

Abstract
A spindle of fast network oscillations precedes the ischaemia-induced rapid depolarisation in the rat hippocampus in vivo. However, this oscillatory pattern could not be reproduced in slices and the underlying mechanisms remain poorly understood. We have found that anoxia-induced network oscillations (ANOs, 20–40 Hz, lasting for 1–2 min) can be reproduced in the intact hippocampi of postnatal day P7–10 rats in vitro, and we have examined the underlying mechanisms using whole-cell and extracellular field potential recordings in a CA3 pyramidal layer.
ANOs were generated at the beginning of the anoxic depolarisation, when pyramidal cells depolarised to subthreshold values. Maximal power of the ANOs was attained when pyramidal cells depolarised to −56 mV; depolarisation above −47 mV resulted in a depolarisation block of pyramidal cells and a waning of ANOs.
A multiple unit activity in extracellular field recordings was phase locked to the negative and ascending phases of ANOs. Pyramidal cells recorded in current-clamp mode generated action potentials with an average probability of about 0.05 per cycle. The AMPA receptor-mediated EPSCs and the GABA receptor-mediated IPSCs in CA3 pyramidal cells were also phase locked with ANOs.
ANOs were prevented by tetrodotoxin and glutamate receptor antagonists CNQX and APV, and were slowed down by the allosteric GABAA receptor modulator diazepam. In the presence of the GABAA receptor antagonist bicuculline, ANOs were transformed to epileptiform discharges.
In the presence of the A1 adenosine receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), the anoxia induced an epileptiform activity and no ANOs were observed.
In normoxic conditions, a rise of extracellular potassium to 10 mm induced an epileptiform activity. Increasing extracellular potassium in conjunction with a bath application of the adenosine A1 receptor agonist cyclopentyladenosine induced oscillations similar to ANOs.
Multisite recordings along the septo-temporal hippocampal axis revealed that ANOs and anoxic depolarisation originate in the temporal part, and propagate towards the septal pole at a speed of 1.9 mm min−1.
ANOs were observed starting from P7, i.e. at a developmental stage when the effects of GABA change from depolarisation to hyperpolarisation.
These results suggest that the synchronisation of anoxia-induced oscillations relies on synaptic mechanisms; that the inhibition by GABA and adenosine sets the tune for a generation of oscillations and prevents an epileptiform activity; and that a synchronous GABAergic inhibition is instrumental in a phase locking neuronal activity similarly to other types of oscillatory activities in the gamma frequency range.
Glucose oxidation is the principal source of energy for the brain. Deprivation of oxygen and glucose during brain ischaemia results in a progressive decrease of intracellular energy metabolites and complex alterations in energy-dependent processes (Kass & Lipton, 1982; Katsura et al. 1993; for reviews see Hansen, 1985; Katsura et al. 1994; Martin et al. 1994; Lipton, 1999). With regard to brain activity, two principal phases can be distinguished in the pathogenesis of brain ischaemia: a phase of ‘adaptation’ and a phase of anoxic depolarisation (AD).
During the adaptation phase, the brain switches to an energy-saving mode and the effects of anoxia are reversible. To save energy, the neuronal activity is rapidly blocked and intracellular stores of energy are exclusively used to maintain the vital functions – to keep the ionic gradients and to maintain membrane potential. Studies of mechanisms underlying the depression of the neuronal activity during the adaptation phase have emphasised the role of two factors: adenosine-dependent reduction of glutamatergic transmission (Fowler, 1989; Zeng et al. 1992; Gribkoff & Bauman, 1992; Katchman & Hershkowitz, 1993; Khazipov et al. 1995a; Croning et al. 1995; Brundege & Dunwiddie, 1997; Dunwiddie & Fredholm, 1997) and neuronal hyperpolarisation due to an activation of potassium conductances (Hansen et al. 1982; Fujiwara et al. 1987; Leblond & Krnjevic, 1989; Fujimura et al. 1997; Yamamoto et al. 1997; Erdemli et al. 1998). The effects of anoxia during the adaptation phase are reversible, and re-oxygenation recovers brain functions. The transition to the second phase, AD, occurs when the intracellular stores of energy metabolites become exhausted, and there is a rapid fall in the ATP concentration and a cessation of ATP-dependent ionic transporters including Na+,K+-ATPase. This results in major disturbances of the ionic gradients, including the accumulation of extracellular potassium (Hansen et al. 1982; Müller & Somjen, 2000), the entry of calcium and the activation of intracellular cascades leading to neuronal damage (Lipton, 1999; Lee et al. 1999). As a result of ionic changes, neurons first depolarise gradually, then in an accelerated manner, and finally lose membrane potential. The late rapid phase of collective neuronal depolarisation can be reflected as a negative shift of extracellular field potential – a rapid AD (Balestrino & Somjen, 1986; Sick et al. 1987; Korf et al. 1988; Somjen et al. 1989; Lauritzen & Hansen, 1992; Katayama et al. 1992; Kral et al. 1993; Xie et al. 1995) that has many common features with the spreading depression phenomenon described by Leão (1947). If re-oxygenation is delayed for several minutes after AD, brain functions do not recover (Tanaka et al. 1997; Fairchild et al. 1988; Balestrino et al. 1989; Dzhala et al. 2000).
