

Abstract
The effects of the IP3-receptor antagonist 2-aminoethyldiphenyl borate (2-APB) on the Ca2+ release-activated Ca2+ current (ICRAC) in Jurkat human T cells, DT40 chicken B cells and rat basophilic leukaemia (RBL) cells were examined.
2-APB elicited both stimulatory and inhibitory effects on Ca2+ influx through CRAC channels. At concentrations of 1–5 μm, 2-APB enhanced Ca2+ entry in intact cells and increased ICRAC amplitude by up to fivefold. At levels ≥ 10 μm, 2-APB caused a transient enhancement of ICRAC followed by inhibition.
2-APB altered the kinetics of fast Ca2+-dependent inactivation of ICRAC. At concentrations of 1–5 μm, 2-APB increased the rate of fast inactivation. In contrast, 2-APB at higher concentrations (≥ 10 μm) reduced or completely blocked inactivation.
2-APB inhibited Ca2+ efflux from mitochondria.
2-APB inhibited ICRAC more potently when applied extracellularly than intracellularly. Furthermore, increased protonation of 2-APB at low pH did not affect potentiation or inhibition. Thus, 2-APB may have an extracellular site of action.
Neither ICRAC activation by passive store depletion nor the effects of 2-APB were altered by intracellular dialysis with 500 μg ml−1 heparin.
ICRAC is present in wild-type as well as mutant DT40 B cells lacking all three IP3 receptor isoforms. 2-APB also potentiates and inhibits ICRAC in both cell types, indicating that 2-APB exerts its effects independently of IP3 receptors.
Our results show that CRAC channel activation does not require physical interaction with IP3 receptors as proposed in the conformational coupling model. Potentiation of ICRAC by 2-APB may be a useful diagnostic feature for positive identification of putative CRAC channel genes, and provides a novel tool for exploring the physiological functions of store-operated channels.
Depletion of Ca2+ from the endoplasmic reticulum (ER) activates Ca2+ entry across the plasma membrane in a variety of cell types, a process known as store-operated Ca2+ entry. Imaging and patch-clamp studies indicate that the store-operated Ca2+ entry mechanism exists in a wide range of tissues (Parekh & Penner, 1997; Putney, 1997; Lewis, 1999). Nevertheless, detailed information about the properties of the channels underlying store-operated Ca2+ entry is available only for a few cell types such as Jurkat T cells, mast cells and RBL cells. The store-operated current in these cells is highly selective for Ca2+ and is called the calcium release-activated Ca2+ current, or ICRAC. The hallmarks of ICRAC include an extremely high selectivity for Ca2+ over monovalent cations, an extremely low apparent unitary conductance for Ca2+, voltage-independent gating, inward rectification and multiple modes of inactivation by Ca2+ (reviewed by Parekh & Penner, 1997; Lewis, 1999). CRAC channels play essential roles in controlling gene expression during the activation of T cells by antigen (Partiseti et al. 1994; Fanger et al. 1995) and in triggering secretion of inflammatory mediators from mast cells stimulated by allergens (Zhang & McCloskey, 1995).
Despite the wealth of electrophysiological data on the properties of ICRAC, neither the mechanism(s) that links store depletion to activation of the current nor the molecular identity of the channels is well understood. Hypotheses presented to explain the mechanism of activation of ICRAC by store depletion have included activation by calcium influx factor, vesicle-mediated insertion of active channels in the membrane and physical coupling with IP3 receptors in the ER membrane (for reviews, see Putney & McKay, 1999; Prakriya & Lewis, 2001a). The last hypothesis, known as the conformational coupling hypothesis (Berridge, 1995), has received support from observations that activation of Ca2+ entry requires close apposition of the ER, where IP3 receptors are localized, and the plasma membrane (Boulay et al. 1999; Patterson et al. 1999; Ma et al. 2000). Further evidence comes from reports of binding and functional interactions between transient receptor potential (TRP)3 channels and IP3 receptors (Kiselyov et al. 1998, 1999a; Boulay et al. 1999). Although the evidence that trp3 encodes a store-operated channel is controversial (Zitt et al. 1997; Hofmann et al. 1999; Ma et al. 2000), it has been reported that an IP3 receptor antagonist, 2-aminoethyldiphenyl borate (2-APB), blocks not only TRP3-mediated but also native store-operated Ca2+ influx in HEK cells (Ma et al. 2000).
2-APB has been described as a non-competitive antagonist of the IP3 receptor and has been shown to inhibit IP3 receptor-induced [Ca2+]i elevations in a variety of cell types (Sugawara et al. 1997; Ma et al. 2000). Inhibition of store-operated Ca2+ entry by 2-APB has been taken as evidence for a role of IP3 receptors in the activation of store-operated channels and therefore as proof for conformational coupling between store-operated channels and IP3 receptors (Ma et al. 2000; van Rossum et al. 2000). However, 2-APB is not yet well characterized in terms of its possible sites of action, and more recent work suggests that 2-APB may inhibit ICRAC in RBL cells independently of the IP3 receptor (Braun et al. 2001). A more thorough characterization of the effects of 2-APB on ICRAC and other Ca2+ transport pathways is needed to test the potential utility of this compound in studying the activation and regulation of store-operated channels such as the CRAC channel.
To explore the possible role of IP3 receptors in the induction and maintenance of ICRAC, we examined the effects of 2-APB in Jurkat T cells, DT40 B cells and RBL cells. We found that 2-APB has complex effects on ICRAC. At low concentrations (≤ 5 μm), 2-APB enhances the size of ICRAC and speeds up fast Ca2+-dependent inactivation of the channels. At higher concentrations, 2-APB inhibits ICRAC while also removing Ca2+-dependent inactivation. These complex effects on ICRAC also occur in mutant DT40 cells that lack IP3 receptors. Thus, our results indicate that 2-APB elicits multiple effects on ICRAC gating by a mechanism that is independent of IP3 receptors. These results have been reported previously in abstract form (Prakriya & Lewis, 2001b).
METHODS
Cells
Jurkat T cells (Jurkat E6–1 cell line) were grown in medium consisting of RPMI 1640 supplemented with 10 % fetal calf serum (FCS), 2 mm glutamine and penicillin-streptomycin. DT40 B cells and DT40 IP3 receptor triple-knockout cells (RIKEN Cell Bank, Ibaraki, Japan) were maintained in a medium containing RPMI 1640 supplemented with 10 % FCS, 5 % chicken serum, 4 mm glutamine, 50 μmβ-mercaptoethanol and penicillin-streptomycin. RBL-2H3 cells were grown in DMEM supplemented with 10 % FCS, 4 mm glutamine and penicillin-streptomycin. All cells were maintained in log-phase growth at 37 °C in 6 % CO2.
Solutions and chemicals
The extracellular solution contained (mm): 155 NaCl, 4.5 KCl, 1 MgCl2, 10 d-glucose and 5 Na-Hepes (pH 7.4) with 2 mm CaCl2 in Ca2+ imaging experiments, and 20 mm CaCl2 or 20 mm BaCl2 in patch-clamp experiments. Ca2+-free solutions contained 1 mm EGTA and 3 mm MgCl2. The intracellular (pipette) solution contained (mm): 150 caesium methanesulfonate, 10 Cs-Hepes, 10 EGTA and 3 MgCl2 (pH 7.3). In some experiments, a weakly buffered internal solution containing 1.2 mm EGTA was used. To isolate ICRAC from inward rectifier K+ currents in RBL-2H3 cells, 10 mm CsCl2 was added to the standard extracellular solution. In experiments involving caged IP3, 15 μm NPE-IP3 (Calbiochem) was included in the patch pipette, and the internal solution contained 1.2 mm EGTA + 0.705 mm CaCl2 (calculated free [Ca2+]= 150 nm) to prevent store depletion prior to IP3 release.
2-APB was the kind gift of Dr K. Mikoshiba (Tokyo University, Japan). In some experiments, 2-APB purchased from Sigma Chemical Co. (St Louis, MO, USA) was used; the drugs from both sources had identical effects on ICRAC. Stock solutions of 2-APB, thapsigargin (TG; Sigma) and oligomycin (Sigma) were prepared in DMSO at concentrations of 20, 1 and 2 mm respectively. Antimycin A1 (Sigma) was dissolved in ethanol at 2 mm. The drugs were diluted to the concentrations indicated in the figure legends and applied to the cells using a multibarrel local perfusion pipette with a common delivery port. The solution exchange time was measured to be < 2 s, based on the rate at which the K+ current reversal potential changed when the external solution was switched from 2 mm K+ to 150 mm K+.
[Ca2+]i imaging
Cells were loaded for 15 min at 37 °C with 1 μm fura-2 AM (Molecular Probes, OR, USA) dissolved in Jurkat medium, then were washed with Jurkat or DT40 medium and stored in the dark until ready for use. Fura-2-loaded cells were plated on glass coverslips coated with poly-d-lysine and were preincubated for several minutes in an external solution containing 0.5 mm Ca2+. Coverslips were mounted on the stage of a Zeiss Axiovert 35 microscope equipped with a ×40 Achrostigmat objective (NA 1.3). Imaging protocols and equipment were as described previously (Hoth et al. 1997). Briefly, cells were alternately illuminated at 350 and 380 nm using band-pass filters (Chroma Technology) and the fluorescence emissions at wavelengths greater than 480 nm were captured with an intensified CCD camera (Hamamatsu, Japan) and digitized and analysed using a VideoProbe imaging system (ETM Systems, Irvine, CA, USA). The 350 nm/ 380 nm ratio images were recorded at intervals of 5 s. [Ca2+]i was calculated from the relationship [Ca2+]i=K(R – Rmin)/(Rmax–R), where the values of K, Rmin and Rmax were determined from an in situ calibration of fura-2 in Jurkat T cells loaded by intracellular dialysis (Lewis & Cahalan, 1989). Rmin and Rmax are the fluorescence ratios determined with internal solutions containing 10 mm EGTA and 10 mm CaCl2, respectively; K was determined using an internal solution containing 180 nm free [Ca2+]i.
