not shown). The combined application of NBQX (1-3 μm), AP5 (50 μm) or CGP 40116 (50 μm) and picrotoxin (50 μm) inhibited the fast EPSP and GABAA receptor-mediated IPSP (IPSPA) leaving a slow GABAB receptor-mediated IPSP (IPSPB). Increasing the stimulus intensity 2- to 10-fold and delivery of a single stimulus, or a short train of stimuli (2-10 stimuli at 20 Hz), evoked an IPSPB followed by a much slower EPSP in 34 neurones in which this was attempted (Figs 1B, 2, 5 and 6). This slow EPSP could be evoked reproducibly, in isolation, every 8-10 min. Its magnitude and duration could be increased by increasing the stimulus intensity or applying physostigmine (n = 5, data not shown) and reduced by applying atropine (n = 4; Fig. 1A). As such, this component of synaptic transmission will be referred to as an EPSPM to indicate its dependence on mAChR activation.
Figure 1. Effects of atropine and (-)-baclofen on the slow EPSPM.
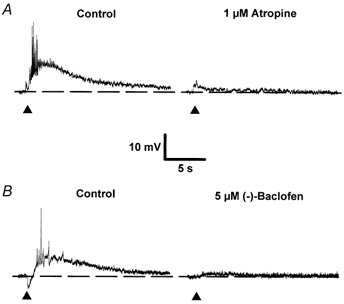
A and B, single sweeps illustrating the effects of 1 μm atropine (A) and 5 μm (-)-baclofen (B) on slow EPSPs evoked in two separate neurones. The initial membrane potential of both cells was -64 mV. In B and in all subsequent figures, unless stated otherwise, traces are individual synaptic responses recorded intracellularly in response to a single stimulus delivered in the stratum oriens in the presence of 1-3 μm NBQX, 50 μm CGP 40116 and 50 μm picrotoxin. A, responses evoked in the additional presence of 1 μm CGP 55845A. Each sweep was taken at the same membrane potential achieved using DC injection to compensate for any drug-induced changes. Filled triangles mark the time of afferent stimulation. In all synaptic traces shown, action potentials are attenuated due to low sampling frequency.
Figure 2. The (-)-baclofen-induced depression is reversed by CGP 55845A.
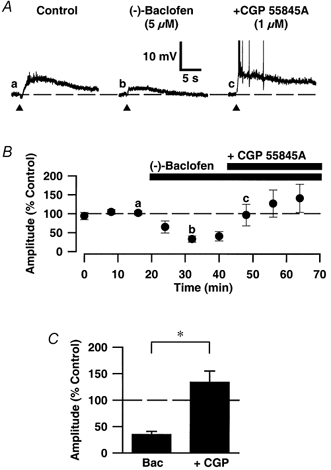
A, synaptic traces of EPSPMs recorded: a, in the presence of AMPA, NMDA and GABAA receptor antagonists (Control); b, in the additional presence of 5 μm (-)-baclofen; and c, subsequent co-application of 1 μm CGP 55845A. The membrane potential of the cell was -64 mV. The graph (B) shows a plot of the mean peak amplitudes of successive EPSPMs, for pooled data, to illustrate the temporal profile of the depressant effect of (-)-baclofen on the EPSPM and its reversal by CGP 55845A. Amplitudes are expressed as a percentage of the mean amplitudes of the three EPSPMs prior to application of (-)-baclofen. C, bar graph illustrating pooled data for the effects of 5-10 μm (-)-baclofen (Bac; n = 7) and 5-10 μm (-)-baclofen + 1 μm CGP 55845A (+ CGP; n = 7) on the EPSPM. The amplitudes of EPSPMs are expressed as a percentage of the mean value of the control EPSPMs. Note that (-)-baclofen significantly depressed the EPSPM whilst responses following co-application of (-)-baclofen + CGP 55845A were not significantly different from control responses. Data are means ±s.e.m.; * statistically significant compared with (-)-baclofen group (P < 0.05). In this and subsequent figures, the bars above the graph (B) indicate the duration for which the drug was applied.
Figure 5. The effects of CGP 55845A alone on the EPSPM and IPSPB.
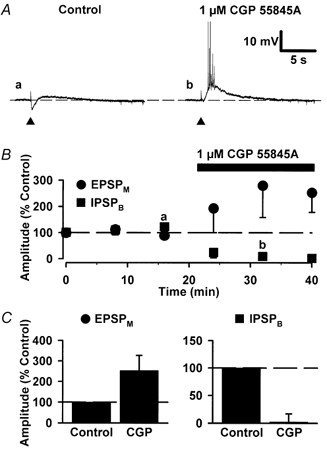
A, synaptic traces of representative EPSPMs recorded in control (a) and in the presence of 1 μm CGP 55845A (B). Note that following stimulation, the neurone responds with a hyperpolarization (IPSPB) followed by a small EPSPM, and following the addition of CGP 55845A the IPSPB is inhibited and the EPSPM enhanced. The membrane potential of this neurone was maintained at -64 mV using DC injection. B, mean peak amplitude of successive EPSPMs and IPSPBs, for pooled data (n = 3), versus time illustrating the enhancement of the EPSPM and the depression of the IPSPB by CGP 55845A. The bar above the graph indicates the duration for which CGP 55845A was applied. C, bar graphs illustrating pooled data for the effect of 1 μm CGP 55845A on the amplitude of the EPSPM and IPSPB (n = 3).