While neuronal activity is strongly reduced during the adaptation phase and is completely lost after AD, it transiently recovers at the beginning of AD. Although this transient ‘paradoxical’ recovery of the neuronal activity has been observed both in vivo and in vitro, the results differ. In the rat hippocampus in vivo, it has been found that ischaemia-induced AD is preceded by a spindle of fast (20–50 Hz) network oscillations (Freund et al. 1989). However, studies in the hippocampal slice preparation in vitro failed to reproduce the anoxia-induced oscillatory pattern. Instead, only the ‘paradoxical’ recovery of the evoked glutamatergic postsynaptic responses has been observed at the beginning of AD (Fowler, 1992; Sick et al. 1987; Zhu & Krnjevic, 1999). The inability to reproduce the in vivo oscillatory pattern in the in vitro conditions has limited the study of the neuronal mechanisms underlying anoxia-induced oscillations.
Interestingly, brief periods of oscillatory activity in the gamma frequency range have also been observed at the beginning of the spreading depression induced in vivo by an injection of a high-potassium solution (Herreras et al. 1994; Bragin et al. 1997), or following an epileptiform activity induced by a tetanic stimulation (Bragin et al. 1997). Pharmacological analysis of these oscillatory activities suggested that neuronal synchronisation is achieved via non-synaptic mechanisms, most likely via gap junctions (Herreras et al. 1994).
In the present study, we used in vitro preparations of the intact hippocampus of P7–10 rats (Khalilov et al. 1997a) to study neuronal mechanisms of the anoxia-induced oscillations. The level of maturity of the rat hippocampus at P7–10 can be compared to the prenatal period of the primate hippocampus (Romijn et al. 1991). The intact hippocampus preparation offers several advantages over slices, in particular an intact neuronal network, to study physiological (Khalilov et al. 1997a; Leinekugel et al. 1998) and paroxysmal network driven activities (Khalilov et al. 1997b,1999a, b; Luhmann et al. 2000), including those induced by anoxia (Dzhala et al. 1999, 2000). The main finding was that the ischaemia-induced oscillatory in vivo pattern can be reproduced in the intact hippocampus in vitro. Using whole-cell and extracellular field recordings we provide evidence that the synchronisation of anoxia-induced oscillations relies on synaptic mechanisms; that inhibition by GABA and adenosine sets the tune for a generation of oscillations and prevents an epileptiform activity; and that the synchronous GABAergic inhibition is instrumental in a phase locking neuronal activity similar to other types of oscillatory activities in the gamma frequency range.
METHODS
Experimental system
Intact hippocampi of (P0–10) male Wistar rats were prepared as described previously (Khalilov et al. 1997a). All animal-use protocols conformed to the French Public Health Service policy and the INSERM guidelines on the use of laboratory animals. Animals were anaesthetised with chloral hydrate (350 mg kg−1, intraperitoneally) and decapitated. After dissection, hippocampi were kept in an oxygenated (95 % O2-5 % CO2) artificial cerebrospinal fluid (ACSF) of the following composition (mm): NaCl 126, KCl 3.5, CaCl2 2.0, MgCl2 1.3, NaHCO3 25, NaH2PO4 1.2 and glucose 11 (pH 7.4) at room temperature (20–22 °C) at least 2 h before use. For recordings, the hippocampi were placed into a conventional fully submerged chamber superfused with ACSF (32 °C) at a rate of 8–9 ml min−1. The hippocampi were placed on a plastic mesh with the ventral side down, allowing easy access to the CA3 and CA1 regions. Then the hippocampi were fixed to the silgard bottom using several entomological needles. Episodes of anoxia were induced by perfusion with ACSF saturated with 95 % N2-5 % CO2. Sucrose at an equimolar concentration was substituted for glucose in the anoxic ACSF to aggravate the metabolic stress.
Electrophysiological recordings and data analysis
Whole-cell and extracellular field recordings were performed in the CA3 area in the middle part of hippocampus except for the study of the longitudinal propagation. Whole-cell recordings were made using an Axopatch 200A (Axon Instruments, USA) amplifier. Patch electrodes were made from borosilicate glass capillaries (GC150F-15, Clark Electromedical Instruments) and had a resistance of 5–8 MΩ when filled with a solution containing (mm): potassium gluconate 135, CaCl2 0.1, MgCl2 2, Na2ATP 2, EGTA 1 and Hepes 10, pH 7.25, osmolarity 280 mosm l−1. The liquid junction potential was measured (+11 mV) and all voltages reported are corrected values.
Extracellular field potentials were recorded using glass microelectrodes (GC120TF-10, Clark Electromedical Instruments) filled with ACSF using custom made amplifiers in quasi-DC mode (bandpass 0.04 Hz to 4 kHz). The recordings were performed in the pyramidal cell layer of the CA3 subfield. Tungsten bipolar electrodes placed in the stratum radiatum of the CA3 area were used to evoke synaptic responses. The stimulation parameters were 30–50 V, 20 ms delivered at 0.033 Hz. Synaptic responses were acquired into the memory of a personal computer using an analog-to-digital converter (Digidata 1200; Axon Instruments). Axoscope (Axon Instruments), Acquis (Gerard Sadoc, France) and Origin 5.0 (Microcal Software, USA) programs were used for the acquisition and analysis. Group measures are expressed as means ±s.e.m.; error bars also indicate s.e.m. The statistical significance of differences was assessed with Student's t test. The level of significance was set at P < 0.05.
Chemicals
Tetrodotoxin, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), 2-amino-5-phosphonovaleric acid (APV), bicuculline and diazepam were from Tocris Neuramin (Bristol, UK); 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) and cyclopentyladenosine (CPA) were from RBI (Natick, MA, USA).
