

Abstract
The whole-cell configuration of the patch-clamp technique was used to study the modulation of giant depolarizing potentials (GDPs) by nicotinic acetylcholine receptors (nAChRs) in CA3 hippocampal neurons in slices from postnatal day (P) 2–6 rats.
Bath application of nicotine increased GDP frequency in a concentration-dependent manner. For example, nicotine (0.5–1 μm) enhanced GDP frequency from 0.05 ± 0.04 to 0.17 ± 0.04 Hz. This effect was prevented by the broad-spectrum nicotinic receptor antagonist dihydro-β-erythtroidine (DHβE, 50 μm) and partially antagonized by methyllycaconitine (MLA, 50 nm) a competitive antagonist of α7 nAChRs. GDP frequency was also enhanced by AR-17779 (100 μm), a selective agonist of α7 nAChRs.
The GABAA receptor antagonist bicuculline (10 μm) and the non-NMDA glutamate receptor antagonist DNQX (20 μm) blocked GDPs and prevented the effects of nicotine on GDPs. In the presence of DNQX, nicotine increased GABA-mediated synaptic noise, indicating that this drug may have a direct effect on GABAergic interneurons.
Bath application of edrophonium (20 μm), a cholinesterase inhibitor, in the presence of atropine (1 μm), increased GDP frequency, indicating that nAChRs can be activated by ACh released from the septo-hippocampal fibres. This effect was prevented by DHβE (50 μm).
In the majority of neurons tested, MLA (50 nm) and DHβE (50 μm) reduced the frequency of GDPs with different efficacy: a reduction of 98 ± 11 and 61 ± 29 % was observed with DHβE and MLA, respectively. In a subset of cells (40 % in the case of MLA and 17 % in the case of DHβE) these drugs induced a twofold increase in GDP frequency.
It is suggested that, during development, nAChRs modulate the release of GABA, assessed as GDPs, through distinct nAChRs. The rise of intracellular calcium via nAChRs would further strengthen GABA-mediated oscillatory activity. This can be crucial for consolidation of synaptic contacts and for the fine-tuning of the developing hippocampus.
Early in postnatal development, spontaneous network-driven oscillatory events, termed giant depolarizing potentials (GDPs), occur synchronously over the entire hippocampus (Ben-Ari et al. 1989; Strata et al. 1997; Menendez de la Prida et al. 1998). These events, which can be recorded either in hippocampal slices (Ben-Ari et al. 1989) or in the intact hippocampus (Khalilov et al. 1997), are generated by the interplay between GABA and glutamate (Ben-Ari et al. 1997; Bolea et al. 1999). As in many other brain structures (Wu et al. 1992; Serafini et al. 1995; Chen et al. 1996; Kaneda et al. 1996; Owens et al. 1996), at early stages of development GABA, acting on GABAA receptors, depolarizes and excites neuronal membranes via an outward flux of chloride (Cherubini et al. 1991; Rivera et al. 1999). Although GDPs are GABAA mediated, as shown in previous studies from this and other laboratories (Ben ari et al. 1989; Gaiarsa et al. 1991; Strata et al. 1995; Khazipov et al. 1997; Bolea et al. 1999) they need a glutamatergic drive for their induction. Thus, glutamate released from glutamatergic terminals (presumably from pyramidal cells to interneurons, Khazipov et al. 1997) triggers the release of GABA from GABAergic interneurons. Glutamate acts mainly on α-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA) receptors as demonstrated by the increase in GDP frequency evoked by cyclothiazide, a selective blocker of AMPA receptor desensitization, and the block of GDP induction by GYKI 53655, a selective and potent AMPA receptor antagonist (Bolea et al. 1999). Co-operation between GABA and glutamate is essential for generating a positive feedback responsible for synchronization of a large population of neurons. This synchronized activity is thought to promote functional maturation of precursor networks (Goodman & Shatz, 1993). This can be achieved via an elevation of intracellular calcium ([Ca2+]i) following calcium entry through voltage-activated calcium channels and/or NMDA receptors (Leinekugel et al. 1995; Canepari et al. 2000).
The hippocampus receives a large cholinergic innervation from the septo-hippocampal pathway that originates in the basal forebrain, from the medial septal nucleus and from the diagonal band of Broca (Kasa, 1986). This projection, which is crucial for maintaining higher cognitive functions (Dutar et al. 1995), is already functional during the first postnatal week. Thus, GDPs can be modulated by endogenous acetylcholine released from cholinergic terminals, via activation of muscarinic receptors (Avignone & Cherubini, 1999).
In the present work the whole-cell configuration of the patch-clamp technique was used to study the effects of nicotinic acetylcholine receptor (nAChR) activation on spontaneous GDPs recorded in the CA3 region of the hippocampus of neonatal rats. In many brain regions, nAChRs have been reported to control neurotransmitter release (McGehee et al. 1995; Gray et al. 1996; Alkondon et al. 1997; Lena & Changeux, 1997; Wonnacott, 1997; Guo et al. 1998). In particular, in the hippocampus, an area involved in learning and memory processes, activation of nAChRs has been shown to enhance the release of both glutamate and GABA from presynaptic nerve terminals (Gray et al. 1996; Alkondon et al. 1997, 1999; Radcliffe & Dani, 1998; Radcliffe et al. 1999).
We have found that activation of α7 and non-α7 subtypes of nAChRs by endogenously released ACh or by nicotinic agonists can induce either a potentiating or a depressant effect on GDP frequency.
METHODS
Slice preparation
Experiments were performed on hippocampal slices obtained from postnatal day (P) 2–6 Wistar rats (P0 being the day of birth) according to the methods previously described (Avignone & Cherubini, 1999). Briefly, animals were decapitated after being anaesthetized with intraperitoneal injection of urethane (2 g kg−1). This procedure is in accordance with the regulations of the Italian Animal Welfare Act and was approved by the local authority veterinary service. The brain was quickly removed from the skull and the hippocampi were dissected free. Transverse 400 μm-thick slices were cut with a vibrotome and maintained at room temperature (22–24 °C) in oxygenated artificial cerebrospinal fluid (ACSF) containing (mm): NaCl 130, KCl 3.5, NaH2PO4 1.2, MgCl2 1.3, CaCl2 2, NaHCO3 25, glucose 10 (pH 7.3), saturated with 95 % O2-5 % CO2. After incubation in ACSF for at least 1 h, an individual slice was transferred to a submerged recording chamber, continuously superfused at 32 °C with oxygenated ACSF at a rate of 2–3 ml min−1.
