

Abstract
Contemporary stable isotope methodology was applied in combination with muscle biopsy sampling to accurately quantify substrate utilisation and study the regulation of muscle fuel selection during exercise.
Eight cyclists were studied at rest and during three consecutive 30 min stages of exercise at intensities of 40, 55 and 75 % maximal workload (Wmax). A continuous infusion of [U-13C]palmitate and [6,6-2H2]glucose was administered to determine plasma free fatty acid (FFA) oxidation and estimate plasma glucose oxidation, respectively. Biopsy samples were collected before and after each exercise stage.
Muscle glycogen and plasma glucose oxidation rates increased with every increment in exercise intensity. Whole-body fat oxidation increased to 32 ± 2 kJ min−1 at 55 % Wmax, but declined at 75 % Wmax (19 ± 2 kJ min−1). This decline involved a decrease in the oxidation rate of both plasma FFA and triacylglycerol fat sources (sum of intramuscular plus lipoprotein-derived triacylglycerol), and was accompanied by increases in muscle pyruvate dehydrogenase complex activation and acetylation of the carnitine pool, resulting in a decline in muscle free carnitine concentration.
We conclude that the most likely mechanism for the reduction in fat oxidation during high-intensity exercise is a downregulation of carnitine palmitoyltransferase I, either by this marked decline in free carnitine availability or by a decrease in intracellular pH.
Fat and carbohydrate are the principal substrates that fuel aerobic ATP synthesis in human skeletal muscle. The relative utilisation of fat and carbohydrate during exercise can vary enormously and depends strongly on exercise intensity. An early stable isotope tracer study (Romijn et al. 1993), which used assumptions to estimate indirectly the oxidation rate of plasma free fatty acids (FFAs), suggested that plasma FFAs provide the majority of the substrate oxidised by skeletal muscle during low- and moderate-intensity (25 and 65 % of maximal oxygen uptake capacity, V̇O2,max) exercise. In addition, it was suggested that whole-body fat and plasma FFA oxidation rates declined during high-intensity exercise (85 % V̇O2,max), as muscle glycogen became the main fuel source utilised (Romijn et al. 1993). The primary aim of the present study was, therefore, to quantify accurately the oxidation rates of plasma FFA, triacylglycerol (TG; sum of intramuscular and lipoprotein-derived TG), plasma glucose and muscle glycogen during steady-state exercise at three intensities (40, 55 and 75 % of maximal workload, Wmax) using contemporary stable isotope tracer methodology (i.e. using direct measurements of plasma FFA oxidation and validated assumptions for plasma glucose oxidation).
Understanding the regulation of fuel selection in human skeletal muscle is important, especially as fuel use abnormalities are present in metabolic diseases like type 2 diabetes (Kelley & Simoneau, 1994; Martin et al. 1995) and obesity (Colberg et al. 1995). However, the mechanisms that regulate the relative contribution of carbohydrate and fat during exercise have not been fully elucidated, and remain open to discussion. Randle et al. (1963) proposed the ‘glucose–FFA cycle’ in an effort to explain the reduction in muscle carbohydrate oxidation rate in the presence of high plasma FFA levels in resting muscle. It was proposed that an increased availability of plasma FFAs could stimulate fat oxidation and decrease carbohydrate oxidation by suppressing pyruvate dehydrogenase complex (PDC) activation (via a rise in the mitochondrial acetyl-CoA/CoA ratio) and by decreasing glycolytic flux (via the inhibitory effect of high citrate concentrations on phosphofructokinase activity). According to this concept, the relative utilisation of fat and carbohydrate is determined primarily by the availability of plasma FFAs. We therefore investigated whether the rate of appearance (Ra) of plasma FFAs and their concentration declined during high-intensity exercise. Earlier studies (Dyck et al. 1993; Romijn et al. 1995) failed to find evidence that plasma FFA availability limited fat oxidation during high-intensity exercise.
More recently, it was hypothesised that high glycolytic flux rates during high-intensity exercise could limit long-chain fatty acid oxidation (Dyck et al. 1993; Sidossis et al. 1997; Odland et al. 1998). High glycolytic flux is known to lead to the accumulation of acetyl-CoA (Constantin-Teodosiu et al. 1991; Dyck et al. 1993), and via subsequent increases in the cytosolic production of malonyl-CoA has been proposed to inhibit carnitine palmitoyltransferase I (CPT I), and thereby limit long-chain fatty acid entry into mitochondria. However, it has been found that muscle malonyl-CoA concentrations do not increase in rat and human skeletal muscle during high-intensity exercise (Rasmussen & Winder, 1997; Odland et al. 1998; Dean et al. 2000); this mechanism is therefore unlikely to lead to a reduction in fat oxidation.
A third mechanism that could explain the decrease in fat oxidation that occurs during high-intensity exercise involves the availability of muscle carnitine. Carnitine is a co-factor that is required for the transport of long-chain fatty acids across the inner mitochondrial membrane and is therefore essential for the oxidation of fatty acids in the mitochondria. This functional relationship between carnitine concentration and fat oxidation capacity is not new and has been previously described in vitro (Fritz, 1955; Cederblad et al. 1976). Furthermore, other studies have demonstrated a continuous accumulation of muscle acetylcarnitine with increasing exercise intensity (Harris et al. 1987; Hiatt et al. 1989). These findings are consistent with the concept that carnitine acts as a sink for acetyl group storage during conditions such as a high flux through the PDC reaction (i.e. when the rate of acetyl group production exceeds its rate of utilisation by the tricarboxylic acid (TCA) cycle; Sahlin, 1990; Constantin-Teodosiu et al. 1991; Timmons et al. 1996, 1998; Howlett et al. 1998, 1999). Our third aim, therefore, was to determine whether the decline in free carnitine concentration that is observed during incremental steady-state exercise, which occurs as a result of acetylation of the carnitine pool, is of sufficient magnitude to limit CPT I activity and fat oxidation.
We tested our objectives during incremental exercise in trained human subjects using an integrated approach, combining contemporary tracer methodology (in order to quantify the components of whole-body substrate utilisation) and muscle biopsy sampling (in order to quantify muscle intermediary metabolism). The information gained from this study is important to our understanding of the mechanisms responsible for the integration of carbohydrate and fat oxidation during exercise, and to our understanding of the metabolic defect(s) that cause(s) the impairment of fat oxidation in subjects with type 2 diabetes (Kelley & Simoneau, 1994; Martin et al. 1995) and obesity (Colberg et al. 1995).
METHODS
Subjects
Eight male cyclists (age, 22.1 ± 0.7 years; body mass, 74.5 ± 2.2 kg; V̇O2,max, 5.48 ± 0.16 l min−1; and Wmax, 413 ± 9 W) participated in this study. Subjects were informed about the nature and risks of the experimental procedures before their informed consent to participate was obtained. This study was approved by the local ethical committee and conformed to the standards set by the Declaration of Helsinki.
Pre-testing
Wmax and V̇O2,max were measured on an electronically braked cycle ergometer (Lode Excalibur; Groningen, The Netherlands) during an incremental exhaustive exercise test (Kuipers et al. 1985) 1 week before the first experimental trial.
