

Abstract
Diffusion-mediated changes in ion channel function within blood vessels have not been demonstrated directly in a patch-clamp study. Here, we examined the hypothesis that endothelium-derived diffusible bioactive substances would modify endothelin-1 (ET-1)-evoked membrane currents in smooth muscle cells situated within intact arterioles.
In pieces of arterioles dissected from the rat cerebral pial membrane, patch electrodes were placed on single smooth muscle cells identified under the microscope. Under perforated patch-clamp conditions, ET-1 evoked an oscillatory inward current at negative potentials in such cells in the presence of the gap junction disrupter 18α-glycyrrhetinic acid. ET-1 also elicited an oscillation superimposed on a membrane depolarization in current-clamp mode.
The oscillatory current exhibited an outwardly rectifying current-voltage relationship, a sensitivity to niflumic acid, a requirement for inositol 1,4,5-trisphosphate (IP3)- and caffeine-sensitive Ca2+ stores and for external Ca2+ and a rank order of anion permeabilities characteristic of Ca2+-activated Cl− currents (ICa(Cl)).
This oscillatory response was inhibited by bradykinin (an effect distinct from the electrical propagation of hyperpolarization) and this effect was attenuated by the NO-synthase inhibitor Nω-nitro-l-arginine and by the NO scavenger oxyhaemoglobin but not by the cyclo-oxygenease inhibitor indomethacin. 8-Bromoguanosine 3′,5′-cyclic monophosphate (8-Br-cGMP) and nitroprusside closely mimicked the effect of bradykinin.
The present patch-clamp study has revealed diffusion-mediated cell-to-cell interaction in an intact blood vessel: bradykinin appears to cause NO to move from endothelium to smooth muscle, there to inhibit an ET-1-evoked oscillatory ICa(Cl) via the NO-cGMP pathway.
The physiological control of cerebral blood flow has been under investigation for a number of years, as have pathological conditions affecting the cerebral circulation (for review see Sobey & Faraci, 1998). The influence of vasoactive substances on ion channels seems likely to be particularly important in regulating both membrane potential and the contractile state of the cerebral arteries. In fact, intercellular communication via a diffusible substance has been demonstrated to alter both the vascular contractility and membrane potential of smooth muscle cells in other types of arterioles (Tare et al. 1990). Patch-clamp studies on ion channels have mostly been performed using single isolated cells. However, interactions within the intact vasculature, whether mediated by electrical events or bioactive substances, are difficult to clarify using such preparations. Recently, Quinn & Beech (1998) reported a modified patch-clamp method that they applied to smooth muscle cells within an intact arteriolar preparation from rabbit brain. Using a development of this method, we recently demonstrated the presence of intercellular electrical communication between adjacent smooth muscle cells and between smooth muscle and endothelial cells in situ in rat intact cerebral arterioles (Yamazaki & Kitamura, 2001).
Studies of the influence of bioactive substances on ion channels in intact blood vessels may provide a new understanding of the pathophysiological implications of changes in the integrity of different types of cells. A well-known cerebrovascular dysfunction is delayed cerebral vasospasm, which frequently occurs after subarachnoid haemorrhage (SAH). The underlying mechanism is not fully understood, although the vasospasm appears to be induced by several factors that cause an imbalance between vasoconstrictor and vasodilator responses (Faraci & Heistad, 1998; Sobey & Faraci, 1998). Endothelin (ET), a potent, long-lasting vasoactive substance, may play a major role in constriction in the cerebral artery (Sobey & Faraci, 1998). Injury to the endothelium also appears likely to contribute to an enhancement of vasospasm since decreased nitric oxide (NO) production is known to weaken vasodilator responses (Faraci & Heistad, 1998; Sobey & Faraci, 1998). Hence, the interaction between such opposing effects in smooth muscle and endothelial cells is a fascinating subject, and one that we thought possible to investigate by applying the patch-clamp method directly to cells in situ within intact arterioles.
In the present study, we examined the hypothesis that ET-evoked changes in membrane currents in cerebral arteriolar smooth muscle cells may be modified by endothelial cells through diffusion-mediated interaction. Our results demonstrate that in rat intact cerebral arterioles, NO acts as a diffusible messenger following its release by bradykinin, possibly from the endothelium, to inhibit two ET-1-induced phenomena in smooth muscle cells: namely, an oscillatory Ca2+-activated Cl− current (ICl(Ca)) and oscillations on the membrane potential in smooth muscle cells.
METHODS
Male Wistar rats (3.5–4.5 weeks old) were anaesthetized with sodium pentobarbitone (60 mg kg−1i.p.). The procedure was approved by the Animal Research Committee of Fukuoka Dental College. The brain was removed and the pial membrane was dissected free from the entire surface of the cerebrum. Thereafter, pial tissue was incubated for 20 min at 37 °C in Hanks’ solution containing collagenase (650 units ml−1; Sigma) and papain (1.7–3.0 units ml−1; Sigma). The Hanks’ solution contained (mm); NaCl 137, KCl 5.4, CaCl2 0.01, NaH2PO4 0.34, K2HPO4 0.44, d-glucose 8, Hepes 5 (adjusted to pH 7.3). Pieces of arteriole were washed with fresh Hanks’ solution and kept at 4 °C until use.
Small pieces of arteriole were placed gently on the bottom of a chamber sited on the stage of an inverted microscope (Diaphot-300; Nikon, Tokyo, Japan). Once the tissues had adhered to the bottom of the chamber (under their own weight), superfusion with bath solution was begun at 1.5–2.0 ml min−1. Each arteriolar preparation used in this study had a mean diameter of ∼30 μm and a mean length of ∼250 μm. Each preparation contained a monolayer of smooth muscle cells (arranged regularly and perpendicularly to the long axis of the arteriole). The smooth muscle layer enclosed the endothelial cell layer, the cells of which were oriented parallel to the long axis of the vessel. We were able to identify single smooth muscle cells and, under direct visual control, place a patch electrode (see below) on the selected cell. To reduce the electrical connection between vessel-wall cells, a gap junction inhibitor, 18α-glycyrrhetinic acid (α-GA), was present throughout the experiments, unless otherwise indicated. The standard bath solution contained (mm): NaCl 150, KCl 5, CaCl2 2, MgCl2 1, d-glucose 11, Hepes 5 (adjusted to pH 7.4, 310 mosmol kg−1). In experiments performed to test anion selectivity, the bath solution contained (mm): NaX 150, Ca(OH)2 2, Mg(OH)2 1, d-glucose 11, Hepes 5 (adjusted to pH 7.4, 301–304 mosmol kg−1) where X− denotes SCN−, I−, Br−, Cl− or methanesulphonate (MeS−). The pipette solution for perforated whole-cell recording contained (mm): KCl 125, MgCl2 4, EGTA 0.02, Hepes 10, amphotericin B (240 μg ml−1) (adjusted to pH 7.3, 300 mosmol kg−1). Osmolarity was adjusted by adding mannitol to the solution. The patch pipettes had a tip resistance of 2–4 MΩ. Ag-AgCl wires immersed in the bath and pipette solutions were connected to a patch-clamp amplifier (Axopatch 200A; Axon Instruments, Foster City, CA, USA). Changes in junctional potentials created between the pipette and the bath solutions were corrected. Experiments were performed at room temperature and currents were filtered at a frequency of 1 kHz and digitized on-line at 5 kHz using an IBM-compatible computer and pCLAMP 8.0 software (Axon Instruments). Data are expressed as means ±s.e.m. (n, number of observations). Statistical analysis was performed using Student's paired or unpaired t test when two groups were compared, and by an analysis of variance (ANOVA) followed by a post hoc Bonferroni correction for more than three groups. A P value of less than 0.05 was considered significant.