Patch-clamp recording
Patch-clamp experiments were conducted in the standard whole-cell recording configuration using recording electrodes that were pulled from 100 μl pipettes, coated with Sylgard and fire polished to a final resistance of 2–5 MΩ. Data were collected with an Axopatch 200 amplifier (Axon Instruments, Foster City, CA, USA) interfaced to an ITC-16 input/output board (Instrutech, Port Washington, NY, USA) and a Macintosh G3 computer. Stimulation, data acquisition and analysis were performed using in-house software developed on the Igor Pro platform (Wavemetrics, Lake Oswego, OR, USA). The command voltage protocol usually consisted of a 100 ms step from the holding potential to −100 mV followed immediately by a 100 ms ramp from −100 to +60 mV, applied every 1–2 s. The holding potential was +30 mV unless otherwise indicated. Current was sampled at a rate of 5 kHz and filtered at 500 Hz offline. All data were corrected for leak currents that were collected using the same voltage stimulus in a Ca2+-free bath solution, after ICRAC induction and before data collection. Unless otherwise indicated, all average results are presented as the mean value ±s.e.m.
Two methods were typically employed to deplete intracellular Ca2+ stores and activate ICRAC. With the first method, stores were passively depleted by dialysing the cells with an internal recording solution containing either 10 or 1.2 mm EGTA. With the second method, cells were preincubated in the recording chamber with Ca2+-free Ringer solution containing 1 μm TG for 5–10 min before seal formation. In the experiments of Fig. 9C, ICRAC was activated by the release of caged IP3; NPE-IP3 was photolysed by a 10 s exposure to unattenuated light from a xenon light source (Lambda-10, Sutter Instruments) passed through a 360 nm interference filter (Chroma Technology).
Figure 9. Heparin does not interfere with the activation of ICRAC by passive store depletion or with the effects of 2-APB.
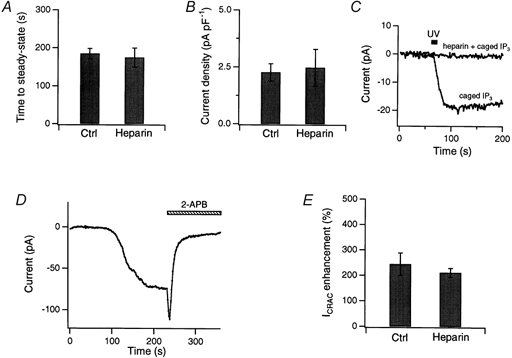
In all experiments except as noted, 500 μg ml−1 heparin was included in the recording pipette, and ICRAC was induced through passive depletion of internal stores by 10 mm EGTA in the patch pipette. A and B, heparin affects neither the rate of ICRAC activation (A) nor its maximal amplitude at steady state (B). The time to steady state was measured as the time to reach 95 % of the maximal current amplitude. Current amplitude was normalized to cell capacitance (average Cm= 9.95 pF). Heparin-treated cells (n = 6) are compared with matched control cells (n = 7). C, heparin loading via the patch pipette effectively inhibited IP3 receptors. In control Jurkat cells lacking heparin, ICRAC was activated by a 10 s UV flash to release caged IP3 (15 μm). With 500 μg ml−1 heparin in the pipette, uncaging IP3 failed to activate ICRAC. Results are representative of four cells loaded with caged IP3 alone and three cells with caged IP3 and heparin. D, 40 μm 2-APB first potentiated and then inhibited ICRAC in a Jurkat cell loaded with heparin via the patch pipette. Shown here are the peak currents during steps to −100 mV from +30 mV. E, summary of the potentiating effects of 5 μm 2-APB in heparin-treated (n = 6) and matched control (n = 5) cells.
RESULTS
Inhibition of store-operated Ca2+ influx and mitochondrial Ca2+ efflux by 2-APB
The effects of 2-APB on Ca2+ influx through CRAC channels were first examined in Ca2+ imaging experiments using fura-2-loaded Jurkat T cells. TG (1 μm), a membrane-permeant inhibitor of the SERCA Ca2+-ATPase in the ER (Thastrup et al. 1989), was applied for ∼500 s in a Ca2+-free solution to deplete intracellular Ca2+ stores and activate CRAC channels (Fig. 1). Subsequent restoration of Ca2+ to the bath produces a rapid elevation of [Ca2+]i due to influx through open CRAC channels, followed by a gradual decline to a high steady-state [Ca2+]i plateau, as the rates of Ca2+ influx and Ca2+ clearance reach a new balance (Hoth et al. 1997). As shown in Fig. 1A, application of 40 μm 2-APB rapidly abolished the [Ca2+]i elevation and brought [Ca2+]i back to baseline within 120 s, similar to its reported effects on store-operated Ca2+ entry in other cell types (Ma et al. 2000; van Rossum et al. 2000). In addition, application of 40 μm 2-APB in a Ca2+-free solution eliminated the subsequent rise in [Ca2+]i upon Ca2+ re-addition (data not shown), indicating that the inhibitory effect required neither external Ca2+ nor elevated [Ca2+]i. Thus, these results confirmed that 2-APB very effectively blocks [Ca2+]i elevations produced by Ca2+ influx through CRAC channels.
Figure 1. Effects of 2-APB on Ca2+ entry through CRAC channels and on Ca2+ release from mitochondria.
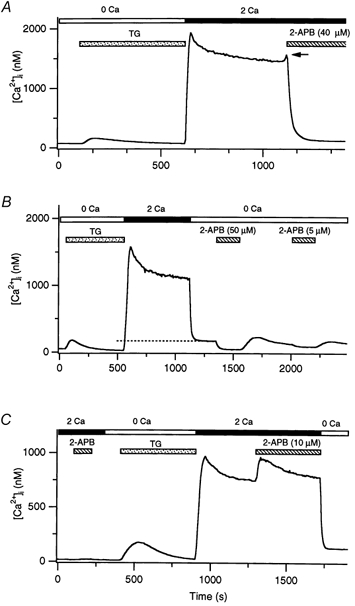
In each panel, TG (1 μm; stippled bar) was applied in Ca2+-free Ringer solution to irreversibly deplete stores and activate CRAC channels in Jurkat T cells. Subsequent addition of 2 mm Ca2+ to the bath (filled bar) elicited a rise in [Ca2+]i. Each trace represents the average of 150–200 cells. A, a high concentration of 2-APB (40 μm; hatched bar) inhibited Ca2+ influx through CRAC channels. The arrow marks a transient enhancement of [Ca2+]i prior to inhibition. B, 2-APB inhibits Ca2+ release by mitochondria. After Ca2+ influx through CRAC channels was terminated by removing extracellular Ca2+, [Ca2+]i fell to a low elevated plateau that reflected the export of Ca2+ from mitochondria (dashed line). Addition of 50 μm 2-APB to the Ca2+-free solution rapidly and reversibly reduced [Ca2+]i to baseline due to inhibition of mitochondrial Ca2+ export. A lower concentration of 2-APB (5 μm) produced a smaller effect. C, a low concentration of 2-APB (10 μm) did not elevate [Ca2+]i in resting cells before internal stores had been depleted by TG. However, after ICRAC activation, 2-APB enhanced the [Ca2+]i rise resulting from influx through CRAC channels.
High doses of 2-APB reduced [Ca2+]i relatively rapidly to baseline values, even when preceded by a long, large [Ca2+]i elevation (Fig. 1A). Previous work has shown that mitochondria take up a substantial amount of Ca2+ during such extended periods of influx, such that when influx is terminated (e.g. by removal of extracellular Ca2+), [Ca2+]i falls to a low plateau value that reflects a balance between Ca2+ pumping across the plasma membrane and Ca2+ efflux from the loaded mitochondria (marked with a dashed line in Fig. 1B; Hoth et al. 1997). This plateau declines with a time constant of hundreds of seconds as the mitochondria slowly unload their Ca2+. The lack of this low [Ca2+]i plateau in the presence of 2-APB suggests that the drug might prevent Ca2+ release from mitochondria. To test this idea, we applied 2-APB in Ca2+-free solution after a period of prolonged [Ca2+]i elevation. Addition of 50 μm 2-APB during the mitochondria-dependent Ca2+ plateau immediately caused [Ca2+]i to decline back to baseline, consistent with an acute inhibition of mitochondrial Ca2+ export (Fig. 1B). In principle, this result could also be produced by an increased rate of Ca2+ extrusion across the plasma membrane; however, 2-APB did not alter the initial rate of [Ca2+]i decline when was first removed (data not shown), demonstrating that the drug does not detectably affect Ca2+ pumps in the plasma membrane. Washout of the drug allowed Ca2+ to recover to the plateau level, often with a small overshoot. This behaviour would be expected from the resumption of mitochondrial export and a delayed approach to a new equilibrium between mitochondrial Ca2+ export and Ca2+ clearance across the plasma membrane. A lower concentration of 2-APB (5 μm) caused less-complete inhibition of mitochondrial export (Fig. 1B). Because a Na+-Ca2+ exchanger in the mitochondrial inner membrane is believed to be responsible for carrying out a significant proportion of Ca2+ export (Gunter et al. 2000), we speculate that 2-APB may inhibit the mitochondrial Na+-Ca2+ exchanger. We did not explore the mechanism of this mitochondrial effect further, but instead focused on the effects of 2-APB on Ca2+ influx.