Patch-clamp whole-cell recordings
Spontaneous GDPs were recorded in the whole-cell configuration of the patch-clamp technique (current-clamp mode) with a standard amplifier (Axoclamp 2B, Axon Instruments, Foster City, CA, USA) from CA3 pyramidal cells. Patch electrodes had a resistance of 4–5 MΩ when filled with an intracellular solution containing (mm): KCl 140, MgCl2 1, EGTA 1, Hepes 10, Mg ATP 2; the pH was adjusted to 7.3 with KOH. Membrane input resistance was measured from the amplitude of electrotonic potentials evoked by passing small hyperpolarizing current pulses (300 ms duration) across the cell membrane.
Drugs
Drugs used were: 6,7-dinitroquinoxaline-2,3-dione (DNQX), methyllycaconitine (MLA) and bicuculline methiodide, all purchased from Tocris Cookson, Bristol, UK; nicotine, α-bungarotoxin (α-Bgt), dihydro-β-erythtroidine (DHβE), atropine, ethyl[m-hydroxyphenil]-dimethylammonium (edrophonium) chloride and tetrodotoxin (TTX) all purchased from Sigma, Milan, Italy; and (-)-spiro[1-azabicyclo[2.2.2]octane-3,5′-oxazolidin-2′-one] (AR-R17779), kindly supplied by Dr M. Keenan, Eli Lilly & Co., Windlesham, UK. All drugs except DNQX were dissolved in ACSF and applied in the bath via a three-way tap system, by changing the superfusion solution to one which differed only in its content of drug(s). DNQX was dissolved in dimethylsulphoxide (DMSO) at a concentration of 20 mm (stock solution). Control experiments using a solution containing 2.5/1000 of DMSO did not affect GDP frequency, spontaneous events, membrane potential or input resistance. The ratio of flow rate to bath volume ensured complete exchange within 1 min.
Data acquisition and analysis
Spontaneous GDP recordings were stored on magnetic tape and transferred to a computer after digitization with an A/D converter (Digidata 1200). Data acquisition was performed using pCLAMP (Axon Instruments) and the amplitude and frequency of GDPs were analysed with Axoscope (Axon Instruments, Foster City, CA). If not otherwise stated, data are expressed as means ±s.d. For each cell, the mean GDPs frequency was calculated in control conditions and during drugs application (starting 2 min after the onset of drug perfusion). Statistical comparisons were made between mean values (control versus drug treatment) using Student's paired t test (P < 0.05 being considered significant).
RESULTS
Activation of nAChRs by nicotine increases the frequency of GDPs in CA3 pyramidal neurons
Experiments where performed on CA3 pyramidal neurons in slices obtained from P2-P6 rats. These neurons exhibited spontaneous GDPs that occurred at a frequency of 0.065 ± 0.008 Hz (n = 102). In order to evaluate whether presynaptic nicotinic receptors are present and functional during the first postnatal week, and whether they can modulate GDP frequency, nicotine was bath applied at concentrations ranging between 0.5 and 10 μm.
Already at P2, nicotine induced a concentration-dependent increase of GDP frequency in 31 of 33 neurons tested (Fig. 1), indicating that nicotinic receptors are already present and functional at this stage of development. Nicotine (0.5–1 μm) significantly (P < 0.005) enhanced GDP frequency from 0.05 ± 0.04 to 0.17 ± 0.04 Hz (n = 21, Fig. 1A–D). Interestingly, the increment in GDP frequency induced by nicotine was inversely proportional to the control level of activity, being the effect of nicotine maximal when the basal GDPs frequency was lower and minimal when this was higher. The effect of nicotine was rapid in onset (1–2 min) and washed out in 5–10 min. Higher concentrations of the drug (10 μm) induced a initial increase in GDP frequency from 0.05 ± 0.02 to 0.36 ± 0.03 Hz (n = 5) followed by a reduction or a complete suppression (Fig. 1E and G). This effect, which recovered in 20–30 min was probably due to nicotinic receptor desensitization and precluded the use of higher nicotine concentrations. It should be stressed that when nicotine at this high concentration was repetitively applied, a response decrement was observed (not shown). In the majority of neurons (23/31) the action of nicotine on GDP frequency was not associated with significant changes in membrane potential. In a subset (8/31) of cells, however, the effect of nicotine (0.5–10 μm) on GDPs frequency was accompanied by a membrane depolarization (5–30 mV). This was probably due to enhanced GABA release since it was absent when nicotine was applied in the presence of bicuculline (3 μm, n = 6) or TTX (3 μm, n = 5, data not shown). Moreover, nicotine did not affect membrane input resistance (n = 5) or the shape of GDPs (see inset of Fig. 1D).
Figure 1. Nicotine increases GDP frequency.

A, representative trace recorded at P3 from a CA3 pyramidal neuron under control conditions and during bath application of nicotine (horizontal bar). B, part of the trace, marked with dots in A, has been enlarged. C, plot of GDP frequency (cell shown in A) before, during and after nicotine application (horizontal bar) as a function of time. Each column represents the number of GDPs recorded in 1 min. The dotted line represents the mean GDP frequency under control conditions. D, individual GDPs, taken at the time indicated (a and b) in A and shown on an expanded time scale. E, in another CA3 pyramidal cell (at P3), the increase in frequency of GDPs produced by nicotine (10 μm) is followed upon wash out by a complete block probably due to nicotinic receptor desensitization. F, part of the trace, marked with dots in E, has been enlarged. G, plot of GDP frequency (cell shown in E) before, during and after nicotine application (horizontal bar) as a function of time. H, changes in GDP frequency as a function of increasing concentrations of nicotine. Error bars represent s.e.m. for the number of cell tested (5–7 cells in each group; *P < 0.05).