Diet and activity prior to testing
All subjects consumed a standardised meal (85 kJ (kg body weight)−1) consisting of (in units of the percentage of the total energy supplied by the entire meal, En%) 69 En% carbohydrate, 15 En% fat and 16 En% protein, the evening before the trials. All subjects were instructed to refrain from heavy physical labour 2–3 days prior to testing.
Experimental trials
Each subject performed two similar trials, separated by at least 7 days. Each trial consisted of 60 min of resting measurements followed by 90 min of cycling exercise divided over three 30 min stages at an exercise intensity of 40, 55 and 75 % Wmax. In one trial, [U-13C]palmitate and [6,6-2H2]glucose were infused and breath samples, blood plasma samples and muscle biopsy samples were collected to quantify the use of different substrate sources and to determine changes in intramuscular levels of various metabolites during exercise at different workloads. The second trial was performed to determine the acetate recovery factor to correct [U-13C]palmitate oxidation rates for the loss of label by way of isotopic exchange reactions in the TCA cycle (Sidossis et al. 1995a,b; Schrauwen et al. 1998). During this trial [1,2-13C]acetate was infused and breath samples were collected. In order to further validate our findings four subjects also volunteered to perform three additional trials. These trials consisted of 60 min of exercise at 40 or 55 % Wmax, or 30 min at 75 % Wmax, and were performed to investigate whether total fat and carbohydrate oxidation rates were different during the sequential exercise stages of the incremental protocol compared to steady-state exercise at each of the individual intensities.
Protocol
Subjects reported at the laboratory at 08.00 h, after an overnight fast. A Teflon catheter (Baxter, Utrecht, The Netherlands) was inserted into an antecubital vein of one arm for blood sampling, and a second catheter was inserted into the contralateral arm for isotope infusion. At 08.30 h a resting blood sample was drawn and expired gases were collected into Vacutainer tubes (Becton Dickinson, France). Subjects were then administered a single intravenous dose of NaH13CO3 (0.06375 mg kg−1), in order to prime the bicarbonate pool, which was followed by a [6,6-2H2]glucose prime (13.5 μmol kg−1). Thereafter, a continuous infusion of [6,6-2H2]glucose and [U-13C]palmitate (or [1,2-13C]acetate in the acetate recovery test) was started (t = 0) via a calibrated IVAC 560 pump (San Diego, CA, USA) and continued for 150 min. At t = 60 min subjects started to exercise on a cycle ergometer at a workload of 40 % Wmax for a 30 min period. At t = 90 and t = 120 min the exercise intensity was increased to 55 and 75 % Wmax, respectively. When the subjects were at rest, oxygen uptake (V̇O2) and carbon dioxide production (V̇CO2) were measured continuously (Oxycon-ß; Mijnhardt, Bunnik, The Netherlands). During exercise, V̇O2 and V̇CO2 were measured every 5 min before sampling breath for the measurement of isotope enrichment. Breath and blood samples were collected at t = 0, 50, 55 and 60 min during rest, at t =80, 85 and 90 min during exercise at 40 % Wmax, at t = 110, 115 and 120 min during exercise at 55 % Wmax), and finally at t = 140, 145 and 150 min during exercise at 75 % Wmax). Percutaneous muscle biopsy samples (Bergstrom, 1975) were taken from the vastus lateralis muscle at rest (t = 60 min) and at the end of each exercise stage (t = 90, 120 and 150 min). Muscle biopsy samples were collected from both legs. The first two biopsy samples were taken from the same incision in one leg, and the second two were collected from the same incision in the contralateral leg. When biopsy samples were taken from the same incision, the first sample was taken from different fibres (distal to the incision with the needle pointing inwards) from the second (proximal to the incision with the needle pointing outwards).
Tracer infusion
Infusion rates of [U-13C]palmitate and [6,6-2H2]glucose averaged 8.2 ± 0.3 and 261 ± 1.9 nmol kg−1 min−1, respectively. At the onset of exercise [U-13C]palmitate infusion rates were doubled (16.5 ± 0.5 nmol kg−1 min−1). In the acetate recovery trial, a corresponding amount of 13C was infused, and [1,2-13C]acetate infusion rates averaged 63.3 ± 1.9 and 126.6 ± 3.7 nmol kg−1 min−1 during the resting and exercise periods, respectively. The palmitate tracer (99 % enriched; Cambridge Isotope Laboratories, Andover, MA, USA) was dissolved in heated sterile water and passed through a 0.2 μm filter into 5 % warm human serum albumin to make a 0.670 mm solution. Both the glucose and acetate tracers (99 % enriched; Cambridge Isotope Laboratories) were dissolved in 0.9 % saline. Palmitate, glucose and acetate tracer concentrations in the infusates averaged 0.77 ± 0.02, 16.75 ± 0.70 and 3.32 ± 0.09 mmol l−1, respectively.
Sample collection and analysis
Blood samples (5 ml) were collected in tubes containing EDTA, and centrifuged at 1000 g at +4 °C for 5 min. Aliquots of plasma were frozen immediately in liquid nitrogen and stored at −40 °C. Glucose (Uni Kit III; Roche, Basel, Switzerland), lactate (Gutmann & Wahlefeld, 1974), FFA (Wako NEFA-C test kit; Wako Chemicals, Neuss, Germany) and free glycerol concentrations (UV method; Boehringer, Mannheim, Germany) were analysed with a COBAS FARA semi-automatic analyser. Muscle biopsy samples (Bergstrom, 1975) were immediately frozen in liquid nitrogen and stored at −80 °C. Muscle biopsy samples were divided into two parts, of which one was freeze-dried, dissected free from visible blood and connective tissue, and powdered. A sample (7–10 mg) of this muscle powder were then extracted with 0.5 m perchloric acid containing 1 mm EDTA. After centrifugation (14 000 g), the supernatant was neutralised with 2.2 m KHCO3 and used for the determination of muscle phosphocreatine, free creatine, their sum (total creatine), glycogen (Harris et al. 1974), citrate (Bergmeyer, 1974), lactate (Harris et al. 1974), free carnitine and acetylcarnitine (Cederblad et al. 1990) concentrations. Muscle free carnitine and acetylcarnitine concentrations were corrected for muscle total creatine concentration for each subject. The remaining portion of frozen wet muscle was used for the determination of PDC activation status (PDCa; Constantin-Teodosiu et al. 1991).
Expired gas samples were analysed for 13C/12C ratio by gas chromatography continuous-flow, isotope-ratio mass spectrometry (GC-IRMS, MAT 252; Finnigan, Bremen, Germany). For determination of plasma palmitate and FFA kinetics, FFAs were extracted from plasma, isolated by thin-layer chromatography, and derivatised to their methyl esters. Palmitate concentration was determined on an analytical gas chromatograph with flame ionisation detection, using heptadecanoic acid as the internal standard, and on average it comprised 20.51 ± 0.27 % of total FFA. The isotope tracer/tracee ratio of [U-13C]palmitate was determined using GC-IRMS (MAT 252; Finnigan). Following derivatisation, plasma [6,6-2H2]glucose enrichment was determined by electron ionisation gas chromatography mass spectrometry (INCOS-XL; Finnigan). Glucose (Uni Kit III) and acetate (kit 148261; Boehringer) concentration in the infusates were determined with the COBAS FARA semi-automatic analyser.