Potentiation of store-operated Ca2+ entry by 2-APB
An unexpected observation was that the inhibition of Ca2+ influx was always preceded by a transient elevation of [Ca2+]i (Fig. 1A, arrow). When the concentration of 2-APB was reduced to 10 μm, the [Ca2+]i rise induced by 2-APB became much more pronounced and sustained (Fig. 1C). Interestingly, this potentiation of Ca2+ influx occurred only after store depletion; no effects were seen after application of dilute 2-APB to resting T cells (Fig. 1C). These results indicate that, in addition to its known ability to inhibit store-operated Ca2+ influx, 2-APB can also potentiate Ca2+ entry in Jurkat T cells.
The progression from potentiation to inhibition of Ca2+ entry as 2-APB concentration is increased is seen most clearly in the dose-response experiment of Fig. 2. Whereas low concentrations of 2-APB (≤ 5 μm) elicited [Ca2+]i elevations that were largely sustained during the 400 s of drug application, high concentrations (≥ 10 μm) produced a transient rise followed by a progressive fall in [Ca2+]i. The rate of both the rise and the decay of [Ca2+]i were accelerated as the concentration of 2-APB was increased.
Figure 2. Potentiation and inhibition of Ca2+ entry by 2-APB are concentration dependent.
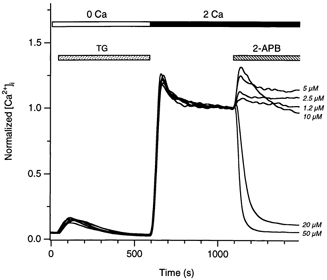
Six representative experiments with increasing concentrations of 2-APB are superimposed. In each experiment, TG followed by re-addition of 2 mm Ca2+ was used to elicit a high [Ca2+]i plateau. As [2-APB] was increased, sustained potentiation of the [Ca2+]i rise gave way to transient potentiation followed by inhibition. Data were normalized to the [Ca2+]i measured immediately before addition of 2-APB, to allow better comparison between the different experiments. The average [Ca2+]i at this time was 1342 ± 20 nm (mean ±s.e.m.).
Potentiation and blockade of ICRAC by 2-APB
Several mechanisms could in principle account for the effects of 2-APB on [Ca2+]i in the imaging experiments shown in Figs 1 and 2. These include potentiation and block of CRAC channels as well as mechanisms unrelated to CRAC channels, such as alteration of the membrane potential and driving force for Ca2+ entry by modulation of K+ channels. To distinguish between these possibilities, we directly examined the effects of 2-APB on ICRAC in whole-cell recordings. ICRAC was induced in these experiments by passively depleting internal Ca2+ stores through intracellular dialysis with 10 mm EGTA in the patch pipette (Hoth & Penner, 1992; Zweifach & Lewis, 1993), and the peak amplitude of ICRAC was measured during repetitive hyperpolarizing pulses to −100 mV.
The effects of 2-APB on the current paralleled those on [Ca2+]i. Thus, 5 μm 2-APB evoked a large sustained increase in the amplitude of the inward current at −100 mV (Fig. 3A, top trace), whilst 20 μm 2-APB induced an initial increase in current followed by a slow decline in ICRAC amplitude (Fig. 3A, middle trace). Increasing the 2-APB concentration to 40 μm increased the rate of potentiation and the subsequent blockade of ICRAC (Fig. 3A, bottom trace). The decay of ICRAC followed an approximately single-exponential time course. No effects were seen in control experiments in which the corresponding concentration of DMSO was applied (data not shown). Therefore, these results suggest that whereas high concentrations of 2-APB block ICRAC, low concentrations activate an additional inward current.
Figure 3. Potentiation and inhibition of ICRAC by 2-APB.
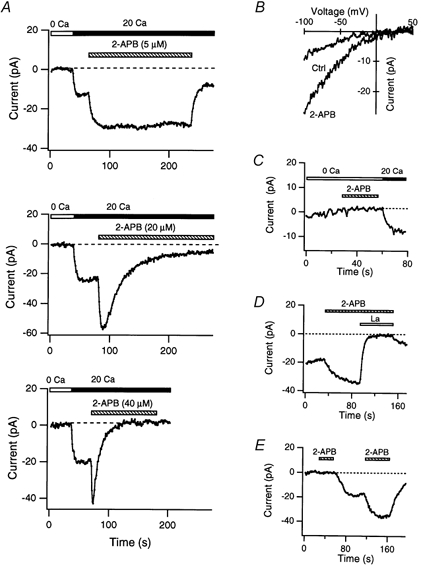
A, effects of 2-APB on ICRAC. Current was activated by depleting Ca2+ stores passively with 10 mm EGTA in the recording pipette. 2-APB at 5 μm (top) enhanced ICRAC amplitude over the duration of drug application, whereas 20 μm (middle) and 40 μm 2-APB (bottom) initially enhanced, then inhibited, ICRAC. Shown here are the peak Ca2+ currents elicited during 200 ms steps to −100 mV from a holding potential of +10 mV. B–E, evidence that the additional inward current activated by 2-APB is ICRAC. B, the current-voltage relation of the inward current activated by store depletion before and after 2-APB (10 μm). C, 2-APB (10 μm) did not activate inward current in Ca2+-free Ringer solution. ICRAC was induced passively as described above. D, the inward current activated by 2-APB (10 μm) is blocked by extracellular La3+ (1.5 μm). E, 2-APB (5 μm) does not elicit inward current before ICRAC is activated. After the delayed induction of ICRAC by intracellular EGTA (1.2 mm), application of 2-APB potentiated ICRAC robustly.
Several lines of evidence indicate that the additional inward current activated by 2-APB is ICRAC itself rather than an unrelated current. First, the I–V profile and prominent inward rectification of the current in 2-APB were identical to that of ICRAC (Fig. 3B). Second, consistent with the high Ca2+ selectivity of ICRAC, the inward current was absent in nominally Ca2+-free external solutions (Fig. 3C). Third, the 2-APB-induced current was completely blocked by low concentrations of La3+ (1.5 μm; Fig. 3D) and SKF 96365 (not shown), two known inhibitors of ICRAC (Franzius et al. 1994; Aussel et al. 1996). Finally, 2-APB only induced inward current subsequent to store depletion and activation of ICRAC by EGTA-containing internal solutions. In the experiment shown in Fig. 3E, brief application of 2-APB (5 μm) before the passive induction of ICRAC had no effect, but after ICRAC was activated, the same dose caused robust potentiation. Taken together with the evidence that a low dose of 2-APB elevated [Ca2+]i only after stores were depleted (Fig. 1C), these data indicate that the transient inward current activated by 2-APB is ICRAC itself. Thus, we conclude that 2-APB elicits both excitatory and inhibitory effects on ICRAC.
The potentiating and inhibitory effects of 2-APB on ICRAC differed in two ways. First, although potentiation of ICRAC was rapidly reversible (Fig. 3A, top trace; average time constant of recovery, τrec= 6.2 ± 1.3 s, n = 6), inhibition of ICRAC in Jurkat cells reversed extremely slowly following removal of the drug (Fig. 3A, bottom trace and Fig. 6A). In four cells exposed to 40 μm 2-APB, washout of the drug caused the current to recover by an average of 22 % after 160 s, suggesting a τrec of > 600 s. A second difference was in the concentration dependence of the two effects. Current potentiation was evident at lower drug concentrations than those that inhibited ICRAC. Unfortunately, an overlap in the effective concentrations for potentiation and inhibition complicated efforts to estimate separately the K1/2 values for the two processes. The concentration dependence of ICRAC enhancement measured directly from the peak current amplitudes before and after exposure to 2-APB is shown in Fig. 4A (filled circles). Potentiation peaked around 5 μm, then decreased with further increases in 2-APB concentration, presumably because of increasing current inhibition. To attempt to correct for the effect of inhibition and estimate the true extent of potentiation by 2-APB at these concentrations, we fitted a single-exponential function to the current decay induced by 2-APB and extrapolated the current amplitude back to the time the drug was first applied (Fig. 4A, open circles). The estimated potentiation at different concentrations of 2-APB was then fitted with a Hill equation, yielding an apparent K1/2 of ∼3 μm for potentiation. By contrast, the apparent K1/2 for ICRAC blockade as measured by inhibition of the peak current was ∼10 μm (Fig. 4B). This sensitivity of CRAC channels to inhibition by 2-APB is identical to that reported by Ma et al. (2000) for inhibition of TRP3-mediated Sr2+ entry in transfected HEK 293 cells. However, it is significantly lower than the IC50 for inhibition of Ca2+ release from IP3 receptors (25–42 μm; Maruyama et al. 1997; Ma et al. 2000).
Figure 6. High concentrations of 2-APB eliminate fast inactivation of ICRAC.
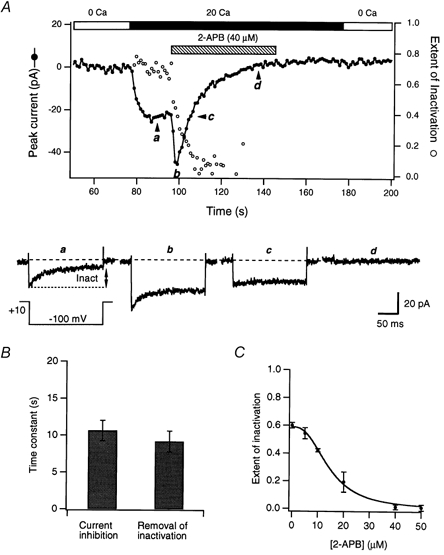
A, plot of peak ICRAC (•) and the extent of inactivation (○) during pulses to −100 mV before and after exposure to 40 μm 2-APB (hatched bar). The extent of inactivation was measured at the end of the 250 ms test pulses (It= 250 ms; see legend to Fig. 5). Examples of currents at various time points are shown below to illustrate the removal of fast inactivation. B, inhibition of ICRAC and removal of fast inactivation occur at a similar rate. The time courses of ICRAC inhibition and reduction in the extent of inactivation were fitted with single-exponential functions and the time constants from the resulting fits are shown (40 μm 2-APB; n = 5). C, dose dependence of the removal of inactivation by 2-APB. The extent of inactivation was measured 100 ms after steps to −100 mV, at a time when the effects of 2-APB had reached steady state. Each point represents the mean of three to six cells.