The potentiating effect of nicotine on GDP frequency was prevented by DHβE (50–100 μm) a competitive broad-spectrum nicotinic receptor antagonist (n = 7/8; Fig. 2A and C) and partially (56 ± 29 %) antagonized by MLA (50 nm; n = 4/5; not shown) which at low concentrations is considered a competitive and selective antagonist of α7 receptors (Palma et al. 1996). Both DHβE and MLA per se were able to modulate GDP frequency (see below).
Figure 2. The effects of nicotine and AR-17779, a selective α7 nAChR agonist, on GDP frequency are prevented by DHβE and MLA, respectively.

A, recordings from a single CA3 pyramidal neuron at P6 in control conditions and during application of nicotine (horizontal bar, left panel). Bath application of DHβE prevents the action of nicotine on GDPs (right panel). Note that DHβE per se reduced GDP frequency. B, in another neuron at P3, bath application of AR-17779 (horizontal bar) markedly enhances the frequency of GDPs (left panel). In the same cell, bath application of MLA (50 nm) prevented the action of AR-17779 on GDPs (right panel). C and D, time course of GDP frequency (under different experimental conditions) for the cells shown in A and B, respectively. Each column represents the number of GDPs recorded in 1 min. The dotted line represents the mean GDP frequency under control conditions. Note that in these cells both DHβE and MLA per se reduced the frequency of GDPs below control level (dotted lines).
Selective activation of α7 subtype of nAChRs by AR-17779 increases the frequency of GDPs
The α7 subtype of nAChR, which is highly expressed in the hippocampus, exerts a crucial role in regulating transmitter release (Gray et al. 1996; Radcliffe & Dani, 1998; Alkondon et al. 1999). In the attempt to clarify the involvement of this receptor in the control of GABA release during postnatal development, we have used a newly synthesized analogue of ACh, AR-17779, which selectively activates α7 receptor types (Mullen et al. 2000).
Bath application of AR-17779 (100 μm) induced a significant (P < 0.05) increase in GDP frequency from 0.05 ± 0.03 to 0.11 ± 0.08 Hz (n = 12/13; Fig. 2B and D). This was sometimes followed by a slight depression in GDP activity immediately after washing, possibly due to nAChR desensitization. The effect of the drug was rapid in onset (the maximum effect was achieved in 1–1.5 min) and full recovery was attained 3–5 min after reintroduction of the control solution. In one case the increase in GDP frequency upon application of AR-17779 was associated with a membrane depolarization (8 mV). In contrast to nicotine, AR-17779 could be repetitively applied to the same neuron two to three times without response decrement. The effects of AR-17779 on GDP frequency were not associated with apparent modifications of basal synaptic activity.
The effects of AR-17779 on GDP frequency were partially (n = 1/6) or fully (n = 4/6) prevented by MLA (50 nm; Fig. 2B and D). In one of six cells MLA failed to prevent the effect of AR-17779. In two cells (from two different slices) the effects of AR-17779 on GDP frequency were also antagonized by the α7 blocker αBgt (20 nm; not shown), further supporting the finding that α7 receptors are present and functional already at early developmental stages.
Nicotine is not able to trigger GDPs in the absence of a glutamatergic drive
GDPs are generated by the interplay between GABA, acting on GABAA receptors, and glutamate, acting mainly on AMPA receptors (Ben Ari et al. 1989; Bolea et al. 1999). In previous experiments from this laboratory it has been demonstrated that in the absence of a glutamatergic drive calcium transients generated in one cell or a few cells by a single evoked GDP or by focal application of GABA propagate to neighbouring neurons (Canepari et al. 2000). In order to check whether nicotine-mediated increase in GABA release is able to trigger GDPs when the glutamatergic drive is blocked, nicotine was applied in the presence of the AMPA-kainate antagonist DNQX (20 μm). Bath application of DNQX completely suppressed spontaneously occurring GDPs in 1–2 min (Fig. 3A). Subsequent application of nicotine (0.5 −1 μm) failed to trigger GDPs (n = 8), but in six of eight cells produced an increase in GABAA-mediated synaptic noise that reached the threshold for action potential generation (see Fig. 3A), indicating that nicotine may have a direct effect on GABAergic interneurons. Bath application of bicuculline (3–10 μm; n = 6) completely abolished GDPs and prevented the effects of nicotine on both GDPs and synaptic noise suggesting, in agreement with previous reports (Ben Ari et al. 1989), that the final pathway for GDP induction is GABAergic (Fig. 3B).
Figure 3. The effects of nicotine on GDPs are prevented by DNQX and bicuculline.

Representative traces recorded at P5 from two different neurons, during superfusion of nicotine (filled horizontal bars, 1 μm) in the presence of DNQX (open horizontal bar, 20 μm, A) or bicuculline (open horizontal bar, 3 μm, B). Note that DNQX and bicuculline block GDPs and prevent the action of nicotine on giant events. However, in DNQX, nicotine induced the appearance of action potentials. Insets, expanded traces taken from A at the times indicated (1 and 2) showing GDPs or single action potentials, respectively.
Activation of nAChRs by endogenous ACh modulates GDPs frequency
It has recently been shown that ACh tonically released from cholinergic fibres may affect GABA release in a developmentally regulated way through the activation of muscarinic receptors (Avignone & Cherubini, 1999), suggesting that the cholinergic projection from the septum is already functional in the immediate postnatal stage. Therefore the following experiments were undertaken to clarify whether tonically released ACh also regulates GABA release via nAChRs. For this purpose, slices were treated with atropine (1 μm) to block muscarinic receptors. At this concentration atropine has minimal inhibitory action on nicotinic currents (Alkondon et al. 2000). Since ACh is rapidly hydrolysed by acetylcholinesterase (AChE), we enhanced the basal level of ACh using the AChE inhibitor edrophonium. In the presence of this compound (20 μm), a clear increase in GDP frequency was found in five of seven neurons tested (Fig. 4A and B). In edrophonium the frequency of GDPs increased from 0.008 ± 0.002 to 0.038 ± 0.011 Hz (mean ±s.e.m.; Fig. 4C), suggesting that endogenous ACh modulates GABA release via nicotinic receptor types. In atropine, basal GDP frequency was reduced to about 60 % of their control value (see Avignone & Cherubini, 1999). The potentiating effect of edrophonium was prevented by DHβE (50–100 μm, n = 3, Fig. 4D and E), indicating that nicotinic receptors were involved in its action. However, the possibility exists that edrophonium, like other anticholinesterase inhibitors, may directly interact with nicotinic receptors (Zwart et al. 2000). Therefore, in order to see whether activation of α7 or β2-containing receptors by endogenous ACh may regulate GABA release, as reported below, slices were treated with the α7 nicotinic receptor antagonist MLA and the broad-spectrum antagonist DHβE.