Calculations
From respiratory measurements (V̇O2, V̇CO2) total fat and carbohydrate oxidation rates were calculated using the non-protein respiratory quotient (Peronnet & Massicotte, 1991):
| (1) |
| (2) |
with V̇O2 and V̇CO2 in litres per minute (l min−1) and oxidation rate in grams per minute (g min−1). Breath and plasma enrichments are expressed as the tracer/tracee ratio (TTR);
| (3) |
where sa denotes the sample and bk the background value.
The rate of appearance (Ra) and rate of disappearance (Rd) of palmitate and glucose were calculated using the single-pool non-steady-state Steele equations (Steele, 1959) adapted for stable isotope methodology as described elsewhere (Wolfe & Jahoor, 1990):
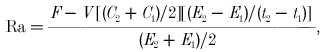 |
(4) |
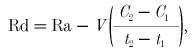 |
(5) |
where F is the infusion rate (μmol kg−1 min−1), V is the distribution volume for palmitate or glucose (40 and 160 ml kg−1, respectively), C1 and C2 are the palmitate or glucose concentrations (mmol l−1) at times 1 (t1) and 2 (t2), respectively, and E1 and E2 are the plasma palmitate or glucose enrichments (TTR) at t1 and t2, respectively. 13CO2 production (Pr13CO2; mol min−1) from the infused palmitate tracer was calculated as:
| (6) |
where TTRCO2 is the breath 13C/12C ratio at a given time point, k is the volume of 1 mol of CO2 (22.4 l mol−1), and Ar is the fractional 13C label recovery in breath CO2 after the infusion of labelled acetate, calculated as:
| (7) |
where F is infusion rate of [1,2-13C]acetate (mol min−1). Plasma palmitate oxidation (Rox; mol min−1) can subsequently be calculated as:
| (8) |
where Rd palmitate is the rate of disappearance of palmitate (mol min−1), F is the palmitate infusion rate (mol min−1) and 16 is the number of carbon atoms in palmitate. Total plasma FFA oxidation was calculated by dividing palmitate oxidation rate by the fractional contribution of plasma palmitate to total plasma FFA concentration. The contribution of plasma FFA oxidation to total fat oxidation was calculated by assuming that the molecular mass of TG equals 860 g mol−1 and that every TG molecule contains three fatty acids. The contribution of fat sources other than plasma FFA was calculated by subtracting plasma FFA oxidation from total fat oxidation.
In a previous study, where we applied both a U-13C-labelled and a 6,6-2H2-labelled glucose tracer (Jeukendrup et al. 1999) during moderate-intensity exercise, it was shown that the Rd of plasma glucose measured with [6,6-2H2]glucose was identical to the oxidation rate of plasma glucose measured as the 13CO2 production during a [U-13C]glucose infusion. Therefore, plasma glucose oxidation rate was calculated as:
| (9) |
Muscle glycogen oxidation was calculated by subtracting plasma glucose oxidation from total carbohydrate oxidation.
Statistics
All data are expressed as means ± s.e.m. To compare tracer kinetics and oxidation rates of the different substrate sources between resting and exercise conditions and between the different exercise stages, we used a two-factor analysis of variance (ANOVA) for repeated measures. Scheffé's post hoc test was applied in cases of a significant (P < 0.05) F ratio to locate the differences. The same statistical methodology was used to compare muscle and mean plasma metabolite concentrations. Simple linear regression was used to investigate the correlation between free carnitine concentration and the relative contribution of fat oxidation to total energy expenditure.
RESULTS
Tracer kinetics
Breath 13CO2/12CO2 ratios, and plasma palmitate enrichment and concentration are shown in Fig. 1A, B and C, respectively. As plasma palmitate concentration showed a small increase with time at each exercise intensity (Fig. 1C), non-steady-state Steele equations were applied. With the exception of Ra palmitate at the 75 % Wmax workload, Ra, Rd and Rox palmitate were significantly higher during exercise compared to resting values (Table 1). Differences in Ra palmitate between exercise stages did not reach statistical significance. Rd and Rox palmitate were significantly lower during the 75 % Wmax compared to the 40 and 55 % Wmax workload, respectively. The percentage of Ra palmitate oxidised (% Raox) was higher during exercise compared to resting values, but no significant differences were observed between exercise stages (Table 1). Plasma glucose enrichment and concentration data are shown in Fig. 2A and B, respectively. Since plasma glucose levels showed a small increase with time, particularly at the highest exercise intensity (Fig. 2B), non-steady-state Steele equations were applied. Ra and Rd glucose showed a non-significant increase from rest to exercise and were significantly increased as exercise intensity was further increased compared to preliminary values at rest and during exercise (Table 1).
Figure 1. Palmitate tracer data.
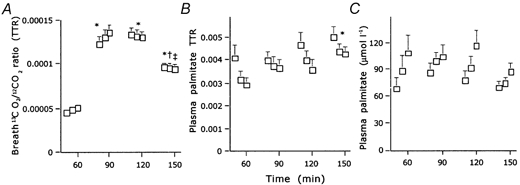
A, breath 13CO2/12CO2 ratio (mean + s.e.m..); B, plasma [U-13C]palmitate enrichment (mean + s.e.m..); C, plasma palmitate level (mean + s.e.m..). TTR, tracer/tracee ratio. * Significantly different from the mean resting value, † significantly different compared to the mean value observed during exercise at 40 % of the maximal workload (Wmax), ‡ significantly different from the mean value observed during exercise at 55 % Wmax (P < 0.05).
Table 1.
Tracer kinetics at rest and during cycling exercise at 40, 55 and 75 % of Wmax
| Rest | 40% Wmax | 55% Wmax | 75% Wmax | ||
|---|---|---|---|---|---|
| Ra | Palmitate | 2.83 ± 0.29 | 4.68 ± 0.38* | 4.53 ± 0.48* | 3.83 ± 0.24 |
| Rd | Palmitate | 2.67 ± 0.27 | 4.60 ± 0.37* | 4.38 ± 0.47* | 3.76 ± 0.22*† |
| Rox | Palmitate | 1.02 ± 0.18 | 3.64 ± 0.41* | 3.86 ± 0.41* | 2.98 ± 0.24*‡ |
| %Raox | Palmitate | 34.5 ± 2.96 | 76.9 ± 4.64* | 85.2 ± 3.47* | 77.9 ± 3.62* |
| Ra | Glucose | 16.9 ± 1.11 | 25.3 ± 2.12 | 38.9 ± 2.10*† | 71.8 ± 4.70*†‡ |
| Rd | Glucose | 16.6 ± 0.93 | 24.3 ± 2.33 | 37.6 ± 1.99*† | 66.8 ± 4.64*†‡ |
Ra, rate of appearance; Rd, rate of disappearance; Rox, rate of oxidation (μmol kg−1 min−1); %Raox, percentage Ra palmitate oxidised. Values are expressed as means ±s.e.m. (n = 8).