Amphotericin B (Wako Pure Chemical, Osaka, Japan), α-GA and niflumic acid (both from Sigma) were freshly dissolved as stock solutions in dimethylsulphoxide (DMSO). The final concentration of DMSO was less than 0.1 %, which, by itself, did not affect currents when applied externally. Oxyhaemoglobin (HbO2) was freshly prepared by reacting O2 and deoxyhaemoglobin, which had been converted from methaemoglobin (Sigma) by means of sodium hydrosulphite (Wako). Endothelin-1 (ET-1; Peptide Institute, Osaka, Japan) was dissolved in 0.1 % acetic acid solution to make a 0.1 mm stock solution. Gap-27 peptide (amino-acid sequence, SRPTEKTIFII; purity = 99.3 %) was synthesized by Sigma Genosys Japan K.K. Some tissues were pretreated with bath solution containing Gap-27 peptide (300 μm) for approximately 30 min. Other compounds were dissolved as stock solutions in deionized water.
RESULTS
The amphotericin B-perforated whole-cell patch-clamp technique was used for the measurement of membrane current and potential in single smooth muscle cells within intact cerebral arterioles. Previously (Yamazaki & Kitamura, 2001), we recorded a sustained current component following a capacitative surge during a brief voltage step (5 ms, from −50 to −40 mV) in such cells. Application of α-GA (25 μm) reduced the sustained component and increased input resistance approximately 2.7-fold. This compound is reported to be a potent gap junction inhibitor (Taylor et al. 1998; Guo et al. 1999). In mesenteric arterioles, β-GA (an isomer of α-GA) has been reported to block the bradykinin-induced hyperpolarization of smooth muscle cells, which is postulated to be due to electrical spread from endothelium to smooth muscle (Yamamoto et al. 1999). Likewise, in the present study, α-GA (25 μm) inhibited the bradykinin-induced hyperpolarization of smooth muscle cells within rat cerebral arterioles (control, hyperpolarization by 17.8 ± 7.7 mV (n = 4); α-GA, depolarization by 11.6 ± 8.9 mV (n = 4); P < 0.05). The data suggest that these compounds are capable of inhibiting electrical coupling between endothelial cells and smooth muscle cells. In most experiments in the present study, therefore, we reduced cell-to-cell electrical coupling by exposing the preparation to α-GA (25 μm) throughout the experiment. Diffusion-mediated intercellular responses were presumed to be intact during such a blockade of gap junctions.
Oscillatory response to ET-1 shown by smooth muscle cells in intact arterioles
Figure 1 depicts the effects of ET-1 (50 nm) on the membrane potential recorded in current-clamp mode and on the membrane current recorded in voltage-clamp mode, both in α-GA-treated smooth muscle cells. Application of ET-1 depolarized the membrane by 11.3 mV (Fig. 1A), an effect that lasted for at least 200 s. The mean membrane potentials obtained from six cells were −29.1 ± 1.8 mV in the absence of ET-1 and −19.4 ± 2.4 mV in its presence (n = 6, P < 0.01). Concomitantly, in all cells tested the membrane potential exhibited frequent transient depolarizations, each followed by a rapid repolarization (Fig. 1A). In the record shown in Fig. 1A, this oscillation had a maximum amplitude of 16.9 mV and a frequency of 0.48 Hz.
Figure 1. ET-1-induced oscillatory depolarization and membrane currents in single smooth muscle cells within rat intact cerebral arterioles.
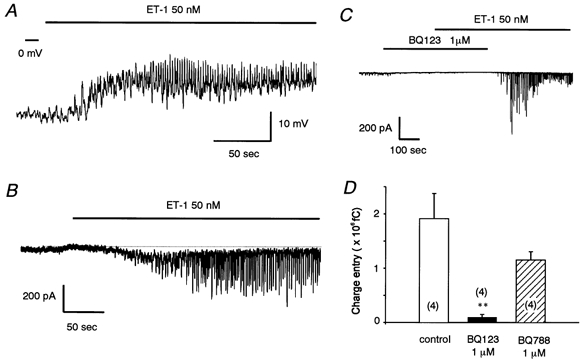
A, ET-1 (50 nm) evoked dual effects on membrane potential in current-clamp mode. An oscillatory change in potential is superimposed on a long-lasting depolarization. B, ET-1 (50 nm) evoked dual effects on membrane currents in voltage-clamp mode (holding potential, −50 mV). Dotted line indicates the zero current level. C, inhibition of ET-1 (50 nm)-induced currents in the presence of the ETAR antagonist, BQ123 (1 μm). D, summary bar graph for the effects of BQ123 (1 μm) and BQ788 (1 μm). The preparation was perfused with the ET-1 antagonist prior to ET-1 (50 nm). Charge entry (Q) was calculated using the following equation: Q =∫I(t)dt, where I(t) is the current flowing at time t. Control data were obtained from the same batch of preparations. **P < 0.01versus control. α-GA (25 μm) was present throughout. Numbers in parentheses indicate the number of observations.
Figure 1B shows an example of a recording of membrane current at −50 mV under voltage-clamp conditions (obtained using a perforated-patch technique). ET-1 (50 nm) caused a slowly developing inward current, with a maximum of 77 pA occurring at 50 s after its onset. The current decayed with a slow time course and stabilized at 225 s or more after its onset. It did not return completely to the resting level as long as ET-1 was present. The mean amplitude of the long-lasting inward current was −4.8 ± 1.3 pA pF−1 at −50 mV, measured at 50 s after its onset (n = 5). Because of its small size (ca. 50 pA), we could not examine the ion selectivity of this current component in detail after distinguishing it from the repetitive component described below. As in the current-clamp situation, a repetitive pattern of inward deflections was observed at negative potentials in voltage-clamp mode. In some cells, the repetitive response was rhythmic and persistent (more than 40 min in duration), so that we had ample time accurately to determine the electrical and pharmacological profiles of the channels involved. Figure 1B depicts an example of such an oscillatory response, in this case with a frequency of 0.46 Hz and a current amplitude of approximately 338 pA. In five preparations, the mean amplitude of the current at −50 mV was −11.9 ± 2.5 pA pF−1 and its frequency was 0.36 ± 0.05 Hz (see Figs 2B and 3B).
Figure 2. I–V relationship and time constant of decay (τ) for the oscillatory currents evoked by ET-1 (50 nm).