RESULTS
Response of hippocampus to anoxia
Perfusion with an anoxic-aglycaemic solution induced a biphasic response in the hippocampal neurons (Fig. 1). The first adaptation phase was characterised by moderate changes in the membrane potential and depression of the synaptic activity. Within 2–3 min after the onset of anoxia, evoked field EPSPs were depressed to 11 ± 2 % of the control values and pyramidal neurons recorded in current-clamp mode were slightly hyperpolarised from −73.5 ± 1.5 to −75.1 ± 1.6 mV (n = 10; P8–9). The second phase was characterised by an anoxia-induced neuronal depolarisation (AD). AD started gradually and progressively evolved to a rapid process resulting in nearly complete loss of membrane potential (−15.5 ± 0.5 mV; n = 10). Simultaneously, a remarkable negative shift of 5.8 ± 1.0 mV and 38 ± 4 s duration (n = 8; 0.04 Hz to 4 kHz band-pass) was observed in extracellular field potential recordings similar to that in the cerebral cortex in vivo (Korf et al. 1988; Katayama et al. 1992; Lauritzen & Hansen, 1992; Xie et al. 1995) as well as in adult (Balestrino & Somjen, 1986; Sick et al. 1987; Somjen et al. 1989; Kral et al. 1993) and neonatal (Luhmann & Kral, 1997) cortical slices. Prior to AD, we observed a partial paradoxical recovery of the evoked field EPSPs to 34 ± 7 % of the control (pre-anoxic) values (n = 8; Fig. 1), that has also been described in slices (Fowler, 1992) and in the intact hippocampus from younger rats (Dzhala et al. 2000). Following AD, neither the membrane potential nor the synaptic responses recovered during re-oxygenation.
Figure 1. Response of hippocampus to anoxia.
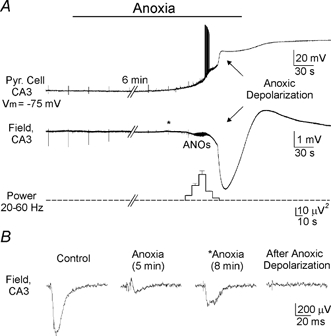
A, perfusion with anoxic-aglycemic solution induces a biphasic response in the intact rat (postnatal day P9) hippocampus: simultaneous whole-cell current clamp (upper trace) and extracellular field potential (lower trace) recordings in the CA3 pyramidal cell layer. During the ‘adaptation’ phase, the synaptic responses evoked by an electrical stimulation (shown in B) are blocked but neurons maintain their membrane potential. The transition to the second phase is manifested by a rapid neuronal depolarisation and a negative deflection in the extracellular field recordings. A rapid anoxic depolarisation is preceded by a phase of oscillations (ANOs) in the gamma-frequency band (power in the 20–60 Hz band is shown on the graph below). Following the anoxic depolarisation, the membrane potential and synaptic responses did not recover. B, evoked field EPSPs are rapidly blocked during anoxia but transiently recover (marked by an asterisk) shortly before the rapid anoxic depolarisation begins.
Anoxia-induced network oscillations (ANOs)
In agreement with a previous in vivo study (Freund et al. 1989), a spindle of high-frequency oscillations was observed at the beginning of AD. Anoxia-induced network oscillations (ANOs) waxed and waned during the gradual phase of AD and usually lasted for 1–2 min (Figs 1–3). The main component of ANOs was in the range of 20–40 Hz (30.4 ± 2.8 Hz; in the middle part of hippocampus); it was nested in slow oscillations of 2–8 Hz (n = 22/25; P8–10; Fig. 2B). In each experiment, ANOs were highly periodic, as evidenced by harmonics in the power spectrum (Fig. 2C). Multiple unit activity (MUA) in the CA3 pyramidal layer was locked to the negative and ascending phase of ANOs (Fig. 2B). Simultaneous recording of the population activity and the CA3 pyramidal neurons in current-clamp mode revealed that neurons generate the action potentials with an average probability of 5.6 ± 2.4 % per cycle (n = 6; Fig. 2B).
Figure 3. ANOs coincide with neuronal depolarisation.
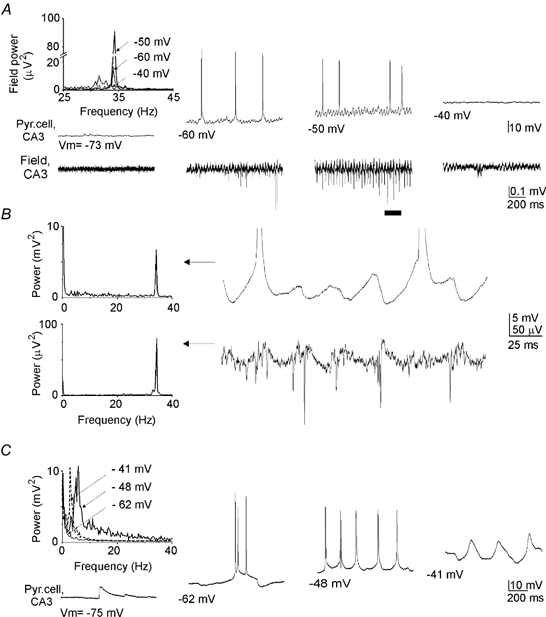
A, parts of records from a CA3 pyramidal cell in current-clamp mode (upper trace, values of membrane potential are indicated below) and extracellular field potential (lower trace) during ANOs. Left top: power spectrum of the field ANOs at different levels of anoxic depolarisation of the pyramidal cell. B, six cycles of ANOs from the traces shown in A (indicated by a bar) on an expanded time scale. Action potentials are truncated. Note that both extracellularly recorded action currents and action potentials in the pyramidal cell are generated at the negative and ascending phases of ANOs. Left, power spectrum of the corresponding traces. Note the maximal power at 34 Hz in both the pyramidal cell and the extracellular field. C, response of the cell shown in A and B to injection of the positive current in the normoxic conditions (before anoxia). Left top, power spectrum of the membrane potential at different levels of depolarisation. Note that the maximal power of oscillations is attained at 5.5 Hz.