Figure 6. Kinetic comparison of the IPSPB with the EPSPM.
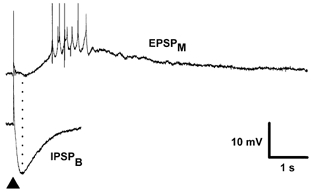
Traces are an EPSPM (top) recorded in the presence of GABA and glutamate receptor antagonists NBQX, CGP 40116, picrotoxin and CGP 55845A and an IPSPB (bottom) in the presence of NBQX, CGP 40116, picrotoxin and the GABA uptake inhibitor NNC 05-711. The membrane potentials of the neurones are -64 mV and -62 mV, respectively. Note that even in the presence of an inhibitor of GABA uptake, the isolated IPSPB peaks prior to the start of the depolarizing EPSPM and that the hyperpolarizing response is all but complete by the peak of the EPSPM. The dotted line indicates the time of the peak of the isolated IPSPB, which was evoked in the presence of the GABA uptake inhibitor.
effects of gabab receptor activation on the EPSPM
To establish whether GABAB receptor activation affected the EPSPM the effect of the selective GABAB receptor agonist (-)-baclofen was tested. At 5-10 μm (-)-baclofen caused a postsynaptic hyperpolarization (9 ± 2 mV) and a decrease in cell input resistance of 12 ± 3 % (n = 7). Comparison of EPSPMs prior to (-)-baclofen application and following DC injection to compensate for this hyperpolarization revealed that (-)-baclofen had depressed both the EPSPM as well as the IPSPB preceding it (Fig. 1B). This effect was maintained for the period of the agonist application and was reversible on washout (not shown). In addition, the magnitude of the (-)-baclofen-induced depression of the EPSPM was not statistically different from its depression of pharmacologically isolated IPSPAs and AMPA receptor-mediated EPSPs (EPSPAs; not illustrated). Thus, 5 μm (-)-baclofen depressed the peak amplitudes of EPSPMs, EPSPAs and IPSPAs to 30 ± 15 % (n = 3), 36 ± 16 % (n = 4) and 12 ± 3 % (n = 5) of control, respectively.
To confirm that the depressant action of (-)-baclofen on the EPSPM was mediated by GABAB receptor activation we tested next the ability of the selective GABAB receptor antagonist CGP 55845A to reverse this effect. In all cells tested (n = 7) CGP 55845A (1 μm) fully reversed all the effects of (-)-baclofen (barring the depression of the IPSPB, which was directly inhibited by this antagonist). Thus, in the presence of 5-10 μm (-)-baclofen alone the peak amplitude of the EPSPM was 35 ± 10 % of control compared with 134 ± 21 % of control in the combined presence of 5-10 μm (-)-baclofen and 1 μm CGP 55845A (n = 7; Fig. 2).
is the (-)-baclofen-induced depression of the epspm mediated pre- or postsynaptically?
Having established a depressant action of GABAB receptors on mAChR-mediated postsynaptic responses evoked by afferent stimulation, we addressed next whether these effects were mediated pre- or postsynaptically. To do this we tested the effect of (-)-baclofen on the postsynaptic depolarization and increase in cell input resistance evoked by brief bath applications of carbachol (3 μm for 30-60 s). A low concentration of carbachol was chosen to produce relatively small depolarizations so as to maximize the probability of observing an inhibitory influence of (-)-baclofen.
Repeated applications of carbachol caused consistent and reversible depolarizations (11 ± 4 mV; Fig. 3) that were associated with increases in cell input resistance (9 ± 1 %, n = 4); effects that were comparable to those associated with the EPSPM and that, like the EPSPM, were abolished by atropine (1-5 μm, n = 3; not illustrated). Application of (-)-baclofen (10 μm) caused a hyperpolarization (11 ± 4 mV) that was associated with a small decrease in cell input resistance (13 ± 3 %). In addition, (-)-baclofen caused a 28 ± 11 % inhibition of carbachol-induced responses which was only partially reversed by CGP 55845A (1 μm; n = 4; Fig. 3B), possibly because of the gradual run-down of carbachol-induced responses on repeated applications. Irrespective of this, the depressant effect of (-)-baclofen was significantly smaller than its effect on the EPSPM (P < 0.05; Fig. 3B).
Figure 3. The effects of (-)-baclofen on postsynaptic responses evoked by carbachol.