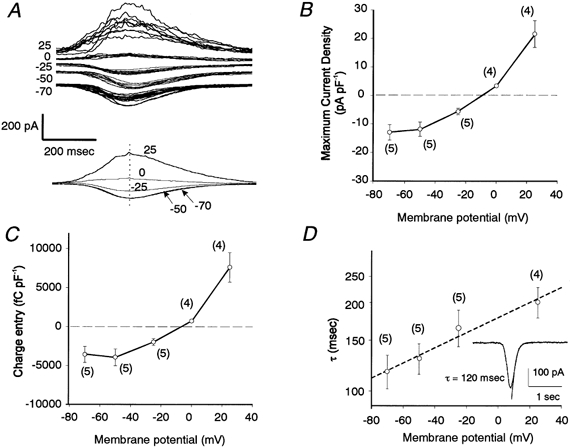
A, superimposed current traces (n = 5–12, upper traces) and averaged traces (lower traces) at the indicated membrane potentials (mV). Time to peak is matched for each trace. B and C, peak current density and charge entry during a single deflection, respectively. D, relationship between values of τ and membrane potential shown on a semi-logarithmic scale. Values on y-axis are shown as a linear sequence to facilitate understanding. The dashed line was drawn using least-squares linear regression analysis (slope = 0.0059, y intercept = 5.18, r = 0.38). Inset, a typical current trace obtained at a holding potential of −70 mV, together with the curve drawn by exponential fitting. α-GA (25 μm) was present throughout.
Figure 3. Voltage dependence of the frequency of the oscillatory currents evoked by ET-1 (50 nm).
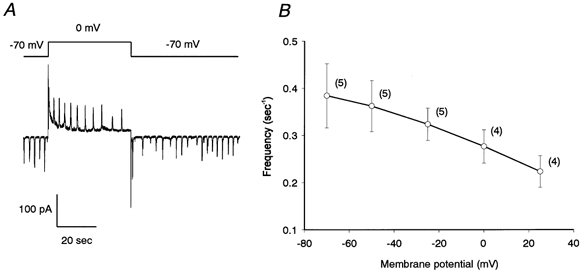
A, membrane potential affects both the direction of the oscillatory currents and their time interval. Note that the time interval becomes progressively longer (shorter) when the holding potential is switched to 0 mV (back to −70 mV). B, relationship between frequency (Hz) and membrane potential. α-GA (25 μm) was present throughout.
To eliminate the possibility that any side-effect of α-GA was involved in the response of these arterioles to ET-1, we tested another type of gap junction disrupter, Gap-27 peptide (Dora et al. 1999). This peptide increased the input resistance approximately 2.5-fold and ET-1 then evoked repetitive inward currents comparable to those seen in experiments performed with α-GA, the mean amplitude of the current at −50 mV being −12.7 ± 4.0 pA pF−1 (n = 3) and its frequency 0.46 ± 0.05 Hz (n = 3).
To try to determine the ET receptor subtype involved in the ET-1-induced oscillatory response, we used selective blockers (BQ-123 (1 μm) for the ETA receptor (ETAR) and BQ-788 (1 μm) for the ETB receptor (ETBR)). Summated charge entry (current amplitude multiplied by time, Q) was measured over a period of 100 s in the presence of ET-1 (50 nm) to enable us to compare the responses elicited with or without each blocker. Figure 1C depicts a current trace showing reversible blockade by BQ-123. Preincubation with BQ-123 prevented the action of subsequently applied ET-1, the response to ET-1 appearing only after washout of BQ-123. In three other cells, BQ-123 seemed to abolish the ET-1 response permanently, since it did not reappear even after washout of BQ-123. Summarized data (Fig. 1D) showed that BQ-123 significantly diminished the ET-1-induced oscillatory response, whereas BQ-788 only slightly, and non-significantly, reduced the response.
To examine whether or not the ET-1-evoked oscillatory response was highly selective for anions, the reversal potential was estimated using Cl−-deficient solutions containing one of several types of anion. In these, external Cl− was replaced entirely with Br−, I−, SCN− or methanesulphonate (MeS−). A voltage ramp (−100 to 70 mV, 1.4 V s−1) was applied every 1 s to cells treated with ET-1 (50 nm). Pronounced outwardly rectifying currents were obtained during the ramp when the oscillatory current at −50 mV was nearly at its peak, whereas only the background current was observed when the oscillatory current at −50 mV was at a minimum. The former appeared to include the ET-1-sensitive current component. Difference currents were then obtained by subtraction of the latter from the former, and the reversal potentials were estimated to be: Cl−, −7.0 ± 1.1 mV (n = 9); Br−, −26.3 ± 3.9 mV (n = 4); I−, −40.3 ± 1.4 mV (n = 4); SCN−, −60.5 ± 6.0 mV (n = 4) and MeS−, 59.4 ± 3.4 mV (n = 3). According to the Goldmann-Hodgkin-Katz equation, the rank order of permeabilities was SCN− (8.8) > I− (4.0) > Br− (2.3) > Cl− (1.0) > MeS− (0.08).
Characteristics of the ET-1-induced oscillatory Cl− current
Figure 2 shows an analysis of the current-voltage (I–V) relationship and time course of the oscillatory currents at several different holding potentials. Repetitively evoked currents were superimposed, with the times at which the maximum current occurred being matched (Fig. 2A, upper traces). Thereafter, pooled traces were averaged to obtain the mean amplitude and time course of decay (Fig. 2A, lower traces). The values obtained from four to five cells for maximum current density (current divided by membrane capacitance (Cm): I/Cm) were averaged and plotted as a function of membrane potential (Fig. 2B). Another way of expressing the voltage dependency of the current is to measure Q during each event. Pooled data for Q/Cm are plotted as a function of membrane potential in Fig. 2C. Both types of analysis revealed an outwardly rectifying property in the I–V relationship. The I/Cm values obtained for the oscillatory currents were −13.0 ± 2.7 pA pF−1 at −70 mV (n = 5) and 21.6 ± 4.7 pA pF−1 at +25 mV (n = 4) (Fig. 2B) and the reversal potential was estimated to be −10.5 ± 1.8 mV (n = 4). Similarly, the Q/Cm values were −3583 ± 1023 fC pF−1 at −70 mV (n = 5) and 7606 ± 1896 fC pF−1 at +25 mV (n = 4) (Fig. 2C), with an estimated reversal potential of −9.5 ± 3.1 mV (n = 4).
The currents observed at −50 and −70 mV resembled each other in both their developing and decaying phases, although at +25 mV both phases seemed to exhibit a slower time course (Fig. 2A). Analysis of the decaying kinetic of the current showed it to fit an exponential function. When the resultant time constant (τ) values were plotted as a function of membrane potential on a semi-logarithmic scale, a voltage dependency of τ was seen at between −70 mV and +25 mV, the values of τ being greater at depolarized potentials (Fig. 2D). The relationship was exponential since the semi-logarithmic plots were fitted by a linear regression line, described by the equation lnτ= lnτ0+V/h (where τ and τ0 are the time constants at potentials of V and 0 mV, respectively, and h is a constant expressing the voltage dependency of τ). This analysis indicates that τ increases e-fold for a change in membrane potential of 169 mV and that the τ0 value is 177 ms.
Next, we examined the voltage dependency of the oscillatory frequency of the ET-1-induced currents. To perform this analysis, we used only cells in which the current oscillated in a regular fashion. As shown in Fig. 3A, oscillatory inward currents were observed at −70 mV in the presence of ET-1 (50 nm); then, after the voltage was switched to 0 mV, an oscillating current in the opposite direction was superimposed on a slowly declining outward current. The interval increased progressively over the 50 s or so after the voltage change. Then, when the membrane potential was switched back to −70 mV, oscillatory inward currents, although of low frequency initially, returned to the original frequency within 60 s. For quantitative measurements, the membrane potential was initially held at −50 mV; thereafter, several levels of voltage were applied stepwise to cells pretreated with ET-1 (50 nm). Grouped data for the frequency of the currents observed during the steady state are summarized in Fig. 3B. The frequency of the ET-1-induced oscillatory currents was 0.38 ± 0.07 (n = 5) and 0.22 ± 0.03 Hz (n = 4) at −70 and 25 mV, respectively. The voltage dependency of the oscillatory frequency was clearly seen over the range illustrated (between −70 and +25 mV), with the frequency decreasing as the membrane potential was shifted in the positive direction.