Figure 2. Anoxia-induced network oscillations (ANOs).
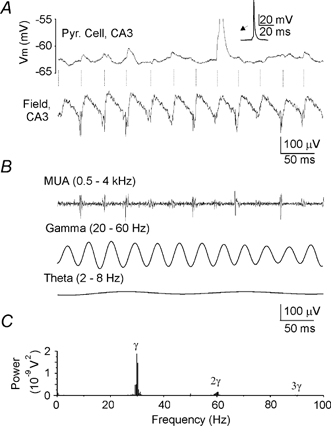
A, current-clamp recordings from a putative CA3 pyramidal cell (upper trace) and the extracellular field potential (lower trace) during ANOs. Note that oscillations of the membrane potential that occasionally trigger action potentials are locked to the negative phase of ANOs. The separation distance between the pyramidal cell and field recording site is < 100 μm. B, extracellular field record from above (A) filtered for action currents, gamma and theta activity. Note that multiple units activity (MUA) is phase locked to the negative phase of gamma cycles during ANOs. C, power spectrum of ANOs determined by fast Fourier transform analysis reveals a peak of oscillations at 30 Hz (γ) and its harmonics (2γ, 3γ).
ANOs coincided with depolarisation of neurons to subthreshold values (Fig. 3A and B). The maximal power of the population activity was observed when the membrane potential attained −55.6 ± 3.8 mV (n = 8). The waning of ANOs was associated with a depolarisation block of pyramidal cells at potentials above −47.0 ± 2.4 mV (n = 8). Interestingly, depolarisation of pyramidal cells in the normoxic conditions by injection of the positive current failed to induce high frequency oscillations of membrane potential in the gamma frequency range, as evidenced by Fourier analysis (Fig. 3C). Maximal power of the intrinsic membrane oscillations was in the theta frequency range (6.4 ± 1.3 Hz; n = 4). Therefore, it is unlikely that the intrinsic oscillations contribute to the main gamma component of ANOs. Yet the intrinsic oscillations can contribute to the slower component of ANOs in the theta frequency range.
Voltage clamping CA3 pyramidal cells at the reversal potential of the GABAA and glutamate receptor-mediated currents revealed that both glutamatergic excitatory and GABAergic inhibitory postsynaptic currents (EPSCs and IPSCs, respectively) are phase locked with population activity (n = 6) (Fig. 4), suggesting that both pyramidal cells and interneurons are activated at the negative and ascending phases of ANOs.
Figure 4. Synaptic correlations of ANOs.
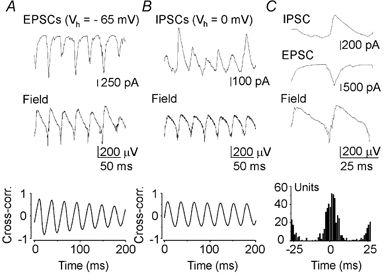
A and B, spontaneous excitatory AMPA receptor-mediated postsynaptic currents (EPSCs) and inhibitory GABAA receptor-mediated IPSCs recorded in CA3 pyramidal cell (upper traces) are phase locked to field ANOs (lower traces). EPSCs were recorded at the reversal potential of the GABAA (Vh =−65 mV) and glutamate receptor-mediated currents (Vh = 0 mV). Bottom, cross-correlation between the current passing through the cell membrane and the extracellular field potential during ANOs. C, synaptic events and the extracellular field potential during a single cycle of ANOs. Below, occurrence of MUA relative to the extracellular field potential for 80 cycles of ANOs. Time zero was set to the negative phase of the extracellular field oscillations.
Pharmacology of ANOs
We further investigated the mechanisms underlying the synchronisation of ANOs by using a pharmacological approach (Fig. 5). The blockade of sodium action potentials by tetrodotoxin (1 μm) completely blocked ANOs, suggesting an involvement of synaptic mechanisms (n = 4). In the presence of the ionotropic glutamate receptor antagonists CNQX (10 μm) and APV (50 μm), only low-amplitude, non-periodic fluctuations have been observed (n = 4), suggesting an involvement of recurrent glutamatergic connections. Diazepam (10 μm), which prolongs the GABAA receptor-mediated IPSCs, significantly slowed down the oscillations to 23 ± 3 Hz (n = 4) and strongly reduced their power from 1092 ± 572 μV2 in control (n = 22) to 32 ± 20 μV2 (n = 4; 30 s long segments; P8–10). The GABAA receptor antagonist bicuculline (10 μm; applied 5–6 min prior to expected AD) transformed the ANOs to hyper-synchronous ictal-like epileptiform discharges (n = 6). Taken together, these results suggest that the synchronisation of neuronal activity during ANOs is determined by glutamatergic and GABAergic synaptic connections. The latter probably contribute to the synchronisation of network discharge by phase locking action potentials via synchronous inhibition mediated by GABAA receptors.
Figure 5. Pharmacology of ANOs.