Significantly different from resting values (P < 0.05)
significantly different from values at 40 % Wmax workload (P < 0.05)
significantly different from values at 55 % Wmax workload (P < 0.05).
Figure 2. Glucose tracer data.
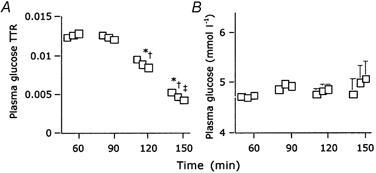
A, plasma [6,6-2H2]glucose enrichment (mean + s.e.m..); B, plasma glucose level (mean + s.e.m..). * Significantly different from the mean resting value, † significantly different from the mean value observed during exercise at 40 % Wmax, ‡ significantly different from the mean value observed during exercise at 55 % Wmax (P < 0.05).
Substrate utilisation
Plasma concentrations of FFA, glycerol and lactate are shown in Fig. 3. Energy expenditure and the use of different substrate sources at rest and during exercise are illustrated in Fig. 4. Oxidation rates of the different substrates during exercise are summarised in Table 2. The 40, 55 and 75 % Wmax workloads applied in this study corresponded to 44 ± 1, 57 ± 1 and 72 ± 2 % V̇O2,max, respectively. At rest, fat oxidation averaged 0.08 ± 0.02 g min−1 and provided 56 ± 11 % of the total energy expenditure. Carbohydrate oxidation rate at rest averaged 0.15 ± 0.04 g min−1 (44 ± 11 %). During exercise at 40 % Wmax, total fat and carbohydrate oxidation rates increased (0.68 ± 0.11 and 1.44 ± 0.35 g min−1, respectively; P < 0.05) and provided 55 ± 9 and 46 ± 9 % of the total energy expenditure, respectively.
Figure 3. Blood plasma metabolites.
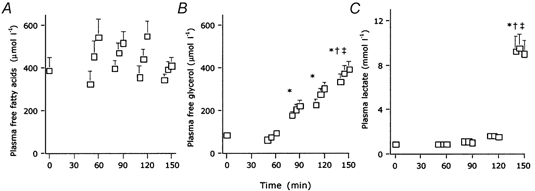
Plasma concentrations of free fatty acids (A), free glycerol (B) and lactate (C; means + s.e.m..) during the rest period (t = 0–60 min), and exercise at 40 %Wmax (t = 60–90 min), 55 % Wmax (t = 90–120 min) and 75 % Wmax (t = 120–150 min). * Mean plasma level significantly different from the mean resting values, † mean plasma levels significantly different from the 40 % Wmax values, ‡ mean plasma levels significantly different from the 55 % Wmax values (P < 0.05).
Figure 4. Energy expenditure and fuel selection.
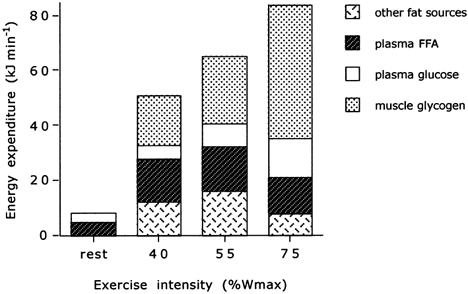
Values are means. FFA, free fatty acid.
Table 2.
Contribution of substrates to total energy expenditure during cycling exercise at 40, 55 and 75 % Wmax
| 40% Wmax | 55% Wmax | 75% Wmax | |
|---|---|---|---|
| Free fatty acids | 0.39 ± 0.04 (31 %) | 0.41 ± 0.04 (25 %) | 0.31 ± 0.02 (15 %)‡ |
| Other fat sources | 0.29 ± 0.05 (24 %) | 0.39 ± 0.03 (24 %) | 0.20 ± 0.04 (9 %)‡ |
| Plasma glucose | 0.33 ± 0.03 (10 %) | 0.51 ± 0.03 (13%)† | 0.90 ± 0.06 (18%)†‡ |
| Muscle glycogen | 1.11 ± 0.12 (35 %) | 1.53 ± 0.13 (38%)† | 3.00 ± 0.10 (58 %)†‡ |
Oxidation rates (g min−1) at 40, 55 and 75 % Wmax are expressed as means ±s.e.m. (n = 8). The percentages given in parentheses indicate the relative contribution of the substrate source to total energy expenditure.
Significantly different from the 40 % Wmax workload (P < 0.05)
significantly different from the 55% Wmax workload (P < 0.05).
Increasing the exercise intensity to 55 % Wmax resulted in a non-significant increase in total fat and a significant increase in total carbohydrate oxidation rates (0.79 ± 0.15 g min−1, or 50 ± 9 %, and 2.04 ± 0.41 g min−1, or 51 ± 9 %, respectively). Plasma glucose and muscle glycogen oxidation rates were significantly higher compared to the 40 % Wmax workload (Table 2). The changes in the oxidation rates of FFAs and other fat sources did not reach statistical significance when compared to the 40 % Wmax workload (Table 2).
Increasing the exercise intensity up to 75 % Wmax resulted in a marked decrease in total fat oxidation, both absolute and relative, compared to that of the other exercise intensities (0.51 ± 0.14 g min−1, or 24 ± 6 %; P < 0.05). This decrease was accounted for by significant decreases in the oxidation rate of both FFAs and other fat sources compared to the 55 % Wmax workload. Total carbohydrate oxidation rate markedly increased (3.91 ± 0.32 g min−1, or 76 ± 6 %; P < 0.05), which was accounted for by significant increases in both plasma glucose and muscle glycogen oxidation rates compared to those at the other workloads (Table 2). Plasma FFA concentration tended to increase during each exercise stage, but did not differ between the different exercise intensities, although it tended to be lower at the highest workload (Fig. 3). Plasma glycerol concentration was higher during exercise compared to resting conditions and was significantly increased at the 75 % Wmax workload compared to that at the 40 and 55 % Wmax workloads. Plasma lactate levels increased substantially at the 75 % Wmax workload (Fig. 3). In the four subjects who performed three additional trials at 40, 55 and 75 % Wmax, we observed no significant differences in total fat oxidation rates compared to the main exercise protocol (0.64 ± 0.06, 0.75 ± 0.10 and 0.35 ± 0.15 g min−1, respectively). The decrease in the contribution of fat oxidation to total energy expenditure at the 75 % Wmax workload tended to be greater under conditions when no preliminary exercise was performed.