Figure 4. Activation of nicotinic receptors by endogenous ACh modulates GDP frequency.

A, recordings from a CA3 pyramidal neuron at P3 in the presence of atropine (1 μm, Control, left panel) and after addition of the AChE inhibitor edrophonium (20 μm, right panel). B, plot of GDP frequency versus time for the cell shown in A. Note the reduction of GDP frequency with atropine. Each column represents the number of GDPs recorded in 1 min. The dotted line represents the mean GDP frequency under control conditions. C, each column represents the mean GDP frequency observed in four neurons during atropine (open column) or atropine plus edrophonium (filled column). Error bars represent s.e.m.D, recordings from the same cell at P3 during application of DHβE (left panel), DHβE plus edrophonium (middle panel) and during wash (right panel). All recordings were made in the presence of atropine (1 μm). E, plot of GDP frequency versus time for the cell shown in D. Each column represents the number of GDPs recorded in 2 min. The dotted line represents the mean GDP frequency under control conditions. The marked increase of GDP frequency during reintroduction of the control solution plus atropine can be mainly attributed to the wash out of DHβE.
Activation of nAChRs by endogenous ACh may up- or down-regulate GABA release
Hippocampal interneurons express different nAChR subunits, in particular α7 which, as already mentioned, is essential for modulating transmitter release (Gray et al. 1996; Radcliffe & Dani, 1998; Alkondon et al. 1999). Bath application of MLA (50 nm, for 4–10 min) modified GDP frequency in 22 of 35 cells tested. This occurred in the absence of any change in membrane potential or input resistance. Of 22 responding neurons, MLA reversibly and significantly (P < 0.005) reduced (61 ± 29 %; n = 13; Figs 5C, 5D and 6) or enhanced (200 ± 35 %; n = 9; Figs 5A, Fig. 5B and 6) GDP frequency. In order to evaluate whether other receptor subtypes (in particular those containing the β2 subunit, Zoli et al. 1998) are also involved in GDP modulation by endogenous ACh, in additional experiments we applied DHβE, a broad-spectrum nAChR antagonist (Zoli et al. 1998). Superfusion of DHβE (50 μm, for 4–10 min) produced an effect in 24 of 28 cells. Among the DHβE-sensitive cells, the majority (n = 20; 83 %) showed almost complete block (98 ± 11 %; Figs 5E, 5F and 6) while the remainder (n = 4; 17 %; Fig. 6) exhibited an increase in GDP frequency (215 ± 35 %; Fig. 6). In three cells, reintroduction of the control solution after DHβE, induced a rebound increase of GDP frequency above control level (data not shown). DHβE did not modify resting membrane potential or input resistance. The effects of both MLA and DHβE were reversible upon wash out and were apparently independent of the age of the rats. When these two antagonists were independently applied to the same neurons they produced effects that were not correlated, being either in the same or opposite direction. This suggests that these drugs act on different receptor subtypes. All together these results indicate that during postnatal development, GABA release is controlled by endogenous ACh via activation of distinct nAChR types. Moreover, while α7 receptor subtypes up- or down-regulate the frequency of GDPs, activation of β2-containing receptors mainly enhances GDP frequency.
Figure 5. Regulation of GDP frequency by MLA and DHβE.

A, traces from the same CA3 pyramidal neuron at P4 in control conditions (left panel) and during bath application of MLA (right panel). B, time course of GDP frequency for the cell shown in A. C, traces from another cell at P5 in control (left panel) and during application of MLA (right panel). D, time course of GDP frequency for the cell shown in C. Note that MLA increases and decreases GDP frequency in A and C, respectively. E, recordings from a CA3 pyramidal cell at P5 in control conditions (left panel) and during bath application of DHβE (right panel). In A, C and E, GDPs are marked by asterisks. F, plot of GDP frequency versus time for the cell shown in E. Each column represents the number of GDPs recorded in 1 min. The dotted line represents the mean GDP frequency under control conditions.
Figure 6. Summary of changes in GDP frequency induced by MLA and DHβE.

A, each column represents the percentage of cells showing an increment (open portion of column) or a decrement (grey portion of column) of GDP frequency induced by MLA (50 nm) or DHβE (50 μm). Numbers inside the columns refer to the number of cells tested. B, changes in GDP frequency during superfusion of MLA (50 nm, open column) or DHβE (50 μm, filled column). Error bars represent s.d.
DISCUSSION
The present experiments clearly show that in the hippocampus α7- and non-α7-containing nACh receptors are present and functional from the first few days of postnatal life. Activation of these receptors either by nAChR agonists or by endogenously released ACh modulates GABA release assayed as GDPs.
nAChRs are present and functional from the first postnatal days
In the hippocampus of adult animals, ligand binding and in situ hybridization studies have shown that the more abundant nAChR subunits are α7 and β2 (Wada et al. 1989; Dineley-Miller & Patrick, 1992; Freedman et al. 1993; Seguela et al. 1993; Deneris et al. 1998; Zoli et al. 1998). These receptor subunits, whose activation by nicotine has been demonstrated to increase the release of GABA and glutamate (Gray et al. 1996; Alkondon et al. 1997, 1999; Radcliffe & Dani, 1998; Radcliffe et al. 1999), are expressed on both GABAergic interneurons and principal cells. While on interneurons they are localized at somato-dendritic preterminal and terminal levels (Alkondon & Albuquerque, 1993; Alkondon et al. 1997, 1998; Jones & Yakel, 1997; Frazier et al. 1998; McQuiston & Madison, 1999; Radcliffe et al. 1999; Ji & Dani, 2000; Alkondon et al. 2000), on pyramidal cells they are mainly present on nerve endings (Gray et al. 1996; Frazier et al. 1998; Radcliffe & Dani, 1998; Radcliffe et al. 1999).