Since the channels mediating the oscillatory response to ET-1 are Cl− dependent and because Ca2+-activated Cl− channels have been reported to play a role in the regulation of vascular tone (Large & Wang, 1996), we next examined whether such Cl− channels might be involved in this phenomenon. Figure 4A demonstrates that niflumic acid (100 μm), which is known to block ICl(Ca), inhibited the ET-1-induced oscillatory response. After washout of this compound, the effect of ET-1 was restored. To investigate whether generation of the current (probably ICl(Ca)) requires intracellular Ca2+ stores, the effects of caffeine, Ruthenium Red, thapsigargin and xestospongin C were examined. In fact, all of these compounds proved able to reduce the oscillatory activity induced by ET-1, suggesting the involvement of internal Ca2+ stores (Fig. 4B). We next observed the ET-1 response using lower concentrations of external Ca2+. When external Ca2+ was reduced from 2 to 1 mm and then to 0.2 mm, the current-inducing effect of ET-1 became progressively weaker (Fig. 4B). Therefore, external Ca2+ would seem to be needed to maintain the oscillatory response.
Figure 4. Blockade of ET-1-evoked oscillatory currents by a Cl− channel blocker and Ca2+ store modulators.
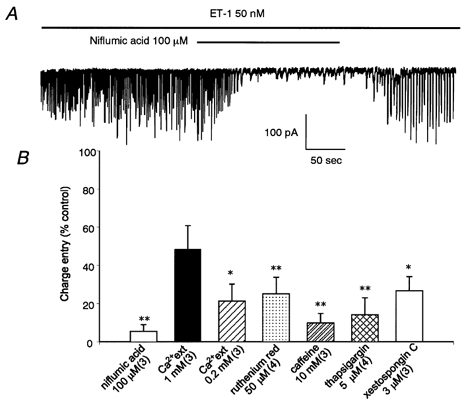
A, niflumic acid (100 μm) abolished the response to ET-1 (50 nm), which was restored after washout of the blocker. Holding potential was −50 mV. B, summary graph for blockade of ET-1-induced oscillatory currents by Ca2+ store modulators and low external Ca2+ (Ca2+ext) concentration. These interventions were performed during the period in which the oscillatory response to ET-1 (50 nm) was at its maximum. Summated charge entry (over 100 s) is normalized with respect to the value obtained before a given intervention ([Ca2+ext]= 2 mm). α-GA (25 μm) was present throughout. *P < 0.05, **P < 0.01versus control value (100 %).
Modulation of the ET-1-induced oscillatory response by bradykinin (through diffusible factors)
We previously reported that α-GA abolishes the bradykinin-induced hyperpolarization of smooth muscle cells (Yamazaki & Kitamura, 2001). This raised the possibility that the electrical response spreads from endothelial cells to neighbouring smooth muscle cells. In Fig. 5A, bradykinin (10 μm) hyperpolarized the membrane of a smooth muscle cell under current-clamp conditions in the absence of α-GA (see Fig. 5Aa and Ab). No regular oscillatory response could be seen during these periods. After the potential had returned almost to the resting level, ET-1 (50 nm) was applied in the continued presence of bradykinin. ET-1 failed to evoke an oscillatory response while the two were present together (Fig. 5Ac), but the characteristic oscillatory response to ET-1 began to appear after the washout of bradykinin (Fig. 5Ad).
Figure 5. ET-1-evoked oscillatory response is modified by intercellular communication between smooth muscle and endothelial cells.
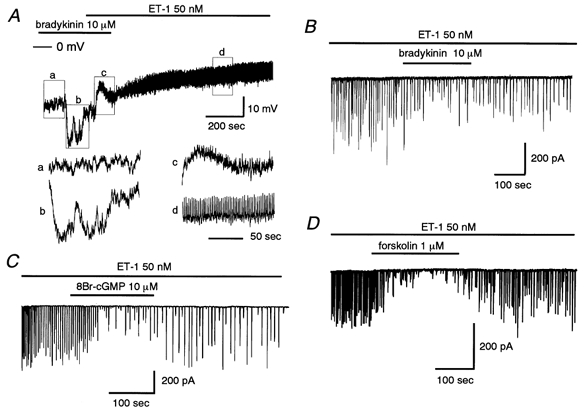
A, hyperpolarizing effect of bradykinin (10 μm) on membrane potential recorded from a smooth muscle cell in the absence of α-GA (25 μm). Expanded sections (a–d) of the trace are also shown. In the continued presence of bradykinin, ET-1 (50 nm) failed to evoke an oscillation of the membrane potential (c), although this response appeared after bradykinin withdrawal (d). B–D, effects of bradykinin (B), 8Br-cGMP (C) and forskolin (D) on ET-1 (50 nm)-induced oscillatory currents at −50 mV. α-GA (25 μm) was present throughout.
The channels involved in the oscillatory action of ET-1 were not blocked within the physiological range of membrane potentials (−50 to −20 mV, see Fig. 2). Thus, we speculated that the inhibitory action of bradykinin on the oscillatory current is not due solely to electrical communication between cells, but at least in part to some mediator(s) transferred from endothelial cells to the smooth muscle cell from which the recording was being made. To eliminate the influence of changes in the membrane potential, the membrane current was measured in voltage-clamp mode. In three of six preparations, bradykinin (10 μm) attenuated the oscillatory ICl(Ca) evoked by ET-1 in the presence of α-GA at a holding potential of −50 mV (Fig. 5B). In the other three preparations, bradykinin initially increased the current amplitude with a reduced oscillation frequency, then progressively suppressed the current amplitude (during a 5–7 min perfusion with bradykinin). Because the regular pattern of the oscillations was mostly lost in the presence of bradykinin, we could not quantify any change in the frequency of the current deflections. Therefore, we calculated total charge entry over 100 s during the inhibitory action of bradykinin, as well as before the drug was applied, and then obtained normalized values (Fig. 6). Bradykinin reduced the charge entry to 22.0 ± 4.0 % of control (n = 6, P < 0.01) and this inhibitory action was significantly attenuated by an NO-synthase inhibitor, Nω-nitro-l-arginine (NNA, 30 μm), but not by a cyclo-oxygenase inhibitor, indomethacin (5 μm) (Fig. 6). Perfusion with HbO2 (30 μm), which is known to scavenge NO or oxidize NO to NO2 outside cells, significantly attenuated the bradykinin effect (Fig. 6). On their own, these inhibitors failed to alter membrane currents in the presence or absence of ET-1 (data not shown).
Figure 6. Summary bar graph showing blockade of the ET-1-induced response by bradykinin and activators of various second messenger pathways.