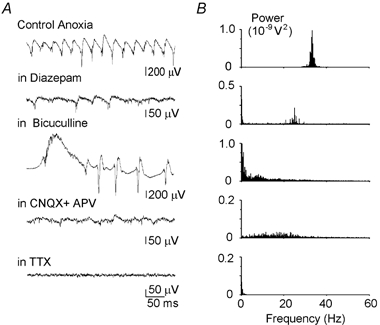
A, hippocampal activity recorded by an extracellular field electrode at the beginning of anoxic depolarisation in control conditions; in the presence of the GABAA receptor modulator diazepam (10 μm); the GABAA receptor antagonist bicuculline (10 μm); the ionotropic glutamate receptor antagonists CNQX (10 μm) and APV (50 μm); and the sodium channel blocker TTX (1 mm). B, corresponding power spectra of the extracellular field potential (30 s long segments; 0.3 Hz spectral resolution).
Pharmacological modulation of synaptic activity not only affected ANOs, but also modified the onset of AD. In control conditions, AD was induced after 7.5 ± 0.6 min of anoxia (n = 11; P9). Suppression of neuronal activity significantly delayed the AD onset: in the presence of CNQX and APV, to 9.9 ± 0.1 min (n = 4); and in the presence of diazepam, to 8.6 ± 0.7 min (n = 4; P9). In contrast, bicuculline-induced hyperactivity was associated with the acceleration of AD onset to 4.4 ± 0.6 min (n = 6; P9). These results are in agreement with our previous report in younger rats (Dzhala et al. 2000).
Roles of potassium and adenosine for the induction of ANOs
What are the factors responsible for the induction of ANOs? To answer this question, we simulated the conditions that are achieved during ANOs in normal ACSF (with oxygen and glucose; Fig. 6). The coincidence of ANOs and neuronal depolarisation indicates that the latter is an important factor in ANO induction. Among various factors, an increase of extracellular potassium concentration [K+]o strongly contributes to AD. Therefore, we first studied the effects of increased [K+]o from 3.5 mm to 10 mm– the level achieved during the gradual phase of AD (Müller & Somjen, 2000) that corresponds to ANOs in the intact hippocampus preparation. However, this failed to induce ANO-like activity in normoxic conditions. Instead, epileptiform discharges were generated (n = 8; see also (Khalilov et al. 1997a). Yet another factor that operates during anoxia is an adenosine A1 receptor-mediated reduction of glutamatergic transmission (Fowler, 1989). To simulate both depolarisation and adenosine-mediated inhibition, we increased [K+]o to 10 mm in the presence of the A1 adenosine receptor agonist CPA (200 nm). In these conditions, network oscillations similar to ANOs have been observed (frequency 24 ± 2 Hz; power 47 ± 19 μV2; n = 5/8, P8–9; 30 s long segments; Fig. 6). In keeping with this, after the blockade of A1 receptors by the selective antagonist DPCPX (200 nm), anoxia induced powerful epileptiform discharges but no ANOs were observed (n = 3; Fig. 6). Therefore, ANOs require both neuronal depolarisation and the A1 adenosine receptor-mediated decrease of glutamatergic transmission.
Figure 6. Roles of potassium and adenosine for ANOs induction.
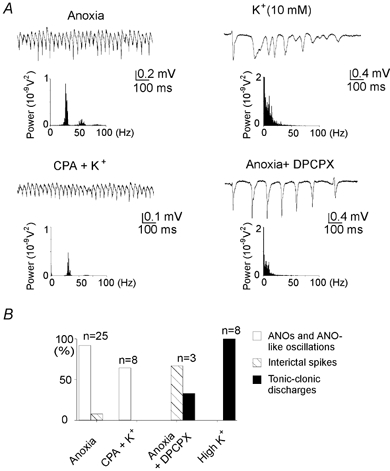
A, extracellular field potential at the beginning of the anoxic depolarisation (ANOs); in normoxic conditions after elevation of the extracellular potassium concentration to 10 mm; in normoxic conditions after co-perfusion with elevated potassium and the A1 adenosine receptor agonist cyclopentyladenosine (CPA, 200 nm); and during anoxia in the presence of the A1 adenosine receptor antagonist DPCPX (100 nm). Note that ANO-like activity can be induced in normoxic conditions by increasing the extracellular potassium concentration in conjunction with the activation of adenosine A1 receptors. B, statistical plot of the occurrence of various types of epileptiform and ANO-like activity in the conditions shown in A.
Longitudinal propagation of ANOs and rapid anoxic depolarisation
Multisite recordings along the septo-temporal axis of the hippocampus (Fig. 7) revealed that the wave of ANOs and AD originate in the temporal part of the hippocampus and propagate towards the septal pole with a speed of 1.9 ± 0.2 mm min−1 (n = 10), which is comparable to the speed of AD propagation in a slice (7 mm min−1 (Aitken et al. 1999). The frequency of ANOs was the highest in the temporal part (30.6 ± 1.2 Hz; n = 16). In the septal part, population activity during anoxia was presented either by ANOs of lower frequency 26.3 ± 1.3 Hz (n = 7/16), or by asynchronous firing (n = 9/16). Cross-correlating the activity recorded along the septo-temporal axis revealed that the actively propagating ANOs (associated with units activity) are synchronised over distances of about 200 μm, although passive propagation could be observed at distances of more than 1 mm (Fig. 7).
Figure 7. Longitudinal propagation of ANOs and anoxic depolarisation.