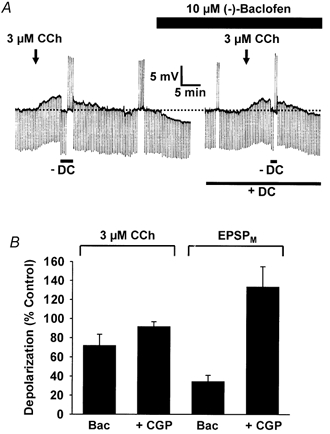
A, a chart record of membrane potential and input resistance (downward voltage deflections to -0.3 nA, 300 ms current steps) with the black arrows and the black bar indicating the period of carbachol (CCh, 3 μm) and (-)-baclofen (10 μm) applications, respectively. Constant DC injection (lower bar) was used in the presence of (-)-baclofen to restore the cell membrane potential to control levels. At the peak of the carbachol-induced depolarization DC injection was adjusted (-DC) to bring the membrane potential in line with that prior to the carbachol application. The transient upward deflections at various time points throughout the chart record are spike frequency adaptation responses to +0.3 nA, 300 ms current steps in the presence and absence of carbachol and (-)-baclofen. The initial membrane potential of this neurone was -64 mV. The bar graph in B shows the peak depolarization induced by carbachol, or evoked by the EPSPM, in the presence of baclofen (Bac) or baclofen + CGP 55845A (+ CGP) plotted as a percentage of that evoked in its absence.
Effects of (-)-baclofen on the reduction in spike frequency adaptation evoked by synaptic activation of mAChRs
As shown previously, stimulation of cholinergic afferents at intensities sub-threshold for activating the EPSPM causes a reduction in spike frequency adaptation in response to depolarizing current steps delivered 2 s after pathway stimulation (Morton & Davies, 1997; Fig. 4). Mechanistically, this mAChR-mediated effect arises from the inhibition of a distinct population of ion channels to that which generates the EPSPM (Madison et al. 1987). As such, a series of experiments were performed to examine whether GABAB receptor activation also affected this kind of cholinergic synaptic response. (-)-Baclofen (10 μm) completely occluded the IPSPB evoked by pathway stimulation (Fig. 4) but only partially inhibited the reduction in spike frequency adaptation evoked by afferent stimulation (n = 5; P < 0.05; Fig. 4B), even when applied at a concentration of 50 μm. Whilst this latter effect was small it was, nevertheless, fully reversed by subsequent application of CGP 55845A (1 μm; n = 5; Fig. 4B).
Figure 4. The effect of CGP 55845A on the (-)-baclofen-induced inhibition of stimulation-evoked reduction in spike frequency.
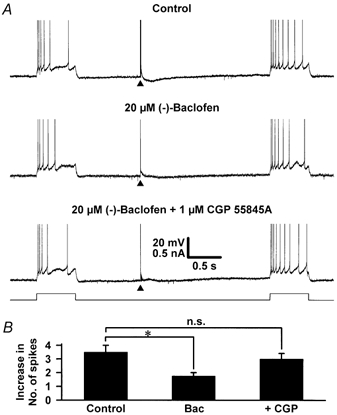
A, continuous records of the membrane potential of a single cell in which a depolarizing current step (+0.2 nA, 600 ms) was delivered 1.0 s prior to, and 2.0 s after pathway stimulation. The stimulation was delivered at an intensity just suprathreshold for activating an EPSPM in control medium containing only the ionotropic glutamate and GABAA receptor antagonists (control), in the additional presence of 20 μm (-)-baclofen and in the additional combined presence of 20 μm (-)-baclofen and 1 μm CGP 55845A. Note that in control medium, pathway stimulation evoked an IPSPB and caused a reduction in spike frequency adaptation. (-)-Baclofen inhibited but did not abolish the stimulation-evoked reduction in spike frequency adaptation. It did, however, occlude the IPSPB. B, pooled data for the difference in the number of action potentials fired during each 600 ms depolarizing step 1.0 s before and 2.0 s after a threshold stimulus in the absence and presence of 20 μm (-)-baclofen (Bac; n = 5) and in the additional presence of 1 μm CGP 55845A (n = 5). The initial membrane potential of the neurone was -65 mV and this was maintained using DC injection. Data are means ±s.e.m.; * significant (P < 0.05); n.s., not significant (compared with control).
Does endogenous GABA inhibit cholinergic synaptic responses?
It is well established that synaptically released GABA (1) activates a late IPSPB in CA1 pyramidal neurones (Dutar & Nicoll, 1988B) and (2) is responsible for activity-dependent depression of both GABA and glutamate receptor-mediated synaptic transmission (Thompson & Gähwiler, 1989; Davies et al. 1990; Nathan & Lambert, 1991; Davies & Collingridge, 1993; Isaacson et al. 1993). As such, we tested next whether CGP 55845A when applied alone affected the EPSPM to establish whether endogenously released GABA might inhibit EPSPMs. CGP 55845A (1 μm) alone abolished the IPSPB preceding the EPSPM and caused a substantial but variable increase in the size of the EPSPM. As such, the peak amplitude of the EPSPM in the presence of CGP 55845A was 253 ± 74 % of control (n = 3; Fig. 5). This result confirmed the small increase in peak amplitude recorded when CGP 55845A had been used to reverse the (-)-baclofen-induced depression of the EPSPM (Fig. 2); the larger effect observed in this current series of experiments stemming from the smaller starting amplitude of EPSPMs and the non-linearity of EPSPM peak amplitude as this rises towards spiking threshold.