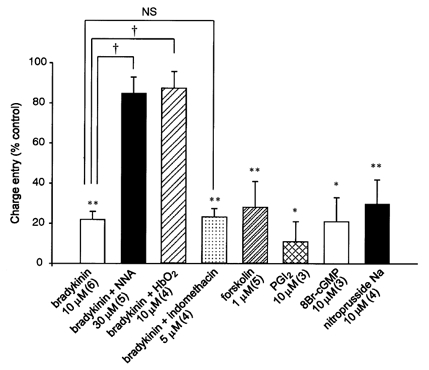
Summated charge entry (over 100 s) is normalized with respect to the value obtained before a given intervention. α-GA (25 μm) was present throughout. *P < 0.05, **P < 0.01versus control value (100 %). †P < 0.001versus bradykinin-treated group. NS, not significant.
To further test for the involvement of a diffusible factor acting on a second messenger pathway, we examined the effect of an endothelium-derived factor (or its donor). For this, we used sodium nitroprusside and PGI2. As shown in Fig. 6, both compounds inhibited the ET-1-evoked oscillatory response. Furthermore, an activator of protein kinase G (PKG), 8-Br-cGMP, and an adenylate-cyclase (AC) activator, forskolin, closely mimicked the actions of PGI2 and sodium nitroprusside. These results indicate that different diffusible factors and the corresponding second messenger pathways can be utilized for modulation of the ET-1-induced response in smooth muscle cells. These data are summarized in Fig. 6, which shows that normalized charge entry was significantly attenuated by forskolin, PGI2, 8Br-cGMP and nitroprusside. Taken together with the result obtained with bradykinin, these observations suggest that the NO-cGMP pathway is the main pathway involved in the bradykinin-induced inhibition of the oscillatory response to ET-1, although stimulation of a PGI2-cAMP pathway is involved as well.
DISCUSSION
The major new findings made in the present study were that in rat cerebral arteriolar smooth muscle cells, ET-1 evoked an oscillatory ICl(Ca) and a resulting fluctuation in the membrane potential and that this response was modulated by NO, which may have been released from adjacent endothelial cells. An illustration of our proposed schema is shown in Fig. 7. The preparation employed in the present study, with cells remaining in situ in the arteriolar wall, allowed us to observe and examine the diffusion-mediated changes in the electrical response induced by a vasoconstrictor in smooth muscle cells.
Figure 7. Schematic illustration showing possible mechanisms for the ET-1-evoked oscillatory ICl(Ca) and for NO-mediated intercellular communication between smooth muscle and endothelial cells.
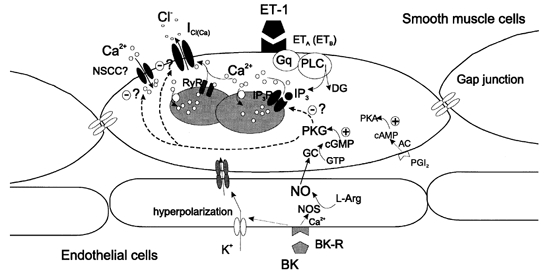
PLC, phospholipase C; Gq, GTP-binding protein; DG, diacylglycerol; NOS, NO synthase; BK, bradykinin; BK-R, bradykinin receptor(s). The PGI2-PKA pathway is unlikely to be involved in the regulation of ICl(Ca) by BK, even though forskolin and PGI2 are capable of modifying ICl(Ca). Question marks denote an undefined target for phosphorylation by PKG as part of the action of BK.
In order to reduce electrical coupling between arteriolar-wall cells, α-GA was used in most of the present experiments. In a recent publication, Coleman et al. (2001) asserted that the effects of GA compounds (α-GA, β-GA and carbenoxolone) are far less than would be required to provide definitive evidence for the involvement of electrical coupling. Among the compounds these authors tested, α-GA was shown to be the least non-selective, although its potency seemed to be limited at a concentration of 50 μm. Nevertheless, the concentrations of α-GA used in the present experiments (25 μm) and in previous studies (50–100 μm) (Taylor et al. 1998; Chaytor et al. 1999) are likely to be high enough effectively to block gap junctions since they are higher than that used in a biochemical study measuring the transfer of citrulline metabolites (Davidson et al. 1986) in which α-GA was shown to inhibit more than 95 % of gap junctions at only 2 μm. We think that α-GA is one of the gap junction inhibitors currently available that is worth testing since: (i) its half-toxic concentration has been reported to be as high as 100 μm (Davidson et al. 1986), (ii) α-GA (25 μm) left resting current or conductance unchanged in isolated rat cerebral arterial smooth muscle cells (n = 4, data not shown), and (iii) another type of gap junction disrupter, Gap-27 peptide mimicked the effect of α-GA.
ET-1-induced ICl(Ca) in rat intact cerebral arterioles
ICl(Ca) has been shown to regulate membrane excitability in vascular smooth muscle cells (for review see Large & Wang, 1996) and to be activated by several vasoactive compounds in a variety of smooth muscle cells (Pacaud et al. 1989; Wang & Large, 1991; Salter & Kozlowski, 1996; Kamouchi et al. 1997; Wang et al. 1997). In the present study, characterization of the oscillatory current revealed that it was entirely of Cl− channel origin and that it was modulated by [Ca2+]i. The oscillatory current appears to be derived entirely from a single type of ion conductance because of the apparent lack of more than a single current component at any holding potential, to judge from its reversal potential. The estimated ion permeabilities for this current gave the rank order SCN− > I− > Br− > Cl− > MeS−, similar to those obtained for histamine- or noradrenaline-induced Cl− currents in different types of smooth muscle cells (Wang & Large 1991, 1993). Intracellular Ca2+ was found to be a prerequisite for producing the Cl− conductance because experimental manipulations that would modify the internal Ca2+ stores all diminished the oscillatory response. Moreover, the current was blocked by niflumic acid, which is a potent inhibitor of ICl(Ca) (for review see Large & Wang, 1996). Taken together, these findings indicate that ICl(Ca) plays the major role in the ET-1-induced oscillatory changes in membrane current and potential.
Modulation of ICl(Ca) by diffusion of NO within arterioles
Of particular interest is the indication in the present study that bradykinin may inhibit the ET-1-evoked oscillatory ICl(Ca) through NO production. A large amount of evidence has accumulated to indicate that bradykinin releases NO and/or PGI2 from endothelial cells and thus decreases vascular tone (D'Orléans-Juste et al. 1989; Frantz et al. 1989; Gambone et al. 1997; Theis et al. 1998; Wahl et al. 1999). Initially, either of these mediators could have been a candidate for involvement in the bradykinin-induced inhibition of the ET-1 response since both PGI2 and an NO donor, nitroprusside (and also forskolin and 8-Br-cGMP) attenuated the ET-1-evoked oscillatory ICl(Ca). However, an inhibitor of NO synthase, NNA, and an NO scavenger, HbO2, but not a cyclo-oxygenase inhibitor, indomethacin, inhibited the effect of bradykinin. On this basis, it is most likely that the diffusible NO-guanylate cyclase (GC)-cGMP-PKG pathway participates in the bradykinin-induced inhibition of the ET-1-evoked oscillatory ICl(Ca). Bradykinin-induced hyperpolarization of endothelial cells has been postulated to propagate electrically to smooth muscle cells through myoendothelial gap junctions, which may account for the action of so-called endothelial-derived hyperpolarizing factor (EDHF) (Yamamoto et al. 1999; Edwards et al. 2000). This mechanism is unlikely to be involved in the present phenomenon since we also observed that bradykinin attenuated the oscillatory response in the presence of α-GA under voltage-clamp conditions. Because of its highly lipophilic nature, NO would diffuse so rapidly through cell membranes from endothelial to smooth muscle cells that it would be an effective intercellular messenger even after blockade of gap junctions. Currently, it is uncertain which type of protein is the target for phosphorylation by PKG (Fig. 7), although there are several possibilities: (1) ICl(Ca) channels, (2) proteins involved in the Ca2+ release and uptake mechanisms, and (3) sarcolemmal Ca2+-permeable channels, which serve to refill the Ca2+ stores (see the paragraphs below). It remains to be determined which is the most likely mechanism.