The increase in GDP frequency due to nicotine, observed in the present experiments, indicates that nAChRs are present and functional already at early stages of hippocampal development and they influence network activity through modulation of GABA release.
As already mentioned, in the neonatal hippocampus GDPs are generated by the synergistic action of glutamate and GABA and therefore the stepping up of GDP frequency induced by nicotine may involve nAChRs localized on GABAergic and/or glutamatergic terminals. Although a direct effect on nAChRs localized on glutamatergic terminals cannot be ruled out, the present experiments do not allow assessment of the relative contribution of glutamate to GDP frequency increase since the induction of GDP is blocked by both AMPA and GABAA receptor antagonists (Ben Ari et al. 1989; Strata et al. 1995). However, it should be stressed that during the early stages of postnatal development glutamatergic activity is scarce due to the late development of glutamatergic pathways (Hosokawa et al. 1994; Tyzio et al. 1999). A direct action of nAChRs on GABAergic interneurons is proved by the observation that nicotine can enhance GABAA-mediated synaptic noise (to the point where the threshold for action potential generation can be reached) when AMPA receptors are blocked with DNQX. Nevertheless, the present data demonstrate that the nAChR-mediated increase of GABA release is insufficient to synchronize the entire network and to induce GDPs.
Nicotine exerts its action mainly on presynaptic receptors since it enhances the frequency of GDPs without changing their shape and without any modification of membrane potential or input resistance of the recorded neuron. It is worth noting that in those cases in which nicotine or AR-17779 produced a membrane depolarization, this was probably due to an increase in GABA release since it was prevented by bicuculline or TTX. In line with the present findings, it has been reported that low concentrations of nicotine increase transmitter release through a presynaptic site of action mainly via high calcium permeable α7 receptors (Gray et al. 1996 but see Vogt & Regehr, 2001).
In contrast, the same concentrations of nicotine were unable to produce any response in postsynaptic α7 receptors expressed on the soma of GABAergic interneurons (due to desensitization of α7 receptors, Alkondon et al. 2000). This apparent discrepancy can be explained by a different degree of desensitisation of pre- versus postsynaptic α7 receptors. This is not the case for high affinity β2-containing receptors expressed at somatic or terminal levels whose activation and desensitization processes follow the same kinetic scheme (Alkondon et al. 2000).
It is possible that preterminal activation of α7 receptors by low concentrations of nicotine contributes also to the increment of GDP frequency. This hypothesis is supported by the observation that a similar effect can be produced by AR-17776, a selective α7 agonist. Moreover, the block of GDPs upon wash out of a higher concentration (10 μm) of nicotine can be attributed to desensitization of both α7- and β2-containing receptors (Alkondon et al. 2000). It follows that rather complex mechanisms underlie the effects of nicotine on GDPs. These are further complicated by the fact that this drug acts on distinct receptor subtypes that are differently expressed on various neurons (Reece & Schwartzkroin, 1991). Therefore, the up-regulation of GABA release by nicotine can be explained either by activation of nAChRs or disinhibition of GABAergic interneurons, following nAChR desensitization (Ji & Dani, 2000). Although a certain degree of receptor desensitization cannot be excluded, the experimental evidence that nicotine action was completely or partially prevented by the broad-spectrum nAChR antagonist DβHE or the selective α7 blocker MLA, respectively, favours the hypothesis that activation of nAChRs plays a major role in the enhancement of GABA release (see paragraph below). Against receptor desensitization is the observation that nicotine-mediated increase in GABA release was not always reproducibly mimicked by the two antagonists tested.
Activation of nAChRs by endogenous ACh up- and down-regulates GDP frequency
The present results confirm and extend previous data on the tonic enhancement of ACh release by the acetylcholinesterase inhibitor edrophonium from postnatal day 2 (Avignone & Cherubini, 1999). However, the relative contribution of nicotinic or muscarinic receptors to this enhancement was not elucidated. In the present study, the fact that edrophonium applied in the presence of atropine increased GDP frequency, and that this effect was prevented using DHβE, indicates that a tonic release of ACh can also facilitate GABA release through the activation of nicotinic receptors.
Although scarce information exists on the distribution of nAChRs in the neonatal brain, in the present study the use of a selective antagonist for α7- and β2-containing receptors has allowed light to be shed on the functional relevance of these subunits in early stages of postnatal hippocampal development. The observation that both MLA and DHβE, selective antagonists of α7- and β2-containing receptors, respectively, enhanced or decreased GDP frequency is not easy to interpret.
As suggested by Ben Ari et al. (1989) the oscillatory activity underlying GDPs might be initiated by the depolarizing action of GABA (released from GABAergic interneurons) on principal cells. This would in turn induce the release of glutamate onto GABAergic interneurons which would depolarize following activation of AMPA receptors. The oscillating GABAergic interneuron would release GABA onto a pyramidal cell and would participate in a positive feedback loop. Therefore, if we assume that nAChR activation facilitates neurotransmitter release, a reduction of GABA or glutamate release by selective α7- or β2-containing nicotinic receptor blockers would interrupt this loop leading to a reduction of GDP frequency (Fig. 7A).
Figure 7. Possible mechanisms underlying α7-mediated up- or down-regulation of GDPs.