Muscle metabolites
Muscle PDC activation and citrate concentration increased in parallel with exercise intensity, and after exercise at 75 % Wmax were significantly greater than the resting values (Table 3). Muscle glycogen concentration decreased with increasing exercise intensity, but was not significantly different from the resting concentration until after exercise at 55 % of Wmax (Table 3). Muscle glycogen concentration following the exercise at 75 % Wmax was significantly lower than after exercise at 40 and 55 % Wmax. There was no significant accumulation of muscle lactate until after exercise at 55 % Wmax (Table 3). Furthermore, muscle lactate concentration was greater after exercise at 75 % Wmax compared with that at rest and at 40 and 55 % Wmax. Muscle acetylcarnitine concentration increased with exercise intensity, and free carnitine decreased in a corresponding manner. As a result, 69 ± 4 % of the total carnitine pool had become acetylated at the highest exercise intensity, compared to 13 ± 3 % at rest. The sum of muscle free carnitine and acetylcarnitine concentration remained unchanged during exercise.
Table 3.
Metabolite concentrations in skeletal muscle at rest and following three 30 min bouts of exercise at 40, 55 and 75 % of Wmax
| Rest | 40% Wmax | 55% Wmax | 75 % Wmax | |
|---|---|---|---|---|
| Glycogen | 526 ± 22 | 414 ± 24 | 386 ± 31* | 200 ± 31*†‡ |
| Lactate | 6.3 ± 0.9 | 11.5 ± 1.7 | 15.6 ± 3.4 * | 29.5 ± 2.4*†‡ |
| Citrate | 0.84 ± 0.09 | 1.06 ± 0.15 | 1.33 ± 0.13 | 1.39 ± 0.11* |
| PDCa | 0.57 ± 0.08 | 1.18 ± 0.10 | 1.23 ± 0.17 | 1.67 ± 0.32* |
| Free carnitine | 15.87 ± 1.17 | 11.25 ± 1.13* | 10.29 ± 0.84* | 5.61 ± 0.76 *†‡ |
| Acetylcarnitine | 2.3 ± 0.4 | 7.3 ± 1.1* | 8.4 ± 1.3* | 12.5 ± 1.1 *†‡ |
| Total carnitine | 18.1 ± 1.1 | 18.5 ± 0.9 | 18.7 ± 0.9 | 18.1 ± 1.0 |
Values are presented as means ±s.e.m. (n = 8) and are expressed as mmol (kg dry muscle)−1 except for the transformation of pyruvate dehydrogenase complex (PDC) into its active form (PDCa), which is given in mmol min−1 (kg wet muscle)−1.
Significantly different from resting values (P < 0.05)
significantly different from the 40 % Wmax workload (P < 0.05)
significantly different from the 55 % Wmax workload (P < 0.05).
DISCUSSION
The present study is the first to make use of a [U-13C]palmitate tracer during incremental exercise in order to estimate directly plasma FFA oxidation from the 13CO2 production in expired air. Careful corrections were made for tracer incorporation into TCA cycle metabolites using an acetate recovery factor (Sidossis et al. 1995a,b; Schrauwen et al. 1998), with measurements being made at each exercise intensity. Romijn et al. (1993) have also presented estimates of the utilisation of different substrate sources as a function of exercise intensity. They, however, used [2H2]palmitate and [6,6-2H2]glucose tracers and assumed that the Rd of both of these tracers equalled the oxidation rate (Rox). Therefore, their reported values for plasma FFA and glucose oxidation rates were maximal estimates, while their reported values for muscle glycogen and TG fat source oxidation rate, both calculated as total macronutrient oxidation minus oxidation of the plasma source, were minimal estimates. The present data show clearly that the Rox of palmitate represented on average 81.7 % of the Rd during exercise (Table 1), implying that plasma FFA oxidation has been overestimated in the past, and conversely that TG oxidation has been underestimated. Furthermore, the data show that the contribution of plasma FFA and TG fat sources was about 50 % each at each exercise intensity. We also correctly assumed that total fat oxidation minus plasma FFA oxidation equals the sum of intramuscular plus lipoprotein-derived TG oxidation, while others (e.g. Romijn et al. 1993, 1995; Sidossis et al. 1997), for reasons that are not immediately obvious, excluded lipoprotein-derived TG as an important energy source during exercise. Finally, we have recently validated the assumption that the Rd of glucose equals its oxidation rate during moderate- to high-intensity exercise (Jeukendrup et al. 1999), indicating that our estimates of blood glucose and muscle glycogen oxidation are about equal to those published previously (Romijn et al. 1993).
In order to eliminate day-to-day variations, we quantified substrate utilisation at different exercise intensities within a single trial. Furthermore, we investigated possible accumulative effects of the incremental protocol on substrate utilisation by asking four of the subjects to perform three additional single trials at 40, 55 and 75 % Wmax. There were no differences in total fat and carbohydrate oxidation rates when comparing these single trials with the incremental protocol. The relative decrease in fat oxidation at 75 % Wmax tended to be even greater when the subjects started exercise at the high-intensity workload immediately after warming up. This indicates that emptying of the intramuscular TG stores did not seem to play a role in the reduction of fat oxidation observed at 75 % Wmax.
In the present study, total fat and carbohydrate oxidation rates increased from rest to exercise (Table 2). However, the relative contribution of fat oxidation to total energy expenditure did not change from resting conditions to exercise, and remained unchanged when exercise was increased up to 55 % Wmax (Fig. 4). No significant changes were found in the relative contribution of the different substrate sources to total energy expenditure. However, as exercise intensity was further increased up to 75 % Wmax, substrate utilisation changed markedly. Total body fat oxidation rate decreased by 34 ± 7 % compared to that at the 55 % Wmax workload (P < 0.05), and this was accounted for by a reduction in the oxidation rate of plasma FFAs and of other fat sources (i.e. the sum of intramuscular and lipoprotein-derived TG; Table 2 and Fig. 4). A reduction in plasma FFA availability did not seem to play a role in reducing fat oxidation rates, as Ra palmitate (Table 1) and the plasma concentrations of palmitate (Fig. 1) and total FFAs (Fig. 3) did not decrease significantly at the highest workload. As blood flow will have been greatest at the highest exercise intensity, this implies that the arterial FFA supply will also have been greatest at this time. In contrast to our data, Romijn et al. (1995) observed a significant reduction in Ra palmitate during high-intensity exercise, which may be explained by the fact that they applied a slightly higher (relative) workload. However, elevation of plasma FFA availability by lipid and heparin infusion only partially restored fat oxidation at the highest exercise intensity (Romijn et al. 1995). Therefore, both studies indicate clearly that the observed decrease in total fat oxidation cannot be accounted for by a decreased plasma FFA availability and that the ‘glucose-FFA cycle’ (Randle et al. 1963) does not operate during high-intensity exercise.
Suggestions have also been made that the transport of FFAs across the plasma membrane limits FFA uptake and oxidation by muscle (Bonen et al. 1999, 2000). Three carrier-mediated transporters for long-chain fatty acids have been identified in the plasma membrane (Bonen et al. 1999). Exercise (muscle contraction) has recently been shown to lead to parallel increases in the translocation of one of these transporters (fatty acid translocase) from endosomal membranes to the plasma membrane, and the fatty acid uptake by giant plasma membrane vesicles in an intensity-dependent manner (Bonen et al. 2000). This recent observation makes it unlikely that the reduction in fat oxidation is caused by a down-regulation of the fatty acid transport capacity during high-intensity exercise. In accordance with this, Kiens et al. (1999) observed an increase in intramyocellular FFA concentration in humans when exercise intensity was increased from 65 to 90 % V̇O2,max, implying that the reduction in fat oxidative capacity during high-intensity exercise is not determined by a reduced intramyocellular availability of fatty acid.