Voltage dependence of ICl(Ca)
The present study revealed several other biophysical characteristics of the oscillatory ICl(Ca). The I–V relationship displayed outward rectification with the holding potential set at a variety of voltages. It has been shown that the I–V relationship for ICl(Ca) is mostly linear when the channel is activated by receptor agonists or by Ca2+ photolytically released from a caged compound (Wang & Large, 1991; Pacaud et al. 1992; Clapp et al. 1996). The voltage dependence of the Cl− conductance has, however, also been reported to involve an increase in mean open time at depolarized potentials (Hogg et al. 1993). Large & Wang (1996) explained these seemingly contradictory results by supposing that this type of voltage dependency is manifest if a small fraction of the channels are open, but not if a large fraction are open.
Results obtained from an analysis of the kinetics of decay also support the voltage dependency of this channel. Spontaneous activity of ICl(Ca), termed spontaneous transient inward currents (STICs), has been documented in several papers (Wang et al. 1992; Hogg et al. 1993, 1994). Localized subsarcolemmal Ca2+ signals (Ca2+ sparks) are known to trigger STICs, although the time course of the local [Ca2+]i increase is faster than that of STICs (Hogg et al. 1993; Nelson et al. 1995). This argues for the notion that the decay of STICs is determined by a factor other than intracellular Ca2+ mobilization (i.e. the closing kinetics of ICl(Ca)). STIC decay has been shown to be fitted monoexponentially with a single τ value, which is indeed similar to the mean open time estimated both from noise analysis (Evans & Marty, 1986) and from single channel recordings (Taleb et al. 1988). The τ values reported previously were 80–100 ms at −50 mV, values consistent with that found for the oscillatory response to ET-1 analysed in the present study. The similar voltage dependence of τ in the case of STICs and the oscillatory ICl(Ca) seems to confirm the close correlation between the channel types involved in these two phenomena.
Utilization of internal and external Ca2+ for ET-1-evoked ICl(Ca)
The present results obtained using specific antagonists for ET receptor types suggest that stimulation mainly of ETARs, and to a lesser extent ETBRs, generates the oscillatory ICl(Ca). Such ET receptor stimulation is generally believed to be linked both to IP3 production (Sugiura et al. 1989; Kasuya et al. 1989) and to Ca2+ mobilization (van Renterghem et al. 1988; Marsden et al. 1989). In fact, the present results suggest that the ET-1-induced oscillatory ICl(Ca) requires both IP3-induced Ca2+ release (IICR) and the Ca2+-uptake mechanism since xestospongin C and thapsigargin both decreased the current. Hyvelin et al. (1998) postulated that the oscillatory pattern of the ET-1-induced Ca2+ response could be related to cyclic Ca2+ release via the IICR channel. Such cyclic Ca2+ release could be due to a biphasic Ca2+ regulation of IP3 receptor (IP3R) activity in smooth muscle cells (Iino, 1990). Additional Ca2+-releasing mechanisms are also likely to help mediate the present phenomenon because the ET-1-induced response was abolished by both caffeine and Ruthenium Red. The ryanodine receptor (RyR) and the Ca2+-induced Ca2+ release (CICR) mechanism are known to contribute to the generation of ICl(Ca) in various types of vascular smooth muscle (Klöckner & Isenberg, 1991; Wang & Large, 1991; 1993; Lamb et al. 1994). Kannan et al. (1997) proposed that while Ca2+ release through the IP3R channel is essential for the initiation of oscillations, Ca2+ release through the ryanodine receptor (RyR) channel is the primary source of Ca2+ oscillations. If this is so, the IP3R and RyR channels are likely to function synergistically to enable a rhythmic pattern of Ca2+ release to be induced by ET-1 (Fig. 7).
Since reducing the extracellular Ca2+ was found to attenuate ICl(Ca), external Ca2+ appears to be required to replenish the internal Ca2+ stores (Amédeéet al. 1990; Wang et al. 1992; Kamouchi et al. 1997; Kannan et al. 1997). Mere recruitment of the released Ca2+ back into the stores may not be sufficient to maintain the oscillatory Ca2+ response since a significant proportion of the Ca2+ released from the internal stores is reported to be removed to the external space (Tepikin et al. 1992). Thus, Ca2+ influx is likely to be important in setting the periodicity of the Ca2+ oscillations. This influx is likely to be accounted for by some of the ET-1-activated Ca2+ entry pathways, such as the non-specific cation channel (NSCC, Salter & Kozlowski, 1998) or the receptor-operated Ca2+-permeable channel (ROCC, Guibert & Beech, 1999). If so, the Ca2+ stores should be more easily replenished at more negative potentials, at which the electrochemical gradient for Ca2+ entry is greater (Wang et al. 1992). This prediction is in good agreement with the present finding that the ET-1-induced oscillatory response has a higher frequency at more negative potentials (Fig. 3).
Pathophysiological implications of communication between endothelium and smooth muscle
The plasma and cerebrospinal fluid levels of ET-1 are known to increase in patients with an SAH (Seifert et al. 1995; Pluta et al. 1997). Furthermore, ET receptor antagonists and ET-converting enzyme inhibitors have both been shown to prevent cerebral vasospasm after SAH (Caner et al. 1996; Zuccarello et al. 1996). Hence, a possible role for ET has been proposed in the pathogenesis of cerebral vasospasm in SAH patients (Sobey & Faraci, 1998). However, a contradictory conclusion has been reached with regard to the possible relationship between the ET level and vasospasm (Seifert et al. 1995; Pluta et al. 1997). Pluta et al. (1997) hypothesized that a lack of availability of endothelial NO allows ET-induced vasospasm to develop after SAH even without a marked increase in ET production during the vasospasm. This scenario is in good agreement with the present argument for NO-mediated intercellular communication between smooth muscle and endothelial cells.
It has been shown that the perivascular concentration of HbO2 is increased 100-fold after SAH (Pluta et al. 1998). We observed in the present study that the inhibitory action of bradykinin was attenuated by perfusion with HbO2. This seems to raise the possibility that HbO2 released from haemolysed erythrocytes may be responsible for the lack of availability of diffusible NO. It would be fascinating to examine whether ET-1- or haemorrhage-evoked constriction of an arteriolar preparation is indeed modified by NO-releasing agents. If the oscillatory current evoked by ET-1 leads to a deterioration of the cerebral circulation, then the present results may represent an important therapeutic target for the treatment of cerebral vasoconstriction. Candidates for the therapeutic agent might be an activator of the NO-PKG pathway, such as an intercellular messenger utilized during the inhibitory action of bradykinin, or an activator of the PGI2-protein kinase A pathway, which is unlikely to be an endogenous messenger in the present preparation.
Acknowledgments
We thank Dr R. Timms for editing the English. This work was supported by a grant-in-aid from the Ministry of Education, Science, Sports and Culture of Japan (Frontier Research Grant), a grant-in-aid for Scientific Research from the Japan Society for the Promotion of Science and by the Uehara Memorial Foundation.