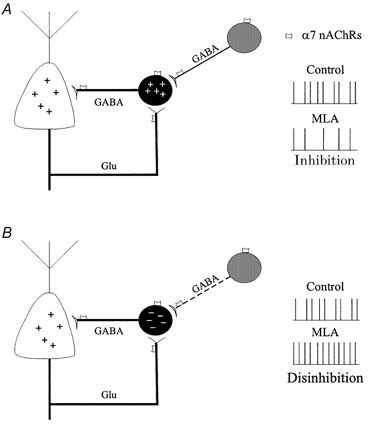
The basic oscillatory unit, depicted in bold (in A and B) is composed of an interneuron (black) and an interconnected pyramidal cell (white). The synergistic action of GABA and glutamate released from GABAergic interneurons and pyramidal cells, respectively, generates a positive feed-back leading to GDP induction. GDPs can only be produced when GABA has a depolarizing action on pyramidal cells through a reverse chloride gradient (positive signs in pyramidal cells). GABAergic interneurons located out of the basic loop (grey) may either reinforce (A) or depress (B) the activity of the network assessed as GDPs. Reinforcement may occur when GABA depolarizes interneurons involved in the basic oscillatory unit (continuos thin line and positive charges in the interneuron). Depression may occur when GABA hyperpolarizes a subset of GABAergic cells which belong to the basic circuit (dashed line and negative charges in the interneuron). Note that α7 receptors are expressed on interneurons and glutamatergic terminals. A reduction of GABA release following α7 receptor block with MLA would exert an inhibitory (A) or disinibitory (B) action on the circuit leading to a decrease or increase in GDP frequency, respectively.
More difficult to explain is the increase in GDP frequency observed in 40 % of slices treated with MLA. This paradoxical effect, which cannot be explained on the basis of simple positive feedback generated by the depolarizing action of GABA, can be generated by negative control exerted by the hyperpolarizing action of GABA in some part of the circuit. The negative control could not occur at the level of principal cells because if that were the case GDPs would not be generated. In contrast a hyperpolarizing action of GABA at the level of a subpopulation of GABAergic interneurons synapsing on principal cells (probably due to changes in the chloride gradient) might account for the enhancement of GDP frequency. The activation of GABA receptors in these cells by GABA released from adjacent interneurons would reduce the release of GABA onto pyramidal cells and GDP frequency. The block of nicotinic receptors (mainly α7, present on interneurons synapsing onto other interneurons expressing hyperpolarizing GABA receptors) with MLA would disinhibit principal cells leading to an increase in GDP frequency (Fig. 7B). Of course, α7 nicotinic receptors are present not only on ‘disinhibitory’ interneurons but also on other cells. Hence, the final observation may result from the balance of inhibitory and excitatory effects at different levels of this complex circuit.
Although this hypothesis is attractive it is difficult to prove experimentally. However, the possibility that nicotinic receptors exert a disinhibitory role on hippocampal principal cells or stratum radiatum interneurons has been already suggested (Alkondon et al. 1999; Ji & Dani, 2000). Disinhibitory interneurons constitute a homogeneous population of calretinin-positive GABAergic cells that are clearly distinct from interneurons projecting to principal cells (Gulyas et al. 1993, 1996). As a matter of speculation it is possible to conceive that during postnatal development these two populations of interneurons undergo different maturational processes.
As an alternative explanation for the antagonist-induced increase in GDP frequency the possibility that, at least in some neurons, activation of nicotinic receptors down-regulates transmitter release cannot be excluded. Block of these receptors with MLA or DHβE would induce an increase in GDP frequency. However, this hypothesis seems unlikely because it is not supported by our experimental data for nicotinic receptor agonists.
Physiological relevance
It has been hypothesized that nicotine enhances synaptic transmission by increasing the probability of transmitter release via a rise in intracellular calcium with a mechanism similar to that of paired pulse facilitation (Gray et al. 1996; but see Vogt & Regehr, 2001). It is reasonable to assume that during development, coincidence detection of two separate stimuli within a short time window, delivered to the synapse via nAChRs and GABA-mediated depolarization, respectively, would induce a large increase in intracellular calcium, thus strengthening synaptic efficacy in an associative manner. In this respect, endogenous ACh released from the septo-hippocampal fibres already functional at early stages of postnatal development would further strengthen GABA release assayed as GDPs by acting on both nicotinic and muscarinic receptor types. These GDPs can be crucial for consolidation of synaptic contacts and for the fine tuning of the developing hippocampus before a complete adult circuit is in place.
Acknowledgments
This work was partially supported by a grant from Ministero dell'Universita’ e Ricerca Scientifica e Tecnologica (MURST, cofinanziamento) and from Regione Friuli Venezia Giulia to E.C. L.M. was recipient of a fellowship from Novartis Pharma.
References
- Alkondon M, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. I. Pharmacological and functional evidence for distinct structural subtypes. Journal of Pharmacology and Experimental Therapeutics. 1993;265:1455–1473. [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Albuquerque EX. alpha-bungarotoxin- and methyllycaconitine-sensitive nicotinic receptors mediate fast synaptic transmission in interneurons of rat hippocampal slices. Brain Research. 1998;810:257–263. doi: 10.1016/s0006-8993(98)00880-4. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Almeida LE, Eisenberg HM, Albuquerque EX. Choline and selective antagonists identify two subtypes of nicotinic acetylcholine receptors that modulate GABA release from CA1 interneurons in rat hippocampal slices. Journal of Neuroscience. 1999;19:2693–2705. doi: 10.1523/JNEUROSCI.19-07-02693.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Almeida LE, Randall WR, Albuquerque EX. Nicotine at concentrations found in cigarette smokers activates and desensitizes nicotinic acetylcholine receptors in CA1 interneurons of rat hippocampus. Neuropharmacology. 2000;13:2726–2739. doi: 10.1016/s0028-3908(00)00156-8. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Barbosa CT, Albuquerque EX. Neuronal nicotinic acetylcholine receptor activation modulates gamma-aminobutyric acid release from CA1 neurons of rat hippocampal slices. Journal of Pharmacology and Experimental Therapeutics. 1997;283:1396–1411. [PubMed] [Google Scholar]