Figure 4. Concentration dependence of potentiation and inhibition of ICRAC by 2-APB.
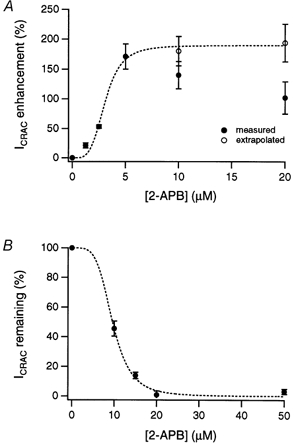
A, extent of ICRAC potentiation as a function of 2-APB concentration. Filled circles show peak currents measured from experiments similar to those in Fig. 3. To correct for the overlap of potentiation and inhibition at 2-APB concentrations ≥ 10 μm, peak currents were also estimated by back-extrapolation (open circles; see text). The dashed line indicates a fit to the Hill equation Y =Ymax/(1 + ([2-APB]/K1/2)nH), where Ymax is the maximal potentiation, K1/2 is the concentration that yields half-maximal potentiation, and nH is the apparent cooperativity of the process. The best fit was obtained with K1/2= 3.1 μm and nH= 4.1. B, degree of ICRAC inhibition as a function of 2-APB concentration. Inhibition was calculated from the steady-state current level relative to the peak current after addition of 2-APB. The dashed line is a fit of the Hill equation, with K1/2= 9.6 μm and nH= 4.2. Experimental conditions were as described for Fig. 3; holding potential =+30 mV. In A and B, each point represents the mean ±s.e.m. of three to five cells.
Low concentrations of 2-APB enhance fast inactivation of ICRAC
Ca2+ entering through CRAC channels causes them to inactivate over tens of milliseconds during large hyperpolarizing pulses (Hoth & Penner, 1993; Zweifach & Lewis, 1995b; Fierro & Parekh, 1999a). Fast inactivation is a biexponential process and is believed to result from Ca2+ binding to multiple intracellular sites located within several nanometers of the channel pore (Zweifach & Lewis, 1995b). Interestingly, 2-APB altered fast inactivation in parallel with its effects on the amplitude of ICRAC. At low concentrations that caused potentiation, 2-APB increased the rate and extent of fast inactivation (Fig. 5). Enhancement of fast inactivation by dilute 2-APB was observed across a wide range of test potentials, where the predominant effect was to accelerate the fastest exponential component while increasing its amplitude. As shown in Fig. 5A, this effect developed with a similar time course to the increase in peak ICRAC (potentiation), suggesting that the two phenomena may be related. At long times (> 150 s), the inactivation rate actually decreased (not shown), perhaps due to the slow removal of inactivation at these low 2-APB concentrations (see below). There are several possible explanations for the speeding of ICRAC inactivation by dilute 2-APB; a likely possibility is that by increasing the activity of CRAC channels, 2-APB enhances the accumulation of Ca2+ at the inactivation sites. This and other alternative mechanisms are discussed below (see Discussion).
Figure 5. Low concentrations of 2-APB accelerate fast inactivation of ICRAC.
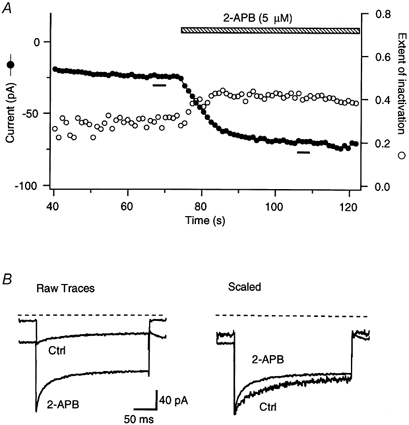
A, peak ICRAC (•) and the extent of inactivation (○) measured during steps to −100 mV. To monitor changes in the speed of inactivation, the extent of inactivation was measured as (Ipeak–It)/Ipeak, where Ipeak is the peak current and It is the current measured 50 ms after each step to −100 mV. Addition of 5 μm 2-APB enhanced the peak current amplitude and increased the extent of inactivation. B, the time course of inactivation in the presence and absence of 2-APB. Traces on the left show the averages of five leak-corrected responses to hyperpolarizations given in A (bars in A) in 20 mm with or without 5 μm 2-APB. The right plot shows the same traces normalized to their peak current. Note that the rate of current decay was enhanced in the presence of 2-APB. Holding potential =+30 mV.
High concentrations of 2-APB remove fast inactivation of ICRAC
In addition to inhibiting ICRAC, a dramatic effect of high concentrations of 2-APB was to eliminate fast inactivation. In the example shown in Fig. 6A, 40 μm 2-APB evoked a rapid transient increase in ICRAC followed by a slow and complete inhibition of the current over the next 40 s. As can be seen in the lower traces, the extent of inactivation during brief voltage pulses from +10 to −100 mV decreased over the duration of exposure to the drug. The removal of inactivation (open circles) appeared to occur in parallel with inhibition of the peak current (filled circles). The exponential time constants for the two effects were similar (Fig. 6B; n = 5 cells), suggesting that the two could be coupled. The simplest hypothesis would be that the inhibition of current through CRAC channels reduces inactivation by lessening the accumulation of intracellular Ca2+. This seems unlikely, however. In Fig. 6A, traces a (before 2-APB was applied) and c (collected after 2-APB had potentiated and inhibited ICRAC) show identical peak current amplitudes even though inactivation was almost completely absent in c. Hence, while the removal of inactivation and drug-induced inhibition of the current may be related (see Discussion), inactivation did not disappear because of reduced Ca2+ flux through CRAC channels.
The removal of inactivation by 2-APB measured during drug applications lasting 150 s or longer was dose dependent, increasing in extent from 5 to 50 μm (Fig. 6C). The threshold concentration for this effect appears to be ∼5 μm, as judged from the small delayed decline in the extent of inactivation at this dose of 2-APB (see above).
The potentiation of ICRAC by 2-APB is not due to removal of fast inactivation
Given that 2-APB was able to inhibit fast Ca2+-dependent inactivation of ICRAC (Fig. 6), we considered the possibility that a reduction of steady-state inactivation might contribute to potentiation of the current. As a first test, we measured the degree of inactivation under steady-state conditions in the presence and absence of 2-APB (Fig. 7). Following a 1 s conditioning step to varying potentials, ICRAC peak amplitude was measured in response to a step to −100 mV. ICRAC amplitude was roughly constant following conditioning steps to +20 mV and above, but declined following more negative conditioning steps to a level of ∼40 % at −80 mV and below. A dose of 2-APB (5 μm) that caused a severalfold potentiation of ICRAC reduced this inactivation by only ∼20 % at the most negative potentials. Therefore, it seems unlikely that the modest effect of dilute 2-APB on inactivation makes a large contribution to potentiation. This conclusion was tested further by investigating whether 2-APB could potentiate ICRAC under conditions that prevented fast inactivation. The degree of potentiation by 2-APB was similar at holding potentials of −50 or +50 mV (Fig. 7B), even though steady-state inactivation was prominent at −50 mV but absent at +50 mV (see Fig. 7A). A third test exploited the fact that Ba2+ currents through CRAC channels do not show fast inactivation (Zweifach & Lewis, 1995b). 2-APB potentiated Ba2+ currents through CRAC channels to at least as great a degree as it did Ca2+ currents (Fig. 7C and D). These results show clearly that the removal of inactivation does not explain the potentiation of ICRAC by 2-APB.
Figure 7. Potentiation of ICRAC by 2-APB does not result from the removal of fast inactivation.
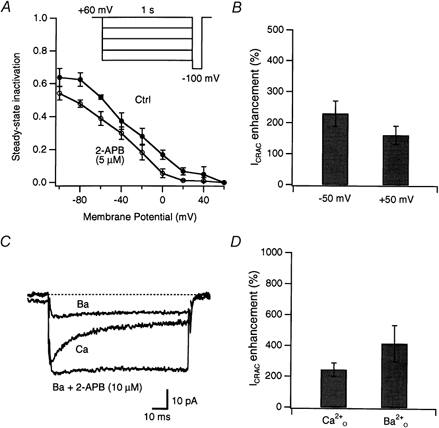
A, 2-APB alters the extent of steady-state inactivation of ICRAC. The extent of inactivation was manipulated by applying a 1 s conditioning step to various potentials (inset) followed by a test pulse to −100 mV where ICRAC amplitude was measured. Steady-state inactivation at each potential, Inact(V), was calculated from the relation Inact(V) = 1 − (Icond/I60 mV), where Icond is the peak current during a test pulse to −100 mV after a conditioning step to potential V, and I60 mV is the peak current at −100 mV following a conditioning step to +60 mV. Holding potential =+60 mV; n = 4 cells. B, potentiation of ICRAC by 5 μm 2-APB is only marginally reduced at positive potentials that remove resting inactivation. Peak ICRAC amplitude was measured during 50 ms test pulses to −100 mV from the indicated holding potential; n = 5 cells. C, potentiation of ICRAC by 2-APB persists when Ba2+ is used as the charge carrier. ICRAC was activated by internal dialysis with 10 mm EGTA. Traces show sequential recordings of current from one cell during test pulses to −100 mV in the presence of 20 mm (Ca), then shortly after 20 mm Ba2+ was substituted for (Ba), and finally after 10 μm 2-APB was applied in the presence of Ba2+ (Ba + 2-APB). Even though fast inactivation did not occur in , 2-APB induced robust potentiation. D, summary of potentiation elicited by 10 μm 2-APB in 20 mm (n = 9 cells) or (n = 5 cells).