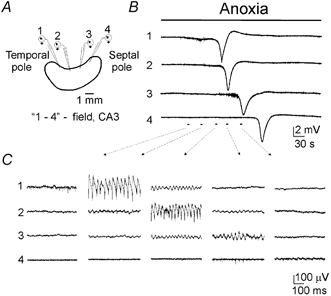
A, scheme of longitudinal 4-site recordings in the intact hippocampus (separation distance 1.2 mm between the electrodes). Electrodes are positioned in the pyramidal layer of the CA3 subfield. B, rapid anoxic depolarisation is first observed in the temporal part (electrode 1) and then it propagates in the septal direction (electrode 4). C, the wave of ANOs that precedes the anoxic depolarisation also originates in the temporal pole and propagates towards the septal pole, providing only local synchrony in the hippocampus.
Developmental changes of ANOs
In the next series of experiments we studied the developmental changes of anoxia-induced network activity (Fig. 8A–C). At birth (P0–1), only single population spikes have been observed. By the end of the first postnatal week, they evolved to more complex epileptiform population bursts. However, ANOs were not observed until P7. From P7 to P10, the intensity and frequency of ANOs progressively increased. Not only did the network patterns change but also the AD onset significantly accelerated with age from 44 ± 7 min at P0 (n = 5) to 6.2 ± 1.0 min at P10 (n = 4; Fig. 8D). This is in keeping with lower vulnerability of the immature brain to anoxia (Hansen, 1977).
Figure 8. Developmental changes of anoxia-induced network activity.
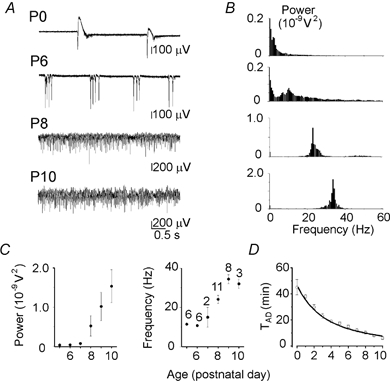
A, hippocampal activity recorded by an extracellular electrode at the beginning of the anoxic depolarisation at different postnatal ages (postnatal days P0–10). Note the emergence of ANOs during the second postnatal week. B, corresponding power-spectra of the extracellular field potential (30 s long segments; 0.3 Hz spectral resolution). C, postnatal changes in the power and frequency of ANOs. D, dependence of onset of the anoxia-induced depolarisation on age.
DISCUSSION
We showed that ANOs – high frequency network oscillations generated at the beginning of AD in the intact hippocampus in vitro– are very similar to the network activity observed at the comparable stage of global ischaemia in vivo (Freund et al. 1989). The main conclusions of the present study are that: (i) anoxia-induced depolarisation and adenosine-mediated reduction of excitatory synaptic transmission are the principal conditions for ANOs; (ii) ANOs are synchronised by glutamatergic and GABAergic synaptic connections, the latter providing synchronous inhibition and phase locking population discharges; (iii) postnatal development of ANOs is probably due to the development of GABAergic inhibition.
Similar to ANOs, network oscillations have been observed in vivo during the phase that precedes the spreading depression induced by injections of high potassium (Herreras et al. 1994; Bragin et al. 1997) or following the epileptiform activity induced by tetanic stimulation (Bragin et al. 1997). This provides additional evidence in favour of the similarities between AD and spreading depression (Leão, 1947). However, injections of 6,7-dinitro-quinoxaline-2,3-dione (DNQX) and 3-(R-)(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP) to block ionotropic glutamate receptors, a cocktail of divalent cations to block voltage-gated calcium channels or tetrodotoxin to block sodium spikes failed to block high potassium-induced population spike bursts (Herreras et al. 1994). Therefore, it has been proposed that the synchronisation of population activity is brought about by way of gap junctions that open during the spreading depression. In contrast, the synchronisation of neuronal activity during ANOs is clearly determined by synaptic mechanisms. This discrepancy can be due to differences in conditions settled during anoxia- and high potassium-induced spreading depressions, differences in the age of animals and experimental conditions (in vitro versus in vivo, the use of anaesthesia in the studies in vivo). An alternative explanation could be that a local blockade of synaptic transmission (Herreras et al. 1994) is not sufficient to block oscillations. It is possible that both synaptic and non-synaptic mechanisms contribute to the synchronisation of ANO-like activities, but their relative importance may depend on experimental conditions. Probably, this issue can be addressed in future when a specific gap junction blocker becomes available.
ANOs are also reminiscent of gamma-frequency oscillations induced in a hippocampal slice by carbachol (Fisahn et al. 1998). Similar features include the pattern of activity (gamma-frequency oscillations nested in theta oscillations), the contribution of the glutamatergic and GABAergic synaptic connections to the synchronisation of oscillations and the probability of pyramidal cells firing (about 0.05 per cycle). This can be due to certain similarities in the conditions achieved during anoxia and in the presence of carbachol. Thus, in both cases there is neuronal depolarisation and reduction of glutamatergic transmission (Scanziani et al. 1995). One can also add the depression of after-hyperpolarisation and the reduction of spike accomodation in both situations (Krnjevic & Leblond, 1989; Azouz et al. 1994).
As for the other types of oscillatory activity in the gamma frequency band (Bragin et al. 1995; Whittington et al. 1995; Fisahn et al. 1998), GABAergic interneurons are instrumental in synchronising the network during ANOs. This is in keeping with the fact that GABAA receptor-mediated IPSCs are relatively resistant to anoxia both in pyramidal cells (Khazipov et al. 1993; Zhu & Krnjevic, 1994; Katchman et al. 1994) and in interneurons (Khazipov et al. 1995b). The fact that diazepam significantly reduced ANO frequency suggests that ANOs are predominantly interneuron network based, as with other transient models of gamma oscillations (Whittington et al. 2000). This is also compatible with the developmental changes of the anoxia-induced network patterns. Indeed, oscillatory activity in the gamma frequency range has not been observed until P7. On the other hand, it is known that around this age, a developmental switch in the action of GABA from depolarising to hyperpolarising occurs in the rat hippocampus (Ben-Ari et al. 1989). Therefore, maturation of ANOs is likely to be determined by the development of GABAergic inhibition. In further studies, it will be interesting to determine whether the maturation of other types of oscillatory activity in the gamma frequency band is also due to developmental changes in the action of GABA. Emergence of theta oscillations – which are associated with gamma oscillations in adult rats in vivo (Bragin et al. 1995) – around postnatal day P8 (Leblanc & Bland, 1979) supports this hypothesis.