- Avignone E, Cherubini E. Muscarinic receptor modulation of GABA-mediated giant depolarizing potentials in the neonatal rat hippocampus. Journal of Physiology. 1999;518:97–107. doi: 10.1111/j.1469-7793.1999.0097r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. Journal of Physiology. 1989;416:303–325. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Khazipov R, Leinekugel X, Caillard O, Gaiarsa JL. GABAA, NMDA and AMPA receptors: a developmentally regulated ‘menage a trois’. Trends in Neurosciences. 1997;20:523–529. doi: 10.1016/s0166-2236(97)01147-8. [DOI] [PubMed] [Google Scholar]
- Bolea S, Avignone E, Berretta N, Sanchez-Andres JV, Cherubini E. Glutamate controls the induction of GABA-mediated giant depolarizing potentials through AMPA receptors in neonatal rat hippocampal slices. Journal of Neurophysiology. 1999;81:2095–2102. doi: 10.1152/jn.1999.81.5.2095. [DOI] [PubMed] [Google Scholar]
- Canepari M, Mammano F, Kachalsky SG, Rahamimoff R, Cherubini E. GABA- and glutamate-mediated network activity in the hippocampus of neonatal and juvenile rats revealed by fast calcium imaging. Cell Calcium. 2000;27:25–33. doi: 10.1054/ceca.1999.0086. [DOI] [PubMed] [Google Scholar]
- Chen G, Trombley PQ, Vanden Pol AN. Excitatory actions of GABA in developing rat hypothalamic neurones. Journal of Physiology. 1996;494:451–464. doi: 10.1113/jphysiol.1996.sp021505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherubini E, Gaiarsa JL, Ben-Ari Y. GABA: an excitatory transmitter in early postnatal life. Trends in Neurosciences. 1991;14:515–519. doi: 10.1016/0166-2236(91)90003-d. [DOI] [PubMed] [Google Scholar]
- Deneris ES, Connolly J, Boulter J. Primary structure and expression of beta2: a novel subunit of neuronal nicotinic acetylcholine receptors. Neuron. 1988;1:45–54. doi: 10.1016/0896-6273(88)90208-5. [DOI] [PubMed] [Google Scholar]
- Dineley-Miller K, Patrick J. Gene transcripts for the nicotinic acetylcholine receptor subunit, beta4, are distributed in multiple areas of the rat central nervous system. Molecular Brain Research. 1992;16:339–344. doi: 10.1016/0169-328x(92)90244-6. [DOI] [PubMed] [Google Scholar]
- Dutar P, Bassant MH, Senut M, Lamour Y. The septohippocampal pathway: structure and function of a central cholinergic system. Physiological Reviews. 1988;75:393–427. doi: 10.1152/physrev.1995.75.2.393. [DOI] [PubMed] [Google Scholar]
- Frazier CJ, Rollins YD, Breese CR, Leonard S, Freedman R, Dunwiddie TV. Acetylcholine activates an alpha-bungarotoxin-sensitive nicotinic current in rat hippocampal interneurons, but not pyramidal cells. Journal of Neuroscience. 1998;18:1187–1195. doi: 10.1523/JNEUROSCI.18-04-01187.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman R, Wetmore C, Stromberg I, Leonard S, Olson L. Alpha-bungarotoxin binding to hippocampal interneurons: immunocytochemical characterization and effects on growth factor expression. Journal of Neuroscience. 1993;13:1965–1975. doi: 10.1523/JNEUROSCI.13-05-01965.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiarsa JL, Corradetti R, Cherubini E, Ben-Ari Y. Modulation of GABA-mediated synaptic potentials by glutamatergic agonists in neonatal CA3 rat hippocampal neurons. European Journal of Neuroscience. 1991;3:301–309. doi: 10.1111/j.1460-9568.1991.tb00816.x. [DOI] [PubMed] [Google Scholar]
- Goodman CS, Shatz CJ. Developmental mechanisms that generate precise patterns of neuronal connectivity. Cell. 1993;72:77–98. doi: 10.1016/s0092-8674(05)80030-3. [DOI] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- Gulyas AI, Hájos N, Freund TF. Interneurons containing calretinin are specialized to control other interneurons in the rat hippocampus. Journal of Neuroscience. 1996;16:3397–3411. doi: 10.1523/JNEUROSCI.16-10-03397.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas AI, Miles R, Hajos N, Freund TF. Precision and variability in postsynaptic target selection of inhibitory cells in the hippocampal CA3 region. European Journal of Neuroscience. 1993;5:1729–1751. doi: 10.1111/j.1460-9568.1993.tb00240.x. [DOI] [PubMed] [Google Scholar]
- Guo JZ, Tredway TL, Chiappinelli VA. Glutamate and GABA release are enhanced by different subtypes of presynaptic nicotinic receptors in the lateral geniculate nucleus. Journal of Neuroscience. 1998;18:1963–1969. doi: 10.1523/JNEUROSCI.18-06-01963.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa Y, Sciancalepore M, Stratta F, Martina M, Cherubini E. Developmental changes in spontaneous GABAA-mediated synaptic events in rat hippocampal CA3 neurons. European Journal of Neuroscience. 1994;6:805–813. doi: 10.1111/j.1460-9568.1994.tb00991.x. [DOI] [PubMed] [Google Scholar]
- Ji D, Dani JA. Inhibition and disinhibition of pyramidal neurons by activation of nicotinic receptors on hippocampal interneurons. Journal of Neurophysiology. 2000;83:2682–2690. doi: 10.1152/jn.2000.83.5.2682. [DOI] [PubMed] [Google Scholar]
- Jones S, Yakel JL. Functional nicotinic ACh receptors on interneurones in the rat hippocampus. Journal of Physiology. 1997;504:603–610. doi: 10.1111/j.1469-7793.1997.603bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M, Farrant M, Cull-Candy SG. Whole-cell and single channel currents activated by GABA and glycine in granule cells of the rat cerebellum. Journal of Physiology. 1996;485:419–435. doi: 10.1113/jphysiol.1995.sp020739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasa P. The cholinergic systems in brain and spinal cord. Progress in Neurobiology. 1986;26:211–272. doi: 10.1016/0301-0082(86)90016-x. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Esclapez M, Medina I, Aggoun D, Lamsa K, Leinekugel X, Khazipov R, Ben-Ari Y. A novel in vitro preparation: the intact hippocampal formation. Neuron. 1997;19:743–749. doi: 10.1016/s0896-6273(00)80956-3. [DOI] [PubMed] [Google Scholar]