This CGP 55845A-induced increase in the EPSPM suggested that endogenous GABA released within the slice was capable of activating GABAB receptors which inhibit the EPSPM. Therefore, the next series of experiments examined whether it was possible to potentiate the depression of the EPSPM that was induced by endogenous GABA within the slice. To do this, the effects of the selective GABA uptake inhibitor NNC 05-0711 were investigated (Suzdak et al. 1992). This compound extends the duration of pharmacologically isolated IPSPBs from a mean duration of 623 ± 132 ms to 1830 ± 235 ms (n = 4), which still falls short of the mean latency to peak amplitude of isolated EPSPMs, which was 2.6 ± 0.4 s (n = 6; Fig. 6). In addition, NNC 05-0711 (10 μm) increased the amplitude of the IPSPB to 241 ± 38 % of control and caused a 58 ± 10 % depression of the EPSPM (n = 4; Fig. 7). In the three cells in which recordings were maintained for long enough, subsequent application of CGP 55845A (1 μm) caused a complete reversal of the NNC 05-711-induced depression of the EPSPM (Fig. 7C).
Figure 7. The effects of an inhibitor of GABA uptake on EPSPMs and IPSPBs.
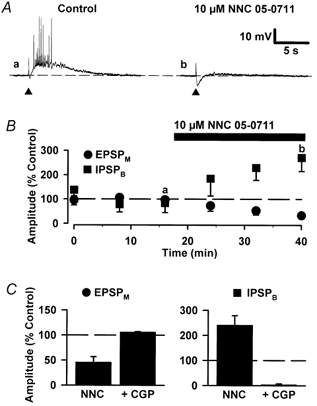
A, synaptic traces of representative IPSPB/EPSPMs recorded in control (a) and in the presence of NNC 05-0711 (10 μm) (b). The membrane potential of this neurone was -64 mV. B, mean peak amplitude of successive EPSPMs and IPSPBs, for pooled data (n = 4), versus time illustrating the depression of the EPSPM and the enhancement of the IPSPB by NNC 05-0711. The bar above the graph indicates the duration for which NNC 05-0711 was applied. C, bar graphs illustrating pooled data for the effects of 10 μm NNC 05-0711 (NNC; n = 4) and 10 μm NNC 05-0711 + 1 μm CGP 55845A (+CGP; n = 3) on the EPSPM and IPSPB. Note that NNC 05-0711 depressed the EPSPM and that CGP 5845A reversed this effect.
DISCUSSION
Here we have demonstrated a GABAB receptor-mediated inhibition of mAChR-mediated synaptic responses in the hippocampal CA1 region. Potentially, this effect may be mediated either pre- or postsynaptically or both. Postsynaptic GABAB receptors classically cause membrane hyperpolarization (Newberry & Nicoll, 1984). However, this does not account for the baclofen-induced depression of the EPSPM since membrane hyperpolarization was routinely compensated for using DC injection. That said, the GABAB receptor-mediated reduction in input resistance could feasibly limit the extent of depolarization during the EPSPM by shunting membrane currents. This possibility, however, is unlikely since NNC 05-0711, which produces no tonic postsynaptic hyperpolarization or decrease in input resistance (but does enhance the IPSPB), also inhibits the EPSPM via GABAB receptor activation. A caveat here, however, is that shunting from the IPSPB before the EPSPM should not preclude full EPSPM expression. In this respect, the IPSPB, even during inhibited GABA uptake, will provide a shunt for up to 1 s with the peak conductance change occurring before the rising phase of the EPSPM (Fig. 6; Morton & Davies, 1997). A similar but opposite argument can be made for the facilitatory action of CGP 55845A on the EPSPM.
If shunting were a major factor limiting mAChR-mediated depolarization, baclofen should produce a substantial inhibition of carbachol-induced depolarizations. Whilst baclofen caused a small inhibition, this was much smaller than its depressant effect on the EPSPM. Based on these data, and the similarity of the cellular mechanisms underlying the EPSPM and carbachol-induced depolarizations, it is tempting to speculate that GABAB receptors act to inhibit ACh release. If true, baclofen might also be expected to inhibit stimulation-induced reductions in spike frequency adaptation afforded by synaptically released ACh. However, the effect of baclofen on this response was less clear than its effect on the EPSPM. In particular, baclofen, even at concentrations (50 μm) that are maximal for activating pre- and postsynaptic GABAB receptors (Thompson & Gähwiler, 1992a), produced only a small reduction in spike frequency adaptation induced by pathway stimulation. However, this effect was statistically significant and antagonized by CGP 55845A, indicating that it was a true GABAB receptor-mediated response presumably reflecting, in part, the reduction in cell input resistance produced by postsynaptic GABAB receptor activation.