References
- Amédeé T, Large WA, Wang Q. Characteristics of chloride currents activated by noradrenaline in rabbit ear artery cells. Journal of Physiology. 1990;428:501–516. doi: 10.1113/jphysiol.1990.sp018224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caner HH, Kwan A-L, Arthur A, Jeng AY, Lappe RW, Kassell NF, Lee KS. Systemic administration of an inhibitor of endothelin-converting enzyme for attenuation of cerebral vasospasm following experimental subarachnoid hemorrhage. Journal of Neurosurgery. 1996;85:917–922. doi: 10.3171/jns.1996.85.5.0917. [DOI] [PubMed] [Google Scholar]
- Chaytor AT, Martin PEM, Evans WH, Randall MD, Griffith TM. The endothelial component of cannabinoid-induced relaxation in rabbit mesenteric artery depends on gap junctional communication. Journal of Physiology. 1999;520:539–550. doi: 10.1111/j.1469-7793.1999.00539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapp LH, Turner JL, Kozlowski RZ. Ca2+-activated Cl− currents in pulmonary arterial myocytes. American Journal of Physiology. 1996;270:H1577–1584. doi: 10.1152/ajpheart.1996.270.5.H1577. [DOI] [PubMed] [Google Scholar]
- Coleman HA, Tare M, Parkington HC. K+ current underlying the action of endothelium-derived hyperpolarizing factor in guinea-pig, rat and human blood vessels. Journal of Physiology. 2001;531:359–373. doi: 10.1111/j.1469-7793.2001.0359i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson JS, Baumgarten IM, Harley EH. Reversible inhibition of intercellular junctional communication by glycyrrhetinic acid. Biochemical and Biophysical Research Communications. 1986;134:29–36. doi: 10.1016/0006-291x(86)90522-x. [DOI] [PubMed] [Google Scholar]
- Dora KA, Martin PEM, Chaytor AT, Evans WH, Garland CJ, Griffith TM. Role of heterocellular gap junctional communication in endothelium-dependent smooth muscle hyperpolarization: inhibition by a connexin-mimetic peptide. Biochemical and Biophysical Research Communications. 1999;254:27–31. doi: 10.1006/bbrc.1998.9877. [DOI] [PubMed] [Google Scholar]
- D'Orléans-Juste Pde, Nucci G, Vane JR. Kinins act on B1 or B2 receptors to release conjointly endothelium-derived relaxing factor and prostacyclin from bovine aortic endothelial cells. British Journal of Pharmacology. 1989;94:920–926. doi: 10.1111/j.1476-5381.1989.tb11903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Thollon C, Gardener MJ, Félétou M, Vilaine J-P, Vanhoutte PM, Weston AH. Role of gap junctions and EETs in endothelium-dependent hyperpolarization of porcine coronary artery. British Journal of Pharmacology. 2000;29:1145–1154. doi: 10.1038/sj.bjp.0703188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MG, Marty A. Calcium-dependent chloride currents in isolated cells from lacrimal glands. Journal of Physiology. 1986;378:437–460. doi: 10.1113/jphysiol.1986.sp016229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraci FM, Heistad DD. Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiological Reviews. 1998;78:53–97. doi: 10.1152/physrev.1998.78.1.53. [DOI] [PubMed] [Google Scholar]
- Frantz E, Soifer SJ, Clyman RI, Heymann MA. Bradykinin produces pulmonary vasodilation in fetal lambs: role of prostaglandin production. Journal of Applied Physiology. 1989;67:1512–1517. doi: 10.1152/jappl.1989.67.4.1512. [DOI] [PubMed] [Google Scholar]
- Gambone LM, Murray PA, Flavahan NA. Synergistic interaction between endothelium-derived NO and prostacyclin in pulmonary artery: potential role for K+ATP channels. British Journal of Pharmacology. 1997;121:271–279. doi: 10.1038/sj.bjp.0701082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guibert C, Beech DJ. Positive and negative coupling of the endothelin ETA receptor to Ca2+-permeable channels in rabbit cerebral cortex arterioles. Journal of Physiology. 1999;514:843–856. doi: 10.1111/j.1469-7793.1999.843ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Martinez-Williams C, Gilbert KA, Rannels DE. Inhibition of gap junction communication in alveolar epithelial cells by 18α-glycyrrhetinic acid. American Journal of Physiology. 1999;276:L1018–1026. doi: 10.1152/ajplung.1999.276.6.L1018. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Wang Q, Large WA. Time course of spontaneous calcium-activated chloride currents in smooth muscle cells from the rabbit portal vein. Journal of Physiology. 1993;464:15–31. doi: 10.1113/jphysiol.1993.sp019622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg RC, Wang Q, Large WA. Effects of Cl channel blockers on Ca-activated chloride and potassium currents in smooth muscle cells from rabbit portal vein. British Journal of Pharmacology. 1994;111:1333–1341. doi: 10.1111/j.1476-5381.1994.tb14891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyvelin J-M, Guibert C, Marthan R, Savineau J-P. Cellular mechanisms and role of endothelin-1-induced calcium oscillations in pulmonary arterial myocytes. American Journal of Physiology. 1998;275:L269–282. doi: 10.1152/ajplung.1998.275.2.L269. [DOI] [PubMed] [Google Scholar]
- Iino M. Biphasic Ca2+ dependence of inositol 1,4,5-triphosphate-induced Ca release in smooth muscle cells of the guinea pig taenia caeci. Journal of General Physiology. 1990;95:1103–1122. doi: 10.1085/jgp.95.6.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamouchi M, Ogata R, Fujishima M, Ito Y, Kitamura K. Membrane currents evoked by histamine in rabbit basilar artery. American Journal of Physiology. 1997;272:H638–647. doi: 10.1152/ajpheart.1997.272.2.H638. [DOI] [PubMed] [Google Scholar]
- Kannan MS, Prakash YS, Brenner T, Mickelson JR, Sieck GC. Role of ryanodine receptor channels in Ca2+ oscillations of porcine tracheal smooth muscle. American Journal of Physiology. 1997;272:L659–664. doi: 10.1152/ajplung.1997.272.4.L659. [DOI] [PubMed] [Google Scholar]
- Kasuya Y, Takuwa Y, Yanagisawa M, Kimura S, Goto K, Masaki T. Endothelin-1 induces vasoconstriction through two functionally distinct pathways in porcine coronary artery: contribution of phosphoinositide turnover. Biochemical and Biophysical Research Communications. 1989;161:1049–1055. doi: 10.1016/0006-291x(89)91349-1. [DOI] [PubMed] [Google Scholar]
- Klöckner U, Isenberg G. Endothelin depolarizes myocytes from porcine coronary and human mesenteric arteries through a Ca-activated chloride current. Pflügers Archiv. 1991;418:168–175. doi: 10.1007/BF00370467. [DOI] [PubMed] [Google Scholar]
- Lamb FS, Volk KA, Shibata EF. Calcium-activated chloride current in rabbit coronary artery myocytes. Circulation Research. 1994;75:742–750. doi: 10.1161/01.res.75.4.742. [DOI] [PubMed] [Google Scholar]