Inhibition and potentiation of ICRAC by 2-APB is not cell specific
Although 2-APB has been reported to block ICRAC in RBL cells (Braun et al. 2001), no information exists about whether it potentiates ICRAC in any cells other than Jurkat human T cells. Therefore, we tested the effects of low and high doses of 2-APB on ICRAC in RBL cells and DT40 cells, a chicken pre-B cell line. TG-treated DT40 cells exhibited inwardly rectifying currents with reversal potentials > +30 mV (Fig. 8A and B). The pharmacological hallmarks of this current were also similar to ICRAC in Jurkat T cells: it was effectively blocked by 20 μm La3+ as well as by 30 μm SKF 963615 (data not shown).
Figure 8. 2-APB potentiates and inhibits ICRAC in DT40 B cells and RBL cells.
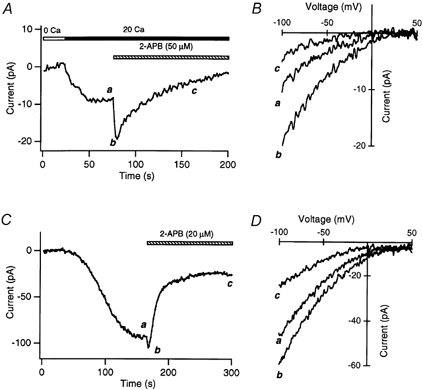
A, effects of 50 μm 2-APB on ICRAC in a DT40 cell. ICRAC was induced by exposing the cell to 1 μm TG for 5 min prior to seal formation; ICRAC during pulses to −100 mV is shown. B, the current-voltage relation of Ca2+ currents before and after exposure to 2-APB, collected at the times shown in A. C, effects of 20 μm 2-APB on ICRAC in an RBL cell. ICRAC was induced by passive depletion of internal stores by 10 mm EGTA in the patch pipette. Note the smaller extent of ICRAC enhancement by 2-APB. D, current-voltage profile of ICRAC in the presence and absence of 2-APB, collected at the times shown in C.
The effects of 2-APB on ICRAC in RBL and DT40 cells were similar to those observed in Jurkat T cells. In both cell types, high doses of 2-APB elicited a transient potentiation of ICRAC, which was then followed by a slow and progressive inhibition that was largely irreversible (Fig. 8). The degree of potentiation in RBL cells tended to be somewhat smaller than that observed in either Jurkat or DT40 cells: 20 μm 2-APB elicited only a 28 ± 8 % increase in ICRAC current amplitude in RBL cells (n = 4 cells), compared with ∼100 % in Jurkat cells (see Fig. 4A). As in Jurkat cells, the high concentration of 2-APB removed fast inactivation of ICRAC in RBL and DT40 cells. Thus, these results indicate that the complex effects of 2-APB on CRAC channel gating were not specific to a single cell type or species.
CRAC channel activation and the effects of 2-APB are independent of IP3 receptors
2-APB has been reported to be a selective antagonist of the IP3 receptor (Maruyama et al. 1997). The inhibition of store-operated Ca2+ influx by 2-APB has therefore been attributed to the inhibition of IP3 receptors, and has been used to support the notion that store-operated channels are activated through conformational coupling to IP3 receptors (Ma et al. 2000). Therefore, a critical question is whether ICRAC is dependent on IP3 receptors, and whether the complex effects of 2-APB on ICRAC are due to the drug acting through IP3 receptors as opposed to other targets, possibly the CRAC channel itself. We examined this issue using three independent approaches. First, we examined the effects of heparin, an IP3 receptor antagonist, on ICRAC to determine whether IP3 binding to its receptor was necessary for ICRAC activation or the effects of 2-APB. Heparin potently inhibits IP3 binding with a Ki of 2–10 μg ml−1 (Supattapone et al. 1988; Parys et al. 1992), and previous reports have indicated that it does not affect passive induction of ICRAC in mast and RBL cells (Hoth & Penner, 1993; Fierro & Parekh, 1999b). Consistent with these reports, we found that, in Jurkat T cells dialysed through the recording pipette with 10 mm EGTA to activate ICRAC, heparin (500 μg ml−1) had no effect on either the current amplitude or time course of induction (Fig. 9A and B). Under these conditions, heparin was clearly effective in inhibiting IP3 receptors because it did block the rapid induction of ICRAC by uncaging intracellular IP3 (Fig. 9C). Importantly, intracellular heparin also failed to affect the ability of 2-APB to either potentiate or inhibit ICRAC (Fig. 9D and E). These results suggest that the binding of background levels of IP3 is not necessary for activation of ICRAC through passive store depletion, nor is it required for the effects of 2-APB on ICRAC.
Next, we investigated whether 2-APB preferentially acts upon ICRAC from the inside or the outside of the cell. A previous report (Braun et al. 2001) showed that 2-APB inhibits ICRAC in RBL cells more effectively when applied outside, and we found a similar result in Jurkat cells. Applying 2-APB intracellularly by including a high concentration of the drug (80 μm) in the patch pipette did not significantly inhibit either the induction or the maintenance of ICRAC (Fig. 10A and B). In contrast, subsequent exposure of the cells to 40 μm 2-APB extracellularly caused significant inhibition (Fig. 10A). The greater potency of extracellular 2-APB appears to support the idea that the site of action is extracellular and therefore independent of IP3 receptors. However, the interpretation of this experiment is complicated by the fact that 2-APB is membrane permeant. It is well established that lipophilic compounds that are known to block ion channels from an intracellular site often act more effectively when applied from the outside (DeCoursey, 1995). This paradox results from the fact that when membrane-permeant compounds are applied through the recording pipette, their concentration at the membrane may be lower than expected because diffusion out of the cell is faster than diffusion out of the pipette (DeCoursey, 1995). The membrane permeability and protonation state of 2-APB under our experimental conditions was not known, but a reduction of extracellular pH would have been expected to increase the protonation of 2-APB and reduce the concentration of the more membrane-permeant, non-protonated form. If the site of action were intracellular, then increased protonation of 2-APB would impair access to the site and result in a reduced effect. Therefore, we examined the effects of extracellular pH on potentiation and inhibition by bath-applied 2-APB. Reducing extracellular pH from 7.4 to 6.4 partially inhibited ICRAC (Fig. 10C; see also Malayev & Nelson, 1995). However, the degrees of potentiation and inhibition of ICRAC by 2-APB were identical at the two pH values (Fig. 10D). Therefore, these data support the idea that the site of action of 2-APB is extracellular rather than intracellular.
Figure 10. 2-APB may have an extracellular site of action.
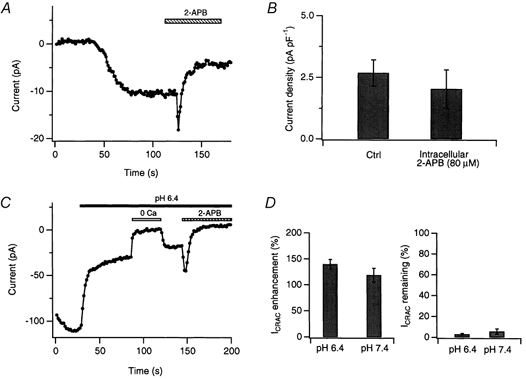
A, intracellular 2-APB is ineffective at inhibiting ICRAC. ICRAC was activated by dialysis with 10 mm EGTA in the presence of 80 μm 2-APB in the recording pipette. Peak ICRAC amplitudes during repeated steps to −100 mV are shown. After ICRAC amplitude reached steady state, application of 40 μm 2-APB via the bath elicited the normal sequence of potentiation and inhibition. B, comparison of the steady-state current amplitudes of ICRAC (normalized to cell capacitance) in cells treated with intracellular 2-APB (80 μm; n = 5 cells) and matched controls (n = 7 cells) in which ICRAC was activated by intracellular EGTA only. C, lowering extracellular pH does not alter the ability of 2-APB to potentiate or inhibit ICRAC. ICRAC was activated by treatment with 1 μm TG immediately prior to seal formation. After allowing the ICRAC amplitude to reach steady state, the external pH was lowered from 7.4 to 6.4, which dramatically reduced the current amplitude. Application of 40 μm 2-APB potentiated and inhibited ICRAC in a manner similar to that at pH 7.4. D, summary of the extent of potentiation and inhibition of ICRAC induced by 40 μm 2-APB at pH 7.4 (n = 5 cells) and pH 6.4 (n = 5 cells).
An extracellular site of action raises the possibility that 2-APB does not exert its effects on ICRAC through the intracellular IP3 receptor. To provide a more direct test of this idea, we examined the effects of 2-APB on ICRAC in mutant DT40 cells in which all three isoforms of the IP3 receptor (IP3R) were knocked out by homologous recombination (Sugawara et al. 1997). These cells have been shown to lack functional type I, II and III IP3 receptors as assayed by their failure to release Ca2+ in response to stimulation of phospholipase C-linked B-cell receptors or muscarinic receptors, and Northern analysis confirmed the absence of the IP3 receptor mRNA sequence in regions including and immediately surrounding the knockout sites (Sugawara et al. 1997). In addition, recent measurements showed that, unlike their wild-type counterparts, the DT40 mutant cells bind essentially no 3H-IP3 (Broad et al. 2001), suggesting that the amino-terminal cytoplasmic domain of the IP3 receptor, which has been proposed to couple to store-operated channels, is also missing. Despite the lack of IP3 receptors, store-operated Ca2+ entry in these cells was shown to be normal (Sugawara et al. 1997). Consistent with these results, we found that the IP3R−/− DT40 mutants exhibited CRAC currents that were virtually indistinguishable from those of control DT40 cells in terms of their size, I–V relationship and Ca2+-dependent fast inactivation (Fig. 11A–C). Thus, ICRAC activation does not appear to require any of the known isoforms of the IP3 receptor.