Interestingly, activation of adenosine A1 receptors, which usually has similar effects to activation of GABAB receptors (Dutar & Nicoll, 1988a; Thompson et al. 1992; Thompson & Gähwiler, 1992a), abolished both EPSPMs and stimulation-induced reduction in spike frequency adaptation (Morton & Davies, 1997). One possible explanation for the differences between the actions of these receptor systems is that presynaptic GABAB receptors, like galanin receptors, are restricted to a select population of cholinergic afferents whereas adenosine A1 receptors are expressed universally. However, this scenario could only exist if release from a relatively small number of afferents is required to inhibit spike frequency adaptation as opposed to generate an EPSPM. Two observations support this concept: (1) much greater intensities of pathway stimulation are required to evoke an EPSPM than are necessary to inhibit spike frequency adaptation (Cole & Nicoll, 1984; Morton & Davies, 1997) and (2) whilst acetylcholine modifies the activity of numerous ion channels (Benson et al. 1988; Colino & Halliwell, 1993; Fraser & MacVicar, 1996; Haj-Dahmane & Andrade, 1999) it is inhibition of a leak K+ channel that is principally responsible for generation of the EPSPM (Madison et al. 1987), this action necessitating higher concentrations of ACh (and therefore potentially ACh release from more afferents) than are required to inhibit the Ca2+-activated K+ channel principally responsible for the loss of spike frequency adaptation (Madison et al. 1987).
Alternatively, it is possible that adenosine A1 and GABAB receptors are co-expressed on all cholinergic terminals but couple to separate effector systems that exhibit different degrees of efficacy for inhibiting ACh release. In this respect, a limited capacity of GABAB receptors to inhibit ACh release may explain the relative lack of supporting neurochemical release data for this effect in the CNS (Wichmann et al. 1987; Taniyama et al. 1992; Ikarashi et al. 1999). That said, (1) GABAB receptors inhibit ACh release in the superior colliculus and (2) it is possible that activation of GABAB receptors may produce complex changes in the release of other neurotransmitters that may lead to an overall negative regulatory effect on the EPSPM.
Leaving presynaptic mechanisms aside, non-electrophysiological postsynaptic interactions between GABAB receptors and mAChRs could conceivably contribute to the baclofen-induced depression of the EPSPM. In particular, the small baclofen-induced inhibition of carbachol-evoked depolarizations may represent a postsynaptic interaction that reflects both shunting and additional receptor crosstalk interactions. In this respect, it is possible that GABAB receptors could inhibit mAChR-mediated responses postsynaptically via interactions with the G-protein(s)- or second messenger-activated signal transduction cascades responsible for the electrophysiological effects of these Gq/11-coupled mAChRs. Indeed, interactions between Gi/o- and Gq/11-coupled receptors, and specifically between GABAB and mACh receptors, have been described (Worley et al. 1987). However, the latter interaction was a mAChR-mediated inhibition of GABAB receptor-mediated responses rather than the converse.
This aside, GABAB receptors promote the ability of a number of G-protein-coupled receptors (e.g. β-noradrenergic receptors) to inhibit the Ca2+-activated K+ channels responsible for spike frequency adaptation (Andrade et al. 1993). Such a scenario would oppose the proposed inhibitory influence of GABAB receptors on ACh release and, therefore, might account for the weak effect of baclofen on this phenomenon. However, this interaction has only been defined for Gs-coupled receptors and not Gq/11-coupled receptors to which the M1 and/or M3 mAChRs responsible for the EPSPM belong (Dutar & Nicoll, 1988b; Pitler & Alger, 1990; Segal & Fisher, 1992). That said, there are many complex examples of receptor crosstalk and, as such, the possibility that postsynaptic interactions between these two receptor systems that account, at least in part, for the inhibitory interactions described above cannot be dismissed. Should crosstalk exist, the differential effects of GABAB receptors on the EPSPM and mAChR-induced inhibition of spike frequency adaptation may result from the different molecular interactions governing each effect as well as the extent of saturation of each effector pathway.
Irrespective of this it is important to establish the physiological relevance of the inhibitory influence of GABAB receptors on the EPSPM. Here synaptic activation of GABAB receptors has been shown to regulate the EPSPM since (1) CGP 55845A increases the amplitude of EPSPMs beyond control levels and (2) NNC 05-0711 (Suzdak et al. 1992) inhibits the EPSPM (as well as increases the IPSPB) in a CGP 55845A-sensitive manner. However, what is the source of the endogenously released GABA? Since neither CGP 55845A nor NNC 05-711 affect neuronal resting membrane properties, both compounds presumably modify the EPSPM by influencing the action of phasically released GABA (Thompson & Gähwiler, 1992b; Roepstorff & Lambert, 1994; Soltesz et al. 1995). This GABA could originate from: (i) release from hippocampal or septohippocampal GABAergic neurones or (ii) direct co-release from cholinergic nerve terminals (Frotscher et al. 1986; Kosaka et al. 1988; Beauleiu & Somogyi, 1991; Bayraktar et al. 1997). However, co-expression of GABA and ACh in septohippocampal as well as intrinsic hippocampal cholinergic afferents is controversial (Frotscher et al. 2000). As such, if GABA is released from non-cholinergic terminals it may operate through a diffusable, action-at-a-distance mechanism. Alternatively, closer structural interactions between GABAergic and cholinergic terminals, whether these be the cholinergic terminals forming synaptic specializations (Umbriaco, et al. 1995) or those mediating untargeted ACh release (Vizi & Kiss, 1998), may exist. In this respect, both diffuse and targeted GABA release can account for synaptically activated GABAB receptor-mediated events. Specifically, GABA spillover to extrasynaptic GABAB receptors, as a result of coincident activation of multiple GABAergic interneurones/release sites (Scanziani, 2000), is a favoured mechanism in the hippocampus. Indeed, this accounts for GABAB receptor-mediated inhibition of mAChR agonist-induced hippocampal rhythmic activity (Scanziani, 2000). However, more detailed physiological and anatomical analyses will be required to determine the source of the released GABA which modulates cholinergic synaptic transmission.