- Large WA, Wang Q. Characteristics and physiological role of the Ca2+-activated Cl− conductance in smooth muscle. American Journal of Physiology. 1996;271:C435–454. doi: 10.1152/ajpcell.1996.271.2.C435. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- Marzden PA, Danthuluri NR, Brenner BM, Ballermann BJ, Brock TA. Endothelin action on vascular smooth muscle involves inositol triphosphate and calcium mobilization. Biochemical and Biophysical Research Communications. 1989;158:86–93. doi: 10.1016/s0006-291x(89)80180-9. [DOI] [PubMed] [Google Scholar]
- Pacaud P, Loirand G, Grégoire G, Mironneau C, Mironneau J. Calcium-dependence of the calcium-activated chloride current in smooth muscle cells of rat portal vein. Pflügers Archiv. 1992;421:125–130. doi: 10.1007/BF00374818. [DOI] [PubMed] [Google Scholar]
- Pacaud P, Loirand G, Lavie JL, Mironneau C, Mironneau J. Calcium-activated chloride current in rat vascular smooth muscle cells in short-term primary culture. Pflügers Archiv. 1989;413:629–636. doi: 10.1007/BF00581813. [DOI] [PubMed] [Google Scholar]
- Pluta RM, Afshar JK, Boock RJ, Oldfield EH. Temporal changes in perivascular concentrations of oxyhemoglobin, deoxyhemoglobin, and methemoglobin after subarachnoid hemorrhage. Journal of Neurosurgery. 1998;88:557–561. doi: 10.3171/jns.1998.88.3.0557. [DOI] [PubMed] [Google Scholar]
- Pluta RM, Boock RJ, Afshar JK, Clouse K, Bacic M, Ehrenreich H, Oldfield EH. Source and cause of endothelin-1 release into cerebrospinal fluid after subarachnoid hemorrhage. Journal of Neurosurgery. 1997;87:287–293. doi: 10.3171/jns.1997.87.2.0287. [DOI] [PubMed] [Google Scholar]
- Quinn K, Beech DJ. A method for direct patch-clamp recording from smooth muscle cells embedded in functional brain microvessels. Pflügers Archiv. 1998;435:564–569. doi: 10.1007/s004240050553. [DOI] [PubMed] [Google Scholar]
- Salter KJ, Kozlowski RZ. Endothelin receptor coupling to potassium and chloride channels in isolated rat pulmonary arterial myocytes. Journal of Pharmacology and Experimental Therapeutics. 1996;279:1053–1062. [PubMed] [Google Scholar]
- Salter KJ, Kozlowski RZ. Differential electrophysiological actions of endothelin-1 on Cl− and K+ currents in myocytes isolated from aorta, basilar and pulmonary artery. Journal of Pharmacology and Experimental Therapeutics. 1998;284:1122–1131. [PubMed] [Google Scholar]
- Seifert V, Löffler B-M, Zimmermann M, Roux S, Stolke D. Endothelin concentrations in patients with aneurysmal subarachnoid hemorrhage. Correlation with cerebral vasospasm, delayed ischemic neurological deficits, and volume of hematoma. Journal of Neurosurgery. 1995;82:55–62. doi: 10.3171/jns.1995.82.1.0055. [DOI] [PubMed] [Google Scholar]
- Sobey CG, Faraci FM. Subarachnoid haemorrhage: what happens to the cerebral arteries? Clinical and Experimental Pharmacology and Physiology. 1998;25:867–876. doi: 10.1111/j.1440-1681.1998.tb02337.x. [DOI] [PubMed] [Google Scholar]
- Sugiura M, Inagaki T, Hare GMT, Johns JA. Endothelin action: inhibition by a protein kinase C inhibitor and involvement of phosphoinositols. Biochemical and Biophysical Research Communications. 1989;158:170–176. doi: 10.1016/s0006-291x(89)80193-7. [DOI] [PubMed] [Google Scholar]
- Taleb O, Feltz P, Bossu JL, Feltz A. Small-conductance chloride channels activated by calcium on cultured endocrine cells from mammalian pars intermedia. Pflügers Archiv. 1988;412:641–646. doi: 10.1007/BF00583766. [DOI] [PubMed] [Google Scholar]
- Tare M, Parkington HC, Coleman HA, Neild TO, Dusting GJ. Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature. 1990;346:69–71. doi: 10.1038/346069a0. [DOI] [PubMed] [Google Scholar]
- Taylor HJ, Chaytor AT, Evans WH, Griffith TM. Inhibition of the gap junctional component of endothelium-dependent relaxations in rabbit iliac artery by 18-α glycyrrhetinic acid. British Journal of Pharmacology. 1998;125:1–3. doi: 10.1038/sj.bjp.0702078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis JG, Toyoda O, Coceani F. Effect of endothelium removal on prostaglandin and nitric oxide function in pulmonary resistance arteries in the lamb. Canadian Journal of Physiology and Pharmacology. 1998;76:182–187. [PubMed] [Google Scholar]
- Tepikin AV, Voronina SG, Gallacher DV, Petersen OH. Pulsatile Ca2+ extrusion from single pancreatic acinar cells during receptor-activated cytosolic Ca2+ spiking. Journal of Biological Chemistry. 1992;267:14073–14076. [PubMed] [Google Scholar]
- Van Renterghem C, Vigne P, Barhanin J, Schmid-Alliana A, Frelin C, Lazdunski M. Molecular mechanism of action of the vasoconstrictor peptide endothelin. Biochemical and Biophysical Research Communications. 1988;157:977–985. doi: 10.1016/s0006-291x(88)80970-7. [DOI] [PubMed] [Google Scholar]
- Wahl M, Gorlach C, Hortobagyi T, Benyo Z. Effects of bradykinin in the cerebral circulation. Acta Physiologica Hungarica. 1999;86:155–160. [PubMed] [Google Scholar]
- Wang Q, Hogg RC, Large WA. Properties of spontaneous inward currents recorded in smooth muscle cells isolated from the rabbit portal vein. Journal of Physiology. 1992;451:525–537. doi: 10.1113/jphysiol.1992.sp019177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Large WA. Noradrenaline-evoked cation conductance recorded with the nystatin whole-cell method in rabbit portal vein cells. Journal of Physiology. 1991;435:21–39. doi: 10.1113/jphysiol.1991.sp018496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Large WA. Action of histamine on single smooth muscle cells dispersed from the rabbit pulmonary artery. Journal of Physiology. 1993;468:125–139. doi: 10.1113/jphysiol.1993.sp019763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Wang Y-X, Yu M, Kotlikoff MI. Ca2+-activated Cl− currents are activated by metabolic inhibition in rat pulmonary artery smooth muscle cells. American Journal of Physiology. 1997;273:C520–530. doi: 10.1152/ajpcell.1997.273.2.C520. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Fukuta H, Hakahira Y, Suzuki H. Endothelium-dependent hyperpolarization and intercellular electrical coupling in guinea-pig mesenteric arterioles. Journal of Physiology. 1999;514:505–513. doi: 10.1111/j.1469-7793.1999.505ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki J, Kitamura K. Ion channel properties and intercellular communication of smooth muscle cells in rat intact cerebral microvessels. Biophysical Journal. 2001;80:395a. [Google Scholar]
- Zuccarello M, Soattin GB, Lewis AI, Breu V, Hallak H, Rapoport RM. Prevention of subarachnoid hemorrhage-induced cerebral vasospasm by oral administration of endothelin receptor antagonists. Journal of Neurosurgery. 1996;84:503–507. doi: 10.3171/jns.1996.84.3.0503. [DOI] [PubMed] [Google Scholar]