Sidossis et al. (1997) showed in the setting of a constant plasma FFA availability that long-chain fatty acid oxidation, but not medium-chain fatty acid oxidation, is inhibited during high-intensity exercise and not during moderate-intensity exercise. These data were taken to suggest that the decrease in fat oxidation during high-intensity exercise was the result of a direct inhibition of long-chain fatty acid entry into the mitochondria (Sidossis et al. 1997). However, the suggestion that inhibition of CPT I, by increasing concentrations of malonyl-CoA, plays a pivotal role in this decrease in fat oxidation (Sidossis et al. 1997) has not been confirmed by measurements of malonyl-CoA concentration in rat (Rasmussen & Winder, 1997; Odland et al. 1998) and human muscle (Odland et al. 1998). More recently, Dean et al. (2000) showed that in human skeletal muscle acetyl-CoA carboxylase activity, and to a lesser extent malonyl-CoA concentration, actually decreases during exercise, with the magnitude of the decrease generally paralleling exercise intensity. Consequently, the observed decrease in fat oxidation during high-intensity exercise cannot be attributed to an increase in muscle malonyl-CoA concentration (Winder & Hardie, 1996; Odland et al. 1998; Winder, 1998; Dean et al. 2000).
We hypothesised that during light- to moderate-intensity exercise (40 % Wmax), flux through the PDC reaction would be less than TCA cycle flux, which would result in minimal acetylation of the carnitine pool. With increasing exercise intensity up to a moderate workload (55 % Wmax), glycolytic and PDC flux would only be marginally greater than the flux through the TCA cycle, resulting in some muscle lactate and acetylcarnitine accumulation. In contrast, during high-intensity exercise (75 % Wmax), flux through the glycolytic pathway and the PDC reaction would greatly exceed flux through the TCA cycle, thereby markedly increasing muscle lactate and acetylcarnitine concentrations and reducing free carnitine availability. With this in mind, we designed an experimental protocol to test the hypothesis that the expected reduction in fat oxidation during high-intensity exercise would coincide with a marked increase in glycolytic and PDC flux, leading to a marked decrease in muscle free carnitine availability. We further hypothesised that this decrease in free carnitine availability could possibly be directly responsible for the decreased fat oxidation, by limiting long-chain fatty acid entry into the mitochondria. In support of this hypothesis, the data from the present study clearly show marked increases in glycolytic and PDC flux (Table 3) with increasing exercise intensity, which are paralleled by a marked decline in muscle free carnitine availability (Table 3) and a concomitant decrease in fat oxidation (Fig. 4). Many of these observations have been published before as isolated findings (Harris et al. 1987; Hiatt et al. 1989; Sahlin, 1990; Constantin-Teodosiu et al. 1991; Dyck et al. 1993; Howlett et al. 1998), but this is the first study to utilise a fully integrated approach measuring both substrate flux using contemporary stable isotope tracer methodology and quantitative intermediary metabolism in muscle.
The key question is whether the observed decrease in whole-muscle free carnitine concentration was sufficient to reduce CPT I activity. The muscle free carnitine concentration in the present study decreased to 5.6 ± 0.8 mmol (kg dry muscle)−1 (Table 3) or 1.25 ± 0.16 mmol (kg wet muscle)−1. In addition, it is likely that during exercise the concentration of free carnitine in type I fibres was much lower than that measured in mixed-fibre muscle (Constantin-Teodosiu et al. 1996). Therefore, muscle carnitine concentration at the highest workload approaches the reported Km of CPT I for free carnitine measured in human skeletal muscle (0.480 mm; McGarry et al. 1989), and probably reached the Km value in type I fibres. We do not know the local carnitine concentration seen by CPT I in the space between the outer and inner mitochondrial membranes (CPT I is located on the outer side of the inner mitochondrial membrane; Zammit, 1999). Nevertheless, most of the carnitine in skeletal and heart muscle seems to be present in the cytosol (Oram et al. 1975; Zammit, 1999), implying that the cytosolic concentration will be close to the whole-tissue concentration. Therefore, the concentration surrounding the CPT I complex may be lower than the cytosolic concentration. In short, the possibility cannot be excluded that during high-intensity exercise the free carnitine concentration declines to a value that could limit CPT I activity and reduce long-chain fatty acid oxidation. Muscle free carnitine concentration in these healthy subjects during high-intensity exercise ranged from 21 to 55 % of the resting concentration. It is worth noting that in patients with myopathic carnitine deficiency (a genetic defect that manifests itself clinically as muscle weakness, exercise intolerance and excessive lipid accumulation in skeletal muscle; Engel & Angelini, 1973; Di Mauro et al. 1980; Siliprandi et al. 1989), the muscle free carnitine concentration ranges from 5 to 32 % of that of controls.
The observed decline in muscle free carnitine and parallel rise in acetylcarnitine concentration seems to be intrinsically related to a decrease in CoASH concentration in muscle, as this has been shown in both human and equine muscle (Carlin et al. 1990; Constantin-Teodosiu et al. 1991). As most of the CoASH is apparently present in the mitochondrial matrix (Oram et al. 1975; Zammit, 1999), this implies that the availability of mitochondrial CoASH could also limit the rate of β-oxidation during high-intensity exercise. However, assuming that PDC was fully activated at the highest workload in the present study, it can be calculated that the entire pool of cellular CoASH would have become acetylated within 1 s of the initiation of contraction. If this were to occur, the immediate result would be the total inhibition of β-oxidation. An interesting alternative mechanism leading to a down-regulation of CPT I at high exercise intensities has recently been proposed by Starritt et al. (2000). A decrease of the pH from 7.0 to 6.8 substantially reduced CPT I activity measured in vitro. However, the relevance of this to the in vivo situation remains to be established.
In conclusion, this study showed that both carbohydrate and fat oxidation rates increased proportionally as the exercise intensity was increased up to a workload of 55 % Wmax. However, as exercise intensity was increased to 75 % Wmax, both muscle glycogen and plasma glucose oxidation rate markedly increased and fat oxidation rate markedly decreased. This decrease in fat oxidation rate involved a significant decline in the oxidation rate of both plasma FFAs and TG fat sources (sum of intramuscular and lipoprotein-derived TG). Furthermore, the decrease in plasma FFA oxidation was not a consequence of a decrease in plasma FFA availability. We propose that a mechanism involving the down-regulation of CPT I, either by a decrease in free carnitine availability or by a decrease in pH, is the most likely candidate to lead to the reduction in fat oxidation during high-intensity exercise. We would like to propose that this mechanism is relevant to the impairment of fat oxidation seen during exercise in diabetic and obese subjects, as the increase in glycolytic flux is excessive in these subjects at relatively low absolute workloads.