In conclusion, activation of GABAB receptors can control the level of activity at cholinergic synapses in the hippocampal CA1 region. As such, within the hippocampus there exists a reciprocal interaction between GABAergic and cholinergic systems whereby mAChRs inhibit GABAergic synaptic function (Freund & Buszáki, 1996; Manuel & Davies, 1998) and GABAB receptors inhibit cholinergic synaptic function. This control can be provided through phasic GABAergic inputs and, as such, differs from the control afforded by adenosine A1 receptors (Morton & Davies, 1997). In particular, the high background levels of adenosine in cerebrospinal fluid, and its more generalized release during insults such as hypoxia, are consistent with the neuroprotective role of this purine. In contrast, the more dynamic regulation of cholinergic synaptic transmission by synaptically released GABA may be important in mnemonic processes for which a role for both receptor systems has been described (Cole & Nicoll, 1983; Decker & McGaugh, 1991; Mondadori et al. 1993; Dutar et al. 1995). However, GABAB receptors may also play a neuroprotective role through modulation of cholinergic activity. Indeed, activation of GABAB receptors inhibits mAChR-induced synchronous glutamatergic activity in both the rat piriform cortex (Libri et al. 1998) and hippocampus (Scanziani, 2000).
Acknowledgments
We thank Dr Mario F. Pozza (Ciba-Geigy, Basle, Switzerland) for CGP 55845A and CGP 40116. The NNC 05-0711 was provided by Dr H. Mengel. This work was supported by the Wellcome Trust and the MRC.
References
- Andrade R. Enhancement of beta-adrenergic responses by Gi-linked receptors in rat hippocampus. Neuron. 1993;10:83–88. doi: 10.1016/0896-6273(93)90244-l. [DOI] [PubMed] [Google Scholar]
- Bayraktar T, Staiger JF, Acsady L, Cozzari C, Freund TF, Zilles K. Co-localization of vasoactive intestinal polypeptide, gamma-aminobutyric acid and choline acetyltransferase in neocortical interneurons of the adult rat. Brain Research. 1997;757:209–217. doi: 10.1016/s0006-8993(97)00218-7. [DOI] [PubMed] [Google Scholar]
- Beauleiu C, Somogyi P. Enrichment of cholinergic synaptic terminals on GABAergic neurons and coexistence of immunoreactive GABA and choline acetyltransferase in the same synaptic terminals in the striate cortex of the cat. Journal of Comparative Neurology. 1991;304:666–680. doi: 10.1002/cne.903040412. [DOI] [PubMed] [Google Scholar]
- Benson DM, Blitzer RD, Landau EM. An analysis of the depolarization produced in guinea-pig hippocampus by cholinergic receptor stimulation. Journal of Physiology. 1988;404:479–496. doi: 10.1113/jphysiol.1988.sp017301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, Higgins AJ. Presynaptic effects of γ-aminobutyric acid in isolated rat superior cervical ganglia. British Journal of Pharmacology. 1979;66:108–109P. [PMC free article] [PubMed] [Google Scholar]
- Cole AE, Nicoll RA. Acetylcholine mediates a slow synaptic potential in hippocampal pyramidal cells. Science. 1983;221:1299–1301. doi: 10.1126/science.6612345. [DOI] [PubMed] [Google Scholar]
- Cole AE, Nicoll RA. Characterisation of a slow cholinergic post-synaptic potential recorded in vitro from rat hippocampal pyramidal cells. Journal of Physiology. 1984;352:173–188. doi: 10.1113/jphysiol.1984.sp015285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colino A, Halliwell JV. Carbachol potentiates Q current and activates a calcium dependent non-specific conductance in rat hippocampus in vitro. European Journal of Neuroscience. 1993;5:1198–1209. doi: 10.1111/j.1460-9568.1993.tb00974.x. [DOI] [PubMed] [Google Scholar]
- Davies CH, Collingridge GL. The physiological regulation of synaptic inhibition by GABAB autoreceptors in rat hippocampus. Journal of Physiology. 1993;472:245–265. doi: 10.1113/jphysiol.1993.sp019945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CH, Davies SN, Collingridge GL. Paired-pulse depression of monosynaptic GABA-mediated inhibitory postsynaptic responses in rat hippocampus. Journal of Physiology. 1990;424:513–531. doi: 10.1113/jphysiol.1990.sp018080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker MW, McGaugh JL. The role of interactions between the cholinergic system and other neuromodulatory systems in learning and memory. Synapse. 1991;7:151–168. doi: 10.1002/syn.890070209. [DOI] [PubMed] [Google Scholar]