Figure 11. Effects of 2-APB are retained in mutant DT40 cells lacking IP3 receptors.
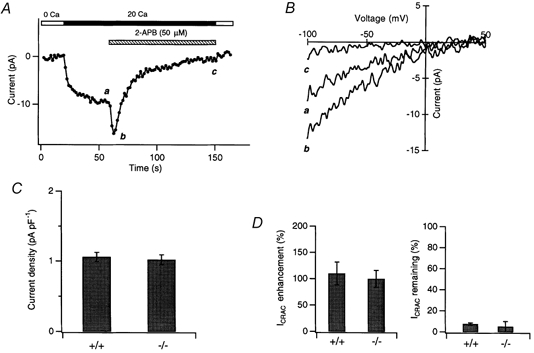
A, ICRAC was induced in a mutant DT40 IP3R−/− cell by application of 1 μm TG 5 min prior to seal formation. After recording traces in 0 and 20 mm solutions, the cell was exposed to 50 μm 2-APB. Shown here are peak current amplitudes recorded during steps to −100 mV. B, responses to voltage ramps recorded in the absence or presence of 2-APB at the times shown in A. C, ICRAC amplitude (at −100 mV, normalized to cell capacitance) in IP3R−/− mutant (−/−; n = 6) and wild-type (+/+; n = 5) DT40 cells. D, comparison of potentiation and inhibition of ICRAC by 50 μm 2-APB in wild-type (n = 5) and IP3R−/− mutant (n = 6) DT40 cells.
Application of 50 μm 2-APB to the IP3R−/− mutant DT40 cells produced a transient elevation of ICRAC followed by a slow inhibition (Fig. 11A). The magnitudes of both the peak potentiation and the steady-state inhibition caused by 2-APB were identical to those seen in the control DT40 and Jurkat T cells (Fig. 11D). In addition, 2-APB diminished the extent of fast inactivation over time, just as it does in control DT40 cells (data not shown). Thus, we conclude that the complex stimulatory and inhibitory effects of 2-APB on CRAC channels occur through a mechanism that is independent of IP3 receptors.
DISCUSSION
In this study, we have described complex effects of 2-APB on the amplitude and kinetic behaviour of ICRAC. These include an increase of current amplitude (potentiation) and speeding of inactivation by low concentrations of 2-APB (< 5 μm), as well as inhibition of the current and removal of fast inactivation by higher concentrations (≥ 5 μm). Importantly, none of these effects appeared to occur through the IP3 receptor, as previously postulated by the conformational coupling hypothesis for store-operated Ca2+ entry. In the sections that follow, we discuss some possible underlying mechanisms and implications of these phenomena.
2-APB exerts complex effects on the amplitude and kinetics of ICRAC
We were surprised to find that 2-APB had much more complex effects on ICRAC than only the slow inhibition reported previously (Braun et al. 2001). Low doses (1–5 μm) reversibly enhanced the amplitude of ICRAC by up to fivefold and slightly increased the rate of fast Ca2+-dependent inactivation. At higher doses (≥ 5 μm) of the drug, the slow inhibition of ICRAC also became apparent, along with the disappearance of fast inactivation. All of these effects were seen in Jurkat, RBL and DT40 cells; thus, they appear to be independent of the cell type (T cells, mast cells and B cells) and species (human, rat and chicken), suggesting that they may apply to CRAC channels in general. Thus, the reason why potentiation was not observed in a previous study of RBL cells (Braun et al. 2001) probably stems not from the cell type but from a combination of other factors. The amplitude of potentiation was less pronounced in RBL cells than in Jurkat or DT40 cells (compare Fig. 8C with Fig. 8A and 3A), and this smaller effect may have been exacerbated in other studies by the use of high concentrations of 2-APB (100 μm), which produce a predominant inhibitory effect, and by pre-application of 2-APB rather than acute application after ICRAC had been activated.
The degree to which these 2-APB effects are specific for CRAC channels is mostly unknown. The inhibitory effects of 2-APB seem to be relatively non-specific, as they apply to IP3 receptors (Sugawara et al. 1997), endogenous store-operated channels in HEK 293 cells (Ma et al. 2000) and the mitochondrial Ca2+ release machinery (Fig. 1B) in addition to ICRAC. However, to our knowledge, 2-APB is the first example of a pharmacological agent that can enhance the activation of ICRAC beyond the level achieved by complete store depletion. Because Ca2+ entry through CRAC channels is a key requisite step for the activation of T cells by antigen, potentiation of ICRAC may be useful as a novel therapeutic approach to boost activation of immune cells in immunodeficient individuals. In addition, potentiation by 2-APB may prove to be a useful diagnostic tool for testing putative CRAC channel genes and probing the physiological functions of CRAC channels in vivo.
Possible mechanisms for the potentiation of ICRAC by 2-APB
In resting cells in which Ca2+ stores were full and which displayed no Ca2+ influx through CRAC channels, low doses of 2-APB were ineffective in raising [Ca2+]i or activating ICRAC. Thus, rather than activating ICRAC by itself, 2-APB appears to boost the activity of CRAC channels once they are activated by store depletion (Fig. 3E). This potentiation does not result from an increased emptying of Ca2+ stores, since 2-APB increases ICRAC severalfold even after treatment with TG, which is known to deplete the ionomycin-releasable stores in Jurkat cells by > 90 % (see Fig. 1A of Hoth et al. 1997).
Although 2-APB does reduce the level of Ca2+-dependent fast inactivation at doses as low as 5 μm, this effect is not responsible for potentiation of the current. Conditions that limit or prevent fast inactivation, including holding potentials more positive than +30 mV or the use of Ba2+ as the charge carrier, did not reduce the amount of potentiation by 2-APB (Fig. 7). In principle, 2-APB could act by interfering with other known forms of ICRAC inactivation, such as slow Ca2+-dependent inactivation (Zweifach & Lewis, 1995a) or kinase-dependent inactivation (Parekh & Penner, 1995). However, this seems unlikely, as both processes would be minimized by our recording conditions (10 mm EGTA and 0 ATP in the recording pipette).
In general, potentiation of ICRAC could occur through increases in the number of activatable channels (N), the single-channel current (i) or the open probability of the channels (Po). Without single-channel recordings, it is difficult to discriminate between these possible mechanisms, but we can make some educated guesses by exploiting the changes in fast inactivation that accompany ICRAC potentiation. Previous work has shown that ICRAC inactivation is influenced by the local [Ca2+]i around individual CRAC channels (Zweifach & Lewis, 1995b). 2-APB at low concentrations does not induce inactivation by itself, since Ba2+ currents do not inactivate even in the presence of the drug (Fig. 7C); instead, a reasonable conclusion is that it increases the speed of fast Ca2+-dependent inactivation, possibly by producing an increase in the local [Ca2+]i around CRAC channels. Fast inactivation is strongly dependent on the amplitude of i but not on N (Zweifach & Lewis, 1995b). Thus, if potentiation were due to an increase in i, then one would expect a significant increase in the extent of inactivation, particularly under conditions where the current increases significantly (e.g. by a factor > 2; see Fig. 5). This was not observed, suggesting that i is probably not affected by 2-APB. In addition, because CRAC channels inactivate independently (Zweifach & Lewis, 1995b), an increase in N would not be expected to affect inactivation. It might be argued that overlap of Ca2+ microdomains between adjacent CRAC channels might increase with N, undermining channel independence and increasing inactivation. To test this possibility, we reduced the current with SKF 96365, reasoning that this would reduce the overlap of microdomains and therefore reduce the inactivation if overlap existed. We found that, in the presence of a potentiating dose of 2-APB, a half-blocking dose of SKF 96365 did not affect the rate of inactivation (data not shown), making the overlap of domains unlikely.
If 2-APB does not influence N or i, the most likely explanation is that 2-APB potentiates ICRAC by increasing Po. The resulting increase in accumulation of intracellular Ca2+ at the inactivation sites would be expected to enhance inactivation, though perhaps to a smaller degree than would be expected from a comparable increase in i. A mechanism of this sort has been proposed to underlie the speeding of inactivation of voltage-gated L-type Ca2+ channels by Bay K 8644, which increases the Po of these channels (Noceti et al. 1998). Further experiments on ICRAC at the single-channel level will be needed to resolve this issue.
Inhibition of ICRAC and the removal of inactivation by 2-APB
At concentrations ≥ 5 μm, 2-APB slowly inhibited ICRAC and removed fast inactivation. The reversal of these effects after 2-APB was washed out of the bath was even slower and was often incomplete. In this sense, the inhibition by 2-APB is similar to that by imidazole compounds like SKF 96365 or econazole (Franzius et al. 1994; Christian et al. 1996). It is not known whether 2-APB (or SKF 96365) acts by blocking the channel pore directly, or by altering channel gating. Our data suggest that 2-APB does not act through a simple 1:1 interaction with the CRAC channel pore. The on-rate for inhibition, calculated from the time constant of ICRAC inhibition in Fig. 6B, and K1/2 for current inhibition (10 μm, Fig. 4B) was 1/500 μm−1 s−1. Thus, a simple 1:1 binding interaction would predict an off-rate of ∼1/50 s−1, or a time constant of recovery of ∼50 s. This is much faster than the observed recovery time (22 % recovery at 160 s; estimated recovery time constant of > 600 s). The discrepancy cannot be explained by slow washout of the drug, as the potentiation effect of 2-APB reversed rapidly after drug removal (Fig. 3A). Instead, these results suggest that the mechanism of inhibition is more complex than simple pore blockade, and that the recovery from inhibition is not limited by the dissociation rate of 2-APB from its target.