References
- Bergmeyer HU. Methods in Enzymatic Analysis. New York: Academic Press; 1974. [Google Scholar]
- Bergstrom J. Percutaneous needle biopsy of skeletal muscle in physiological and clinical research. Scandinavian Journal of Clinical and Laboratory Investigation. 1975;35:609–616. [PubMed] [Google Scholar]
- Bonen A, Luiken JJ, Arumugam Y, Glatz JF, Tandon NN. Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. Journal of Biological Chemistry. 2000;275:14501–14508. doi: 10.1074/jbc.275.19.14501. [DOI] [PubMed] [Google Scholar]
- Bonen A, Miskovic D, Kiens B. Fatty acid transporters (FABPpm, FAT, FATP) in human muscle. Canadian Journal of Applied Physiology. 1999;24:515–523. doi: 10.1139/h99-033. [DOI] [PubMed] [Google Scholar]
- Carlin JI, Harris RC, Cederblad G, Constantin-Teodosiu D, Snow DH, Hultman E. Association between muscle acetyl-CoA and acetylcarnitine levels in the exercising horse. Journal of Applied Physiology. 1990;69:42–45. doi: 10.1152/jappl.1990.69.1.42. [DOI] [PubMed] [Google Scholar]
- Cederblad G, Bylund AC, Holm J, Scherstén T. Carnitine concentration in relation to enzyme activities and substrate utilization in human skeletal muscles. Scandinavian Journal of Clinical and Laboratory Investigation. 1976;36:547–552. doi: 10.3109/00365517609054477. [DOI] [PubMed] [Google Scholar]
- Cederblad G, Carlin JI, Constantin Teodosiu D, Harper P, Hultman E. Radioisotopic assays of CoASH and carnitine and their acetylated forms in human skeletal muscle. Analytical Biochemistry. 1990;185:274–278. doi: 10.1016/0003-2697(90)90292-h. [DOI] [PubMed] [Google Scholar]
- Colberg SR, Simoneau JA, Thaete FL, Kelley DE. Skeletal muscle utilization of free fatty acids in women with visceral obesity. Journal of Clinical Investigation. 1995;95:1846–1853. doi: 10.1172/JCI117864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Carlin JI, Cederblad G, Harris RC, Hultman E. Acetyl group accumulation and pyruvate dehydrogenase activity in human muscle during incremental exercise. Acta Physiologica Scandinavica. 1991;143:367–372. doi: 10.1111/j.1748-1716.1991.tb09247.x. [DOI] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Casey A, Short AH, Hultman E, Greenhaff PL. The effect of repeated muscle biopsy sampling on ATP and glycogen resynthesis following exercise in man. European Journal of Applied Physiology. 1996;73:186–190. doi: 10.1007/BF00262830. [DOI] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Cederblad G, Hultman E. A sensitive radioisotopic assay of pyruvate dehydrogenase complex in human muscle tissue. Analytical Biochemistry. 1991;198:347–351. doi: 10.1016/0003-2697(91)90437-x. [DOI] [PubMed] [Google Scholar]
- Dean D, Daugaard JR, Young ME, Saha A, Vavvas D, Asp S, Kiens B, Kim K, Witters L, Richter EA, Ruderman N. Exercise diminishes the activity of acetyl-CoA carboxylase in human muscle. Diabetes. 2000;49:1295–1300. doi: 10.2337/diabetes.49.8.1295. [DOI] [PubMed] [Google Scholar]
- Di Mauro S, Trevisan C, Hays A. Disorders of lipid metabolism in muscle. Muscle and Nerve. 1980;3:369–388. doi: 10.1002/mus.880030502. [DOI] [PubMed] [Google Scholar]
- Dyck DJ, Putman CT, Heigenhauser GJ, Hultman E, Spriet LL. Regulation of fat-carbohydrate interaction in skeletal muscle during intense aerobic cycling. American Journal of Physiology. 1993;265:E852–859. doi: 10.1152/ajpendo.1993.265.6.E852. [DOI] [PubMed] [Google Scholar]
- Engel AG, Angelini C. Carnitine deficiency of human skeletal muscle with associated lipid storage myopathy, a new syndrome. Science. 1973;179:899–902. doi: 10.1126/science.179.4076.899. [DOI] [PubMed] [Google Scholar]
- Fritz IB. The effect of muscle extracts on the oxidation of palmitic acid by liver slices and homogenates. Acta Physiologica Scandinavica. 1955;34:367–385. doi: 10.1111/j.1748-1716.1955.tb01256.x. [DOI] [PubMed] [Google Scholar]
- Gutmann I, Wahlefeld AW. L-(+)-Lactate, determination with lactate dehydrogenase and NAD. In: Bergmeyer HU, editor. Methods in Enzymatic Analysis. 2. New York: Academic Press; 1974. pp. 1464–1468. [Google Scholar]
- Harris RC, Foster CV, Hultman E. Acetylcarnitine formation during intense muscular contraction in humans. Journal of Applied Physiology. 1987;63:440–442. doi: 10.1152/jappl.1987.63.1.440. [DOI] [PubMed] [Google Scholar]
- Harris RC, Hultman E, Nordesjo LO. Glycogen, glycolytic intermediates and high-energy phosphates determined in biopsy samples of musculus quadriceps femoris of man at rest. Methods and variance of values. Scandinavian Journal of Clinical and Laboratory Investigation. 1974;33:109–120. [PubMed] [Google Scholar]
- Hiatt WR, Regensteiner JG, Wolfel EE, Ruff L, Brass EP. Carnitine and acetylcarnitine metabolism during exercise in humans. Dependence on skeletal muscle metabolic state. Journal of Clinical Investigation. 1989;84:1167–1173. doi: 10.1172/JCI114281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett RA, Heigenhauser GJ, Hultman E, Hollidge-Horvat MG, Spriet LL. Effects of dichloroacetate infusion on human skeletal muscle metabolism at the onset of exercise. American Journal of Physiology. 1999;277:E18–E25. doi: 10.1152/ajpendo.1999.277.1.E18. [DOI] [PubMed] [Google Scholar]
- Howlett RA, Parolin ML, Dyck DJ, Hultman E, Jones NL, Heigenhauser GJ, Spriet LL. Regulation of skeletal muscle glycogen phosphorylase and PDH at varying exercise power outputs. American Journal of Physiology. 1998;275:R418–425. doi: 10.1152/ajpregu.1998.275.2.R418. [DOI] [PubMed] [Google Scholar]
- Jeukendrup AE, Raben A, Gijsen A, Stegen JH, Brouns F, Saris WH, Wagenmakers AJ. Glucose kinetics during prolonged exercise in highly trained human subjects, effect of glucose ingestion. Journal of Physiology. 1999;515:579–589. doi: 10.1111/j.1469-7793.1999.579ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DE, Simoneau JA. Impaired free fatty acid utilization by skeletal muscle in non-insulin-dependent diabetes mellitus. Journal of Clinical Investigation. 1994;94:2349–2356. doi: 10.1172/JCI117600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiens B, Roemen THM, Van Der Vusse GJ. Muscular long-chain fatty acid content during graded exercise in humans. American Journal of Physiology. 1999;276:E352–357. doi: 10.1152/ajpendo.1999.276.2.E352. [DOI] [PubMed] [Google Scholar]