- Dutar P, Bassant M, Senut M, Lamour Y. The septohippocampal pathway: structure and function of a central cholinergic system. Physiological Reviews. 1995;75:393–427. doi: 10.1152/physrev.1995.75.2.393. [DOI] [PubMed] [Google Scholar]
- Dutar P, Nicoll RA. Pre- and postsynaptic GABAB receptors in the hippocampus have different pharmacological properties. Neuron. 1988a;1:585–591. doi: 10.1016/0896-6273(88)90108-0. [DOI] [PubMed] [Google Scholar]
- Dutar P, Nicoll RA. A physiological role for GABAB receptors in the central nervous system. Nature. 1988b;332:156–158. doi: 10.1038/332156a0. [DOI] [PubMed] [Google Scholar]
- Fraser DD, MacVicar BA. Cholinergic-dependent plateau potential in hippocampal CA1 pyramidal neurons. Journal of Neuroscience. 1996;16:4113–4128. doi: 10.1523/JNEUROSCI.16-13-04113.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Buszáki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Schlander M, Leranth C. Cholinergic neurons in the hippocampus. A combined light- and electron-microscopic immunocytochemical study in the rat. Cell Tissue Research. 1986;246:293–301. doi: 10.1007/BF00215891. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Vida I, Bender R. Evidence for the existence of non-GABAergic, cholinergic interneurons in the rodent hippocampus. Neuroscience. 2000;96:27–31. doi: 10.1016/s0306-4522(99)00525-4. [DOI] [PubMed] [Google Scholar]
- Haj-Dahmane S, Andrade R. Muscarinic receptors regulate two different calcium-dependent cation currents in rat prefrontal cortex. European Journal of Neuroscience. 1999;11:1973–1980. doi: 10.1046/j.1460-9568.1999.00612.x. [DOI] [PubMed] [Google Scholar]
- Ikarashi Y, Yuzurihara M, Takahashi A, Ishimaru H, Maruyama Y. Neurochemical determination of the location of NMDA and GABA receptors on rat striatal cholinergic neurons. Brain Research Protocols. 1999;4:378–382. doi: 10.1016/s1385-299x(99)00044-6. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Solís JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- Kosaka T, Tauchi M, Dahl JL. Cholinergic neurons containing GABA-like and/or glutamic decarboxylase-like immunoreactivities in various brain regions. Experimental Brain Research. 1988;70:605–617. doi: 10.1007/BF00247609. [DOI] [PubMed] [Google Scholar]
- Libri V, Constanti A, Postlethwaite M, Bowery NG. Blockade of GABAB receptors facilitates muscarinic agonist-induced epileptiform activity in immature rat piriform cortex in vitro. Naunyn-Schmiedeberg's Archives of Pharmacology. 1998;358:68–174. doi: 10.1007/pl00005239. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Bertram EH, Stringer JL. Functional anatomy of hippocampal seizures. Progress in Neurobiology. 1991;37:1–82. doi: 10.1016/0301-0082(91)90011-o. [DOI] [PubMed] [Google Scholar]
- Madison DV, Lancaster B, Nicoll RA. Voltage-clamp analysis of cholinergic action in the hippocampus. Journal of Neuroscience. 1987;7:733–741. doi: 10.1523/JNEUROSCI.07-03-00733.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuel NA, Davies CH. Pharmacological modulation of GABAA receptor-mediated synaptic potentials in the CA1 region of the rat hippocampus. British Journal of Pharmacology. 1998;125:1529–1542. doi: 10.1038/sj.bjp.0702237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondadori C, Jaekel J, Preiswerk G. CGP36742: the first orally active GABAB blocker improves the cognitive performance of mice, rats and rhesus monkeys. Behavioral and Neural Biology. 1993;60:62–68. doi: 10.1016/0163-1047(93)90729-2. [DOI] [PubMed] [Google Scholar]
- Morton RA, Bulters DO, Davies CH. GABAB receptor-mediated modulation of muscarinic acetylcholine receptor-mediated synaptic responses in the rat hippocampus. British Journal of Pharmacology. 1997;122:274P. [Google Scholar]
- Morton RA, Davies CH. Regulation of muscarinic acetylcholine receptor-mediated EPSPs by adenosine receptors in the rat hippocampus. Journal of Physiology. 1997;502:75–90. doi: 10.1111/j.1469-7793.1997.075bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan T, Lambert JDC. Depression of the fast IPSP underlies paired-pulse facilitation in area CA1 of the rat hippocampus. Journal of Neurophysiology. 1991;66:1704–1715. doi: 10.1152/jn.1991.66.5.1704. [DOI] [PubMed] [Google Scholar]