2-APB inhibited both the amplitude of ICRAC and the extent of fast inactivation with similar time courses, raising the intriguing possibility that these two effects may be causally linked. It is possible, for example, that 2-APB at high concentrations interrupts the coupling between the CRAC channel and a component that confers fast inactivation, and the activity of the CRAC channel declines because this component is necessary for maintaining the channel in an activatable state. Such a mechanism might also explain the kinetic discrepancy discussed above, if recovery of the inactivation component were much slower than the unbinding of 2-APB. Further experiments on the molecular underpinnings of fast inactivation may give clues as to the mechanism of ICRAC inhibition by 2-APB.
IP3 receptors are not needed for ICRAC activation or inactivation
The mechanism underlying the activation of store-operated Ca2+ channels has been an area of active debate and, despite considerable efforts, no consistent picture has emerged. The leading candidate hypotheses include activation by a diffusible factor released from depleted stores, fusion of store-operated channel-laden vesicles with the plasma membrane, and direct activation of store-operated channels by conformational coupling to IP3 receptors in the ER membrane (for reviews, see Putney & McKay, 1999; Prakriya & Lewis, 2001a). The conformational coupling hypothesis has received much recent support from evidence that IP3 receptors bind directly to some members of the TRP family, thought by some to be the molecular correlates of CRAC channels (Kiselyov et al. 1998, 1999a; Boulay et al. 1999). Moreover, inhibitors of IP3 receptors, such as 2-APB and heparin, also inhibit store-operated Ca2+ entry and the currents mediated by the putative store-operated channel TRP3 (Kiselyov et al. 1998; Ma et al. 2000). It has been suggested that fast inactivation of CRAC channels may arise through the inhibitory effects of Ca2+ on IP3 receptors to which they are coupled (Berridge, 1995).
Our data are inconsistent with a necessary role for IP3 receptors in the activation and inactivation of ICRAC. First, we found that heparin failed to affect activation or the maintenance of ICRAC in store-depleted cells, even at a concentration that completely prevented the ability of IP3 to activate ICRAC. A similar result was described recently in RBL cells (Broad et al. 2001). In contrast, currents mediated by recombinant TRP3 channels in excised patches from HEK 293 cells or endogenous ‘CRAC-like’ channels from A431 cells require addition of IP3 and are blocked potently by heparin (Kiselyov et al. 1998, 1999b). Thus, the ability to activate ICRAC through passive depletion, coupled with the inability of heparin to prevent this, suggests that CRAC channels differ from these other channels in not requiring even background levels of IP3 for activation. Second, the activation and properties of ICRAC were apparently normal in DT40 cells in which all three subtypes of IP3 receptor had been knocked out by homologous recombination. These results confirm and extend earlier work by Sugawara et al. (1997) showing that TG was able to induce Ca2+ entry in these cells. Thus, our results show that CRAC channels do not require IP3 receptors or IP3 for their activation. However, these results do not rule out conformational coupling involving other proteins.
2-APB affects CRAC channel gating independently of IP3 receptors
In the mutant DT40 cells lacking all three subtypes of IP3 receptors, 2-APB elicited the same combination of ICRAC potentiation, inhibition and changes in fast inactivation as in wild-type cells. Thus, the actions of 2-APB on ICRAC are probably unrelated to any effect on IP3 receptors. These results therefore show that inhibition of Ca2+ entry by 2-APB cannot be used to test the conformational coupling model for ICRAC activation.
If IP3 receptors are not involved, what then is the target for 2-APB's effects on CRAC channel activity? One possible site of action we considered was mitochondria, as 2-APB inhibited Ca2+ release from these organelles (Fig. 1B). In Jurkat T cells, uptake and release of Ca2+ by mitochondria is required to enable the cells to maintain a high rate of Ca2+ entry through CRAC channels (Hoth et al. 1997). To test the possibility that the effects of 2-APB on ICRAC are an indirect consequence of modulating mitochondrial function, we treated cells with 1 μm antimycin A1 + 1 μm oligomycin to inhibit mitochondrial Ca2+ uptake. The inhibitors did not alter the ability of 2-APB to potentiate and inhibit ICRAC (data not shown). Therefore, the effects of 2-APB on ICRAC appear to be distinct from its effects on mitochondrial Ca2+ transport.
2-APB was more effective at modulating ICRAC when applied extracellularly than when it was applied intracellularly through the recording pipette. A similar result was reported for RBL cells by Braun et al. (2001), who concluded that the drug's site of action was extracellular. The interpretation of this experiment is complicated by several factors. First, 2-APB may exist in multiple forms, including a protonated and unprotonated monomer, as well as a dimer (van Rossum et al. 2000), but the relative efficacy of these various compounds in affecting Ca2+ entry in general is unknown. Second, the ability of lipophilic compounds to interact with intracellular sites when applied intracellularly via a patch pipette is a function of their membrane permeability. Thus, when applied through the recording pipette, the concentration of a hydrophobic form of 2-APB near the plasma membrane may be much lower than expected, due to the fact that the rate-limiting diffusion step for such a hydrophobic compound may be across the tip of the pipette rather than across the plasma membrane. We reasoned that increasing the protonation of the drug should reduce permeability to some degree, and this should reduce the efficacy of extracellular drug if its site of action is intracellular. This approach has been applied previously in studies of membrane-permeant K+ channel blockers. For example, methadone is known to block K+ channels at an intracellular site, yet it is effective only when applied from outside the cell. Acidification of the extracellular medium was shown to inhibit the blocking action of methadone by reducing the concentration of the uncharged, lipophilic species (Horrigan & Gilly, 1996). In our experiments, reducing extracellular pH from 7.4 to 6.4 failed to decrease either potentiation or inhibition by 2-APB, implying that lowering the concentration of the unprotonated (and presumably membrane-permeant) form of 2-APB had no effect. The relative amounts of the protonated and unprotonated forms of 2-APB will depend on its pKa, which is unknown, but is likely to be determined by its ethylamine group (pKa= 10.8; Weast & Lide, 1989). Based on this pKa value, one would predict that only 0.04 % of the 2-APB will be unprotonated at pH 7.4, but that this fraction will decrease by a factor of 10 at pH 6.4. Thus, the lack of an effect from lowering the pH (Fig. 10C) is consistent with an extracellular but not an intracellular site of action. However, given the various complications outlined above, a direct examination of this issue using membrane-impermeant variants of 2-APB will be needed to identify firmly the active species of 2-APB and its site of action.
The opposing effects of 2-APB on ICRAC are reminiscent of the effects of the plant alkaloid ryanodine on the sarcoplasmic reticulum Ca2+ release channel in cardiac muscle. At submicromolar (1–50 nm) concentrations, ryanodine stabilizes the Ca2+ release channel in a subconductance state with a dramatically higher Po, whereas at micromolar (10–1000 μm) concentrations, ryanodine closes the channel (Buck et al. 1992; Humerickhouse et al. 1993). These effects are manifested as increases in Ca2+ flux and muscle contraction by dilute ryanodine and inhibition of Ca2+ flux and muscle contraction by concentrated ryanodine. Binding studies indicate that the Ca2+ release channel has a high-affinity site, thought to mediate the stimulatory effects of ryanodine, and one or more low-affinity sites that mediate channel closure (Sutko et al. 1997). Thus, by analogy, it is possible that 2-APB has two separate binding sites, a high-affinity site that mediates ICRAC potentiation and a low-affinity site that produces inhibition. Alternatively, different species of 2-APB may be responsible for the different effects: monomers, which are more prevalent at low concentrations, may mediate potentiation, whilst dimers, which are favoured at higher concentrations, may be responsible for inhibition. Further studies will be needed to resolve these issues.
In summary, we found that 2-APB elicits complex effects on ICRAC. Low concentrations of 2-APB reversibly enhanced ICRAC, whereas high concentrations also inhibited ICRAC. 2-APB also altered the gating of ICRAC by removing fast Ca2+-dependent inactivation. It was surprising to find that so many different properties of CRAC channels were affected, and an important question is whether the various effects of 2-APB are linked in some way or whether they are truly independent. Given that 2-APB affects the function of so many Ca2+ signalling proteins (IP3 receptors, CRAC channels, TRP channels and the mitochondrial Ca2+ release mechanism), the drug clearly has limited utility as a specific inhibitor of store-operated Ca2+ entry. However, some effects of 2-APB (e.g. potentiation and removal of inactivation) may be sufficiently unique that identification of the parts of the molecule that are responsible for these effects and the proteins with which they interact could provide new clues to the mechanisms underlying activation, deactivation and inactivation of CRAC channels.
Acknowledgments
The authors would like to thank K. Mikoshiba for the generous gift of 2-APB and T. Meyer for the RBL-2H3 cells. We also thank Diana Bautista for early experiments demonstrating the existence of ICRAC in DT40 cells, Jack Waters and Diana Bautista for comments on the manuscript, and members of the Lewis lab for helpful discussions. This work was supported by a postdoctoral fellowship from the Irvington Foundation for Immunological Research to M.P. and NIH grant GM45374 to R.S.L.