In conclusion, ANOs are a particular type of neuronal network activity that is generated during anoxia and manifests a transition from the adaptation phase to the phase of the anoxic depolarisation. At least two factors contribute to the generation of ANOs: neuronal depolarisation and adenosine-mediated reduction of the glutamatergic synaptic transmission. Synchronisation of the neuronal network during ANOs relies on the same synaptic mechanisms contributing to the generation of other types of activity in the gamma frequency band; this includes excitatory glutamatergic connections and GABAergic synapses that phase lock the population discharge via synchronous inhibition. Thus, ANOs are an additional example illustrating a propensity of the hippocampal network to oscillate at gamma frequency.
Acknowledgments
Financial support from INSERM and FRM is acknowledged. We gratefully acknowledge the valuable suggestions made by G. Somjen and G. Buzsaki. We also thank R. Cannon, and O. and G. Medyna for the help in preparing of the manuscript.
References
- Aitken PG, Fayuk D, Somjen GG, Turner DA. Use of intrinsic optical signals to monitor physiological changes in brain tissue slices. Methods. 1999;18:91–103. doi: 10.1006/meth.1999.0762. [DOI] [PubMed] [Google Scholar]
- Azouz R, Jensen MS, Yaari Y. Muscarinic modulation of intrinsic burst firing in rat hippocampal neurons. European Journal of Neuroscience. 1994;6:961–966. doi: 10.1111/j.1460-9568.1994.tb00590.x. [DOI] [PubMed] [Google Scholar]
- Balestrino M, Aitken PG, Somjen GG. Spreading depression-like hypoxic depolarization in CA1 and fascia dentata of hippocampal slices: relationship to selective vulnerability. Brain Research. 1989;497:102–107. doi: 10.1016/0006-8993(89)90975-x. [DOI] [PubMed] [Google Scholar]
- Balestrino M, Somjen GG. Chlorpromazine protects brain tissue in hypoxia by delaying spreading depression-mediated calcium influx. Brain Research. 1986;385:219–226. doi: 10.1016/0006-8993(86)91067-x. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. Journal of Physiology. 1989;416:303–325. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragin A, Jando G, Nadasdy Z, Hetke J, Wise K, Buzsaki G. Gamma (40–100 Hz) oscillation in the hippocampus of the behaving rat. Journal of Neuroscience. 1995;15:47–60. doi: 10.1523/JNEUROSCI.15-01-00047.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragin A, Penttonen M, Buzsaki G. Termination of epileptic afterdischarge in the hippocampus. Journal of Neuroscience. 1997;17:2567–2579. doi: 10.1523/JNEUROSCI.17-07-02567.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundege JM, Dunwiddie TV. Role of adenosine as a modulator of synaptic activity in the central nervous system. Advances in Pharmacology. 1997;39:353–391. doi: 10.1016/s1054-3589(08)60076-9. [DOI] [PubMed] [Google Scholar]
- Croning MD, Zetterstrom TS, Grahame-Smith DG, Newberry NR. Action of adenosine receptor antagonists on hypoxia-induced effects in the rat hippocampus in vitro. British Journal of Pharmacology. 1995;116:2113–2119. doi: 10.1111/j.1476-5381.1995.tb16419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Fredholm BB. Adenosine neuromodulation. In: Jacobsen KA, Jarvis MF, editors. Purinergic Approaches in Experimental Therapeutics. New York: Wiley-Liss; 1997. pp. 359–382. [Google Scholar]
- Dzhala V, Ben-Ari Y, Khazipov R. Seizures accelerate anoxia-induced neuronal death in the neonatal rat hippocampus. Annals of Neurology. 2000;48:632–640. [PubMed] [Google Scholar]
- Dzhala V, Desfreres L, Melyan Z, Ben-Ari Y, Khazipov R. Epileptogenic action of caffeine during anoxia in the neonatal rat hippocampus. Annals of Neurology. 1999;46:95–102. [PubMed] [Google Scholar]
- Erdemli G, Xu YZ, Krnjevic K. Potassium conductance causing hyperpolarization of CA1 hippocampal neurons during hypoxia. Journal of Neurophysiology. 1998;80:2378–2390. doi: 10.1152/jn.1998.80.5.2378. [DOI] [PubMed] [Google Scholar]
- Fairchild MD, Parsons JE, Wasterlain CG, Rinaldi PC, Wallis RA. A hypoxic injury potential in the hippocampal slice. Brain Research. 1988;453:357–361. doi: 10.1016/0006-8993(88)90178-3. [DOI] [PubMed] [Google Scholar]
- Fisahn A, Pike FG, Buhl EH, Paulsen O. Cholinergic induction of network oscillations at 40 Hz in the hippocampus in vitro. Nature. 1998;394:186–189. doi: 10.1038/28179. [DOI] [PubMed] [Google Scholar]
- Fowler JC. Adenosine antagonists delay hypoxia-induced depression of neuronal activity in hippocampal brain slice. Brain Research. 1989;490:378–384. doi: 10.1016/0006-8993(89)90258-8. [DOI] [PubMed] [Google Scholar]