- Kuipers H, Verstappen FT, Keizer HA, Geurten P, Van Kranenburg G. Variability of aerobic performance in the laboratory and its physiologic correlates. International Journal of Sports Medicine. 1985;6:197–201. doi: 10.1055/s-2008-1025839. [DOI] [PubMed] [Google Scholar]
- Martin IK, Katz A, Wahren J. Splanchnic and muscle metabolism during exercise in NIDDM patients. American Journal of Physiology. 1995;269:E583–E590. doi: 10.1152/ajpendo.1995.269.3.E583. [DOI] [PubMed] [Google Scholar]
- McGarry JD, Woeltje KF, Kuwajima M, Foster DW. Regulation of ketogenesis and the renaissance of carnitine palmitoyltransferase. Diabetes Metabolism Reviews. 1989;5:271–284. doi: 10.1002/dmr.5610050305. [DOI] [PubMed] [Google Scholar]
- Odland LM, Howlett RA, Heigenhauser GJ, Hultman E, Spriet LL. Skeletal muscle malonyl-CoA content at the onset of exercise at varying power outputs in humans. American Journal of Physiology. 1998;274:E1080–1085. doi: 10.1152/ajpendo.1998.274.6.E1080. [DOI] [PubMed] [Google Scholar]
- Oram JF, Wenger JI, Neely JR. Regulation of long chain fatty acid activation in heart muscle. Journal of Biological Chemistry. 1975;250:73–88. [PubMed] [Google Scholar]
- Peronnet F, Massicotte D. Table of nonprotein respiratory quotient: an update. Canadian Journal of Sports Science. 1991;16:23–29. [PubMed] [Google Scholar]
- Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose-fatty acid cycle: its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- Rasmussen BB, Winder WW. Effect of exercise intensity on skeletal muscle malonyl-CoA and acetyl-CoA carboxylase. Journal of Applied Physiology. 1997;83:1104–1109. doi: 10.1152/jappl.1997.83.4.1104. [DOI] [PubMed] [Google Scholar]
- Romijn JA, Coyle EF, Sidossis LS, Gastaldelli A, Horowitz JF, Endert E, Wolfe RR. Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. American Journal of Physiology. 1993;265:E380–391. doi: 10.1152/ajpendo.1993.265.3.E380. [DOI] [PubMed] [Google Scholar]
- Romijn JA, Coyle EF, Sidossis LS, Zhang XJ, Wolfe RR. Relationship between fatty acid delivery and fatty acid oxidation during strenuous exercise. Journal of Applied Physiology. 1995;79:1939–1945. doi: 10.1152/jappl.1995.79.6.1939. [DOI] [PubMed] [Google Scholar]
- Sahlin K. Muscle carnitine metabolism during incremental dynamic exercise in humans. Acta Physiologica Scandinavica. 1990;138:259–262. doi: 10.1111/j.1748-1716.1990.tb08845.x. [DOI] [PubMed] [Google Scholar]
- Schrauwen P, Van Aggel Leijssen DPC, Lichtenbelt WDV, Van Baak MA, Gijsen AP, Wagenmakers AJM. Validation of the [1,2-C13]acetate recovery factor for correction of [U-C13]palmitate oxidation. Journal of Physiology. 1998;513:215–223. doi: 10.1111/j.1469-7793.1998.215by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidossis LS, Coggan AR, Gastaldelli A, Wolfe RR. A new correction factor for use in tracer estimations of plasma fatty acid oxidation. American Journal of Physiology. 1995a;269:E649–656. doi: 10.1152/ajpendo.1995.269.4.E649. [DOI] [PubMed] [Google Scholar]
- Sidossis LS, Coggan AR, Gastaldelli A, Wolfe RR. Pathway of free fatty acid oxidation in human subjects: implications for tracer studies. Journal of Clinical Investigation. 1995b;95:278–284. doi: 10.1172/JCI117652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidossis LS, Gastaldelli A, Klein S, Wolfe RR. Regulation of plasma fatty acid oxidation during low- and high-intensity exercise. American Journal of Physiology. 1997;272:E1065–1070. doi: 10.1152/ajpendo.1997.272.6.E1065. [DOI] [PubMed] [Google Scholar]
- Siliprandi N, Sartorelli L, Ciman M, Di Lisa F. Carnitine: metabolism and clinical chemistry. Clinica Chimica Acta. 1989;183:3–11. doi: 10.1016/0009-8981(89)90267-2. [DOI] [PubMed] [Google Scholar]
- Starritt EC, Howlett RA, Heigenhauser GJ, Spriet LL. Sensitivity of CPT I to malonyl-CoA in trained and untrained human skeletal muscle. American Journal of Physiology. 2000;278:E462–468. doi: 10.1152/ajpendo.2000.278.3.E462. [DOI] [PubMed] [Google Scholar]
- Steele R. Influences of glucose loading and of injected insulin on hepatic glucose output. Annals of the New York Academy of Sciences. 1959;82:420–430. doi: 10.1111/j.1749-6632.1959.tb44923.x. [DOI] [PubMed] [Google Scholar]
- Timmons JA, Gustafsson T, Sundberg CJ, Jansson E, Greenhaff PL. Muscle acetyl group availability is a major determinant of oxygen deficit in humans during submaximal exercise. American Journal of Physiology. 1998;274:E377–380. doi: 10.1152/ajpendo.1998.274.2.E377. [DOI] [PubMed] [Google Scholar]
- Timmons JA, Poucher SM, Constantin-Teodosiu D, Worrall V, Macdonald IA, Greenhaff PL. Increased acetyl group availability enhances contractile function of canine skeletal muscle during ischemia. Journal of Clinical Investigation. 1996;97:879–883. doi: 10.1172/JCI118490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder WW. Intramuscular mechanisms regulating fatty acid oxidation during exercise. Advances in Experimental Medicine and Biology. 1998;441:239–248. doi: 10.1007/978-1-4899-1928-1_22. [DOI] [PubMed] [Google Scholar]
- Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. American Journal of Physiology. 1996;270:E299–304. doi: 10.1152/ajpendo.1996.270.2.E299. [DOI] [PubMed] [Google Scholar]
- Wolfe RR, Jahoor F. Recovery of labeled CO2 during the infusion of C-1- vs. C-2-labeled acetate: implications for tracer studies of substrate oxidation. American Journal of Clinical Nutrition. 1990;51:248–252. doi: 10.1093/ajcn/51.2.248. [DOI] [PubMed] [Google Scholar]
- Zammit VA. Carnitine acetyltransferases: functional significance of subcellular distribution and membrane topology. Progress in Lipid Research. 1999;38:199–224. doi: 10.1016/s0163-7827(99)00002-8. [DOI] [PubMed] [Google Scholar]