- Khazipov R, Leinekugel X, Khalilov I, Gaiarsa JL, Ben-Ari Y. Synchronization of GABAergic interneuronal network in CA3 subfield in neonatal rat hippocampal slices. Journal of Physiology. 1997;498:763–772. doi: 10.1113/jphysiol.1997.sp021900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinekugel X, Tseeb V, Ben-Ari Y, Bregestovski P. Synaptic GABAA activation induces Ca2+ rise in pyramidal cells and interneurons from rat neonatal hippocampal slices. Journal of Physiology. 1995;487:319–329. doi: 10.1113/jphysiol.1995.sp020882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lena C, Changeux JP. Role of Ca2+ ions in nicotinic facilitation of GABA release in mouse thalamus. Journal of Neuroscience. 1997;15:576–585. doi: 10.1523/JNEUROSCI.17-02-00576.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- McQuiston AR, Madison DV. Nicotinic receptor activation excites distinct subtypes of interneurons in the rat hippocampus. Journal of Neuroscience. 1999;19:2887–2896. doi: 10.1523/JNEUROSCI.19-08-02887.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendezdela Prida L, Bolea S, Sanchez-Andres JV. Origin of the synchronized network activity in the rabbit developing hippocampus. European Journal of Neuroscience. 1998;10:899–906. doi: 10.1046/j.1460-9568.1998.00097.x. [DOI] [PubMed] [Google Scholar]
- Mullen G, Napier J, Balestra M, Decory T, Hale G, Macor J, Mack R, Loch J, Wu E, Kover A, Verhoest P, Sampognaro A, Phillips E, Zhu Y, Murray R, Griffith R, Blosser J, Gurley D, Machulskis A, Zongrone J, Rosen A, Gordon J. (−)-Spiro[1-azabicyclo[2.2.2]octane-3, 5′-oxazolidin-2′-one], a conformationally restricted analogue of acetylcholine, is a highly selective full agonist at the alpha7 nicotinic acetylcholine receptor. Journal of Medical Chemistry. 2000;43:4045–4050. doi: 10.1021/jm000249r. [DOI] [PubMed] [Google Scholar]
- Owens DF, Boyce LH, Davis MB, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. Journal of Neuroscience. 1996;16:6414–6423. doi: 10.1523/JNEUROSCI.16-20-06414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma E, Bertrand S, Binzoni T, Bertrand D. Neuronal nicotinic α7 receptor expressed in Xenopus oocytes presents five putative binding sites for methyllycaconitine. Journal of Physiology. 1996;491:151–161. doi: 10.1113/jphysiol.1996.sp021203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radcliffe KA, Dani JA. Nicotinic stimulation produces multiple forms of increased glutamatergic synaptic transmission. Journal of Neuroscience. 1998;18:7075–7083. doi: 10.1523/JNEUROSCI.18-18-07075.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radcliffe KA, Fisher JL, Gray R, Dani JA. Nicotinic modulation of glutamate and GABA synaptic transmission of hippocampal neurons. Annals of the New York Academy of Sciences. 1999;868:591–610. doi: 10.1111/j.1749-6632.1999.tb11332.x. [DOI] [PubMed] [Google Scholar]
- Reece LJ, Schwartzkroin PA. Nicotine exerts differential effects on different CA1 hippocampal cell types. Brain Research. 1991;540:287–290. doi: 10.1016/0006-8993(91)90520-6. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. Journal of Neuroscience. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini R, Valeyev AY, Barker JL, Poulter MO. Depolarizing GABA-activated Cl− channels in embryonic rat spinal and olfactory bulb cells. Journal of Physiology. 1995;488:371–386. doi: 10.1113/jphysiol.1995.sp020973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strata F, Atzori M, Molnar M, Ugolini G, Tempia F, Cherubini E. A pacemaker current in dye-coupled hilar interneurons contributes to the generation of giant GABAergic potentials in developing hippocampus. Journal of Neuroscience. 1997;17:1435–1446. doi: 10.1523/JNEUROSCI.17-04-01435.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strata F, Sciancalepore M, Cherubini E. Cyclic AMP-dependent modulation of giant depolarizing potentials by metabotropic glutamate receptors in the rat hippocampus. Journal of Physiology. 1995;489:115–125. doi: 10.1113/jphysiol.1995.sp021035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzio R, Represa A, Jorquera I, Ben-Ari Y, Gozlan H, Antiksztejn L. The establishment of GABAergic and glutamatergic synapses on CA1 pyramidal neurons is sequential and correlates with the development of apical dendrites. Journal of Neuroscience. 1999;19:10372–10382. doi: 10.1523/JNEUROSCI.19-23-10372.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt KE, Regehr WG. Cholinergic modulation of excitatory synaptic transmission in the CA3 area of the hippocampus. Journal of Neuroscience. 2001;21:75–83. doi: 10.1523/JNEUROSCI.21-01-00075.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, Swanson LW. Distribution of alpha 2, alpha 3, alpha 4, and beta 2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. Journal of Comparative Neurology. 1989;284:314–335. doi: 10.1002/cne.902840212. [DOI] [PubMed] [Google Scholar]
- Wonnacott S. Presynaptic nicotinic ACh receptors. Trends in Neurosciences. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- Wu WL, Ziskind-Conhaim L, Sweet MA. Early development of glycine- and GABA-mediated synapses in rat spinal cord. Journal of Neuroscience. 1992;12:3935–3945. doi: 10.1523/JNEUROSCI.12-10-03935.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoli M, Lena C, Picciotto MR, Changeux JP. Identification of four classes of brain nicotinic receptors using beta2 mutant mice. Journal of Neuroscience. 1998;18:4461–4472. doi: 10.1523/JNEUROSCI.18-12-04461.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart R, Van Kleef RG, Gotti C, Smulders CJ, Vijverberg HP. Competitive potentiation of acetylcholine effects on neuronal nicotinic receptors by acetylcholinesterase-inhibiting drugs. Journal of Neurochemistry. 2000;75:2492–2500. doi: 10.1046/j.1471-4159.2000.0752492.x. [DOI] [PubMed] [Google Scholar]