- Newberry NR, Nicoll RA. Direct hyperpolarizing action of baclofen on hippocampal pyramidal cells. Nature. 1984;308:450–452. doi: 10.1038/308450a0. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Activation of a pharmacologically defined M3 muscarinic acetylcholine receptor depolarizes hippocampal pyramidal cells. Brain Research. 1990;534:257–262. doi: 10.1016/0006-8993(90)90137-z. [DOI] [PubMed] [Google Scholar]
- Roepstorff A, Lambert JD. Factors contributing to the decay of the stimulus-evoked IPSC in rat hippocampal CA1 neurons. Journal of Neurophysiology. 1994;72:2911–2926. doi: 10.1152/jn.1994.72.6.2911. [DOI] [PubMed] [Google Scholar]
- Scanziani M. GABA spillover activates postsynaptic GABAB receptors to control rhythmic hippocampal activity. Neuron. 2000;25:673–681. doi: 10.1016/s0896-6273(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Segal M, Fisher A. AF102B, a muscarinic M1 receptor agonist, mimics some effects of acetylcholine on neurons of rat hippocampus slices. European Journal of Pharmacology. 1992;220:103–106. doi: 10.1016/0014-2999(92)90019-z. [DOI] [PubMed] [Google Scholar]
- Soltesz I, Smetters DK, Mody I. Tonic inhibition originates from synapses close to the soma. Neuron. 1995;14:1273–1283. doi: 10.1016/0896-6273(95)90274-0. [DOI] [PubMed] [Google Scholar]
- Suzdak PD, Frederiksen K, Andersen KE, Sørensen PO, Knutsen LJS, Nielsen EB. NNC-711, a novel potent and selective γ-aminobutyric acid uptake inhibitor: pharmacological characterization. European Journal of Pharmacology. 1992;223:189–198. doi: 10.1016/0014-2999(92)90804-d. [DOI] [PubMed] [Google Scholar]
- Taniyama K, Niwa M, Kataoka Y, Yamashita K. Activation of protein-kinase-C suppresses the gamma-aminobutyric acid B receptor-mediated inhibition of the vesicular release of noradrenaline and acetylcholine. Journal of Neurochemistry. 1992;58:1239–1245. doi: 10.1111/j.1471-4159.1992.tb11334.x. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Gähwiler BH. Activity dependent disinhibition III. Desensitization and GABAB receptor-mediated presynaptic inhibition in the hippocampus in vitro. Journal of Neurophysiology. 1989;61:524–533. doi: 10.1152/jn.1989.61.3.524. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Gähwiler BH. Comparison of the actions of baclofen at pre- and post-synaptic receptors in the rat hippocampus in vitro. Journal of Physiology. 1992a;451:329–345. doi: 10.1113/jphysiol.1992.sp019167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SM, Gähwiler BH. Effects of the GABA uptake inhibitor tiagabine on inhibitory synaptic potentials in rat hippocampal slice cultures. Journal of Neurophysiology. 1992b;67:1698–1701. doi: 10.1152/jn.1992.67.6.1698. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Haas HL, Gähwiler BH. Comparison of the actions of adenosine at pre- and post-synaptic receptors in the rat hippocampus in vitro. Journal of Physiology. 1992;451:347–363. doi: 10.1113/jphysiol.1992.sp019168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tóth K, Freund TF, Miles R. Disinhibition of rat hippocampal pyramidal cells by GABAergic afferents from the septum. Journal of Physiology. 1997;500:463–474. doi: 10.1113/jphysiol.1997.sp022033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbriaco D, Garcia S, Beaulieu C, Descarries L. Relational features of acetylcholine, noradrenaline, serotonin and GABA axon terminals in the stratum radiatum of the adult rat hippocampus (CA1) Hippocampus. 1995;5:605–620. doi: 10.1002/hipo.450050611. [DOI] [PubMed] [Google Scholar]
- Vizi ES, Kiss JP. Neurochemistry and pharmacology of the major hippocampal transmitter systems: synaptic and nonsynaptic interactions. Hippocampus. 1998;8:566–607. doi: 10.1002/(SICI)1098-1063(1998)8:6<566::AID-HIPO2>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Wasterlain CG, Fujikawa DG, Penix L, Sankar R. Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia. 1993;3:S37–53. doi: 10.1111/j.1528-1157.1993.tb05905.x. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Illing R-B, Starke K. Evidence for a neurotransmitter function of acetylcholine in rabbit superior colliculus. Neuroscience. 1987;3:991–1000. doi: 10.1016/0306-4522(87)90174-6. [DOI] [PubMed] [Google Scholar]
- Worley PF, Baraban JM, McCarren M, Snyder SH, Alger BE. Cholinergic phosphatidylinositol modulation of inhibitory, G protein-linked neurotransmitter actions: electrophysiological studies in rat hippocampus. Proceedings of the National Academy of Sciences of the USA. 1987;84:3467–3471. doi: 10.1073/pnas.84.10.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]