References
- Aussel C, Marhaba R, Pelassy C, Breittmayer JP. Submicromolar La3+ concentrations block the calcium release-activated channel, and impair CD69 and CD25 expression in CD3- or thapsigargin-activated Jurkat cells. Biochemical Journal. 1996;313:909–913. doi: 10.1042/bj3130909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Capacitative calcium entry. Biochemical Journal. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay G, Brown DM, Qin N, Jiang M, Dietrich A, Zhu MX, Chen Z, Birnbaumer M, Mikoshiba K, Birnbaumer L. Modulation of Ca2+ entry by polypeptides of the inositol 1,4, 5- trisphosphate receptor (IP3R) that bind transient receptor potential (TRP): evidence for roles of TRP and IP3R in store depletion-activated Ca2+ entry. Proceedings of the National Academy of Sciences of the USA. 1999;96:14955–14960. doi: 10.1073/pnas.96.26.14955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun FJ, Broad LM, Armstrong DL, Putney JW., Jr Stable activation of single Ca2+ release-activated Ca2+ channels in divalent cation-free solutions. Journal of Biological Chemistry. 2001;276:1063–1070. doi: 10.1074/jbc.M008348200. [DOI] [PubMed] [Google Scholar]
- Broad LM, Braun F-J, Lievremont J-P, Bird GSJ, Kurosaki T, Putney JW., Jr Role of the phospholipase C-inositol 1,4,5-trisphosphate pathway in calcium release-activated calcium current and capacitative calcium entry. Journal of Biological Chemistry. 2001;276:15945–15952. doi: 10.1074/jbc.M011571200. [DOI] [PubMed] [Google Scholar]
- Buck E, Zimanyi I, Abramson JJ, Pessah IN. Ryanodine stabilizes multiple conformational states of the skeletal muscle calcium release channel. Journal of Biological Chemistry. 1992;267:23560–23567. [PubMed] [Google Scholar]
- Christian EP, Spence KT, Togo JA, Dargis PG, Warawa E. Extracellular site for econazole-mediated block of Ca2+ release-activated Ca2+ current (ICRAC) in T lymphocytes. British Journal of Pharmacology. 1996;119:647–654. doi: 10.1111/j.1476-5381.1996.tb15722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE. Mechanism of K+ channel block by verapamil and related compounds in rat alveolar epithelial cells. Journal of General Physiology. 1995;106:745–779. doi: 10.1085/jgp.106.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanger CM, Hoth M, Crabtree GR, Lewis RS. Characterization of T cell mutants with defects in capacitative calcium entry: genetic evidence for the physiological roles of CRAC channels. Journal of Cell Biology. 1995;131:655–667. doi: 10.1083/jcb.131.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro L, Parekh AB. Fast calcium-dependent inactivation of calcium release-activated calcium current (CRAC) in RBL-1 cells. Journal of Membrane Biology. 1999a;168:9–17. doi: 10.1007/s002329900493. [DOI] [PubMed] [Google Scholar]
- Fierro L, Parekh AB. On the characterisation of the mechanism underlying passive activation of the Ca2+ release-activated Ca2+ current ICRAC in rat basophilic leukaemia cells. Journal of Physiology. 1999b;520:407–416. doi: 10.1111/j.1469-7793.1999.00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzius D, Hoth M, Penner R. Non-specific effects of calcium entry antagonists in mast cells. Pflügers Archiv. 1994;428:433–438. doi: 10.1007/BF00374562. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Buntinas L, Sparagna G, Eliseev R, Gunter K. Mitochondrial calcium transport: mechanisms and functions. Cell Calcium. 2000;28:285–296. doi: 10.1054/ceca.2000.0168. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- Horrigan FT, Gilly WF. Methadone block of K+ current in squid giant fiber lobe neurons. Journal of General Physiology. 1996;107:243–260. doi: 10.1085/jgp.107.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. Journal of Cell Biology. 1997;137:633–648. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. Journal of Physiology. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humerickhouse RA, Besch HR, Jr, Gerzon K, Ruest L, Sutko JL, Emmick JT. Differential activating and deactivating effects of natural ryanodine congeners on the calcium release channel of sarcoplasmic reticulum: evidence for separation of effects at functionally distinct sites. Molecular Pharmacology. 1993;44:412–421. [PubMed] [Google Scholar]
- Kiselyov K, Mignery GA, Zhu MX, Muallem S. The N-terminal domain of the IP3 receptor gates store-operated hTrp3 channels. Molecular Cell. 1999a;4:423–429. doi: 10.1016/s1097-2765(00)80344-5. [DOI] [PubMed] [Google Scholar]
- Kiselyov K, Xu X, Mozhayeva G, Kuo T, Pessah I, Mignery G, Zhu X, Birnbaumer L, Muallem S. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature. 1998;396:478–482. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- Kiselyov KI, Semyonova SB, Mamin AG, Mozhayeva GN. Miniature Ca2+ channels in excised plasma-membrane patches: activation by IP3. Pflügers Archiv. 1999b;437:305–314. doi: 10.1007/s004240050784. [DOI] [PubMed] [Google Scholar]
- Lewis RS. Store-operated calcium channels. Advances in Second Messenger and Phosphoprotein Research. 1999;33:279–307. doi: 10.1016/s1040-7952(99)80014-7. [DOI] [PubMed] [Google Scholar]
- Lewis RS, Cahalan MD. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regulation. 1989;1:99–112. doi: 10.1091/mbc.1.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma HT, Patterson RL, van Rossum DB, Birnbaumer L, Mikoshiba K, Gill DL. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 2000;287:1647–1651. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- Malayev A, Nelson DJ. Extracellular pH modulates the Ca2+ current activated by depletion of intracellular Ca2+ stores in human macrophages. Journal of Membrane Biology. 1995;146:101–111. doi: 10.1007/BF00232684. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. Journal of Biochemistry (Tokyo) 1997;122:498–505. doi: 10.1093/oxfordjournals.jbchem.a021780. [DOI] [PubMed] [Google Scholar]
- Noceti F, Olcese R, Qin N, Zhou J, Stefani E. Effect of Bay K 8644 (−) and the β2a subunit on Ca2+-dependent inactivation in α1C Ca2+ channels. Journal of General Physiology. 1998;111:463–475. doi: 10.1085/jgp.111.3.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Depletion-activated calcium current is inhibited by protein kinase in RBL-2H3 cells. Proceedings of the National Academy of Sciences of the USA. 1995;92:7907–7911. doi: 10.1073/pnas.92.17.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Store depletion and calcium influx. Physiological Reviews. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Partiseti M, Le Deist F, Hivroz C, Fischer A, Korn H, Choquet D. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. Journal of Biological Chemistry. 1994;269:32327–32335. [PubMed] [Google Scholar]
- Parys JB, Sernett SW, Delisle S, Snyder PM, Welsh MJ, Campbell KP. Isolation, characterization, and localization of the inositol 1,4,5- trisphosphate receptor protein in Xenopus laevis oocytes. Journal of Biological Chemistry. 1992;267:18776–18782. [PubMed] [Google Scholar]
- Patterson RL, van Rossum DB, Gill DL. Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell. 1999;98:487–499. doi: 10.1016/s0092-8674(00)81977-7. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Lewis RS. Store-operated calcium channels: properties, functions and the search for a molecular mechanism. In: Maue RA, editor. Molecular Insights into Ion Channel Biology in Health and Disease. Amsterdam: Elsevier Science; 2001a. (in the Press) [Google Scholar]
- Prakriya M, Lewis RS. Blockade and potentiation of ICRAC by 2-APB, an IP3 receptor antagonist. Biophysical Journal. 2001b;80:617a. [Google Scholar]
- Putney JW., Jr . Capacitative Calcium Entry. Austin, TX, USA: Landes Biomedical Publishing; 1997. [Google Scholar]
- Putney JW, Jr, McKay RR. Capacitative calcium entry channels. Bioessays. 1999;21:38–46. doi: 10.1002/(SICI)1521-1878(199901)21:1<38::AID-BIES5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Sugawara H, Kurosaki M, Takata M, Kurosaki T. Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO Journal. 1997;16:3078–3088. doi: 10.1093/emboj/16.11.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supattapone S, Worley PF, Baraban JM, Snyder SH. Solubilization, purification, and characterization of an inositol trisphosphate receptor. Journal of Biological Chemistry. 1988;263:1530–1534. [PubMed] [Google Scholar]
- Sutko JL, Airey JA, Welch W, Ruest L. The pharmacology of ryanodine and related compounds. Pharmacological Reviews. 1997;49:53–98. [PubMed] [Google Scholar]
- Thastrup O, Dawson AP, Scharff O, Foder B, Cullen PJ, Drobak BK, Bjerrum PJ, Christensen SB, Hanley MR. Thapsigargin, a novel molecular probe for studying intracellular calcium release and storage. Agents and Actions. 1989;27:17–23. doi: 10.1007/BF02222186. [DOI] [PubMed] [Google Scholar]
- van Rossum DB, Patterson RL, Ma HT, Gill DL. Ca2+ entry mediated by store depletion, S-nitrosylation, and TRP3 channels. Comparison of coupling and function. Journal of Biological Chemistry. 2000;275:28562–28568. doi: 10.1074/jbc.M003147200. [DOI] [PubMed] [Google Scholar]
- Weast RC. In: Handbook of Chemistry and Physics. Lide DR, editor. Boca Raton, FL, USA: CRC Press; 1989. [Google Scholar]
- Zhang L, McCloskey MA. Immunoglobulin E receptor-activated calcium conductance in rat mast cells. Journal of Physiology. 1995;483:59–66. doi: 10.1113/jphysiol.1995.sp020567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitt C, Obukhov AG, Strubing C, Zobel A, Kalkbrenner F, Luckhoff A, Schultz G. Expression of TRPC3 in Chinese hamster ovary cells results in calcium-activated cation currents not related to store depletion. Journal of Cell Biology. 1997;138:1333–1341. doi: 10.1083/jcb.138.6.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proceedings of the National Academy of Sciences of the USA. 1993;90:6295–6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Slow calcium-dependent inactivation of depletion-activated calcium current. Store-dependent and -independent mechanisms. Journal of Biological Chemistry. 1995a;270:14445–14451. doi: 10.1074/jbc.270.24.14445. [DOI] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Rapid inactivation of depletion-activated calcium current (ICRAC) due to local calcium feedback. Journal of General Physiology. 1995b;105:209–226. doi: 10.1085/jgp.105.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]