- Fowler JC. Escape from inhibition of synaptic transmission during in vitro hypoxia and hypoglycemia in the hippocampus. Brain Research. 1992;573:169–173. doi: 10.1016/0006-8993(92)90128-v. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G, Prohaska OJ, Leon A, Somogyi P. Simultaneous recording of local electrical activity, partial oxygen tension and temperature in the rat hippocampus with a chamber-type microelectrode. Effects of anaesthesia, ischemia and epilepsy. Neuroscience. 1989;28:539–549. doi: 10.1016/0306-4522(89)90003-1. [DOI] [PubMed] [Google Scholar]
- Fujimura N, Tanaka E, Yamamoto S, Shigemori M, Higashi H. Contribution of ATP-sensitive potassium channels to hypoxic hyperpolarization in rat hippocampal CA1 neurons in vitro. Journal of Neurophysiology. 1997;77:378–385. doi: 10.1152/jn.1997.77.1.378. [DOI] [PubMed] [Google Scholar]
- Fujiwara N, Higashi H, Shimoji K, Yoshimura M. Effects of hypoxia on rat hippocampal neurones in vitro. Journal of Physiology. 1987;384:131–151. doi: 10.1113/jphysiol.1987.sp016447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribkoff VK, Bauman LA. Endogenous adenosine contributes to hypoxic synaptic depression in hippocampus from young and aged rats. Journal of Neurophysiology. 1992;68:620–628. doi: 10.1152/jn.1992.68.2.620. [DOI] [PubMed] [Google Scholar]
- Hansen AJ. Extracellular potassium concentration in juvenile and adult rat brain cortex during anoxia. Acta Physiologica Scandinavica. 1977;99:412–420. doi: 10.1111/j.1748-1716.1977.tb10394.x. [DOI] [PubMed] [Google Scholar]
- Hansen AJ. Effect of anoxia on ion distribution in the brain. Physiological Reviews. 1985;65:101–148. doi: 10.1152/physrev.1985.65.1.101. [DOI] [PubMed] [Google Scholar]
- Hansen AJ, Hounsgaard J, Jahnsen H. Anoxia increases potassium conductance in hippocampal nerve cells. Acta Physiologica Scandinavica. 1982;115:301–310. doi: 10.1111/j.1748-1716.1982.tb07082.x. [DOI] [PubMed] [Google Scholar]
- Herreras O, Largo C, Ibarz JM, Somjen GG, Martin DR. Role of neuronal synchronizing mechanisms in the propagation of spreading depression in the in vivo hippocampus. Journal of Neuroscience. 1994;14:7087–7098. doi: 10.1523/JNEUROSCI.14-11-07087.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass IS, Lipton P. Mechanisms involved in irreversible anoxic damage to the in vitro rat hippocampal slice. Journal of Physiology. 1982;332:459–472. doi: 10.1113/jphysiol.1982.sp014424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama Y, Tamura T, Becker DP, Tsubokawa T. Early cellular swelling during cerebral ischemia in vivo is mediated by excitatory amino acids released from nerve terminals. Brain Research. 1992;577:121–126. doi: 10.1016/0006-8993(92)90544-j. [DOI] [PubMed] [Google Scholar]
- Katchman AN, Hershkowitz N. Adenosine antagonists prevent hypoxia-induced depression of excitatory but not inhibitory synaptic currents. Neuroscience Letters. 1993;159:123–126. doi: 10.1016/0304-3940(93)90814-2. [DOI] [PubMed] [Google Scholar]
- Katchman AN, Vicini S, Hershkowitz N. Mechanism of early anoxia-induced suppression of the GABAA-mediated inhibitory postsynaptic current. Journal of Neurophysiology. 1994;71:1128–1138. doi: 10.1152/jn.1994.71.3.1128. [DOI] [PubMed] [Google Scholar]
- Katsura K, Kristian T, Siesjo BK. Energy metabolism, ion homeostasis, and cell damage in the brain. Biochemical Society Transactions. 1994;22:991–996. doi: 10.1042/bst0220991. [DOI] [PubMed] [Google Scholar]
- Katsura K, Rodriguez DTE, Folbergrova J, Bazan NG, Siesjo BK. Coupling among energy failure, loss of ion homeostasis, and phospholipase A2 and C activation during ischemia. Journal of Neurochemistry. 1993;61:1677–1684. doi: 10.1111/j.1471-4159.1993.tb09803.x. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Dzhala V, Ben-Ari Y, Khazipov R. Dual role of GABA in the neonatal rat hippocampus. Developmental Neuroscience. 1999a;21:310–319. doi: 10.1159/000017380. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Dzhala V, Medina I, Leinekugel X, Melyan Z, Lamsa K, Khazipov R, Ben-Ari Y. Maturation of kainate-induced epileptiform activities in interconnected intact neonatal limbic structures in vitro. European Journal of Neuroscience. 1999b;11:3468–3480. doi: 10.1046/j.1460-9568.1999.00768.x. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Esclapez M, Medina I, Aggoun D, Lamsa K, Leinekugel X, Khazipov R, Ben-Ari Y. A novel in vitro preparation: the intact hippocampal formation. Neuron. 1997a;19:743–749. doi: 10.1016/s0896-6273(00)80956-3. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Khazipov R, Esclapez M, Ben-Ari Y. Bicuculline induces ictal seizures in the intact hippocampus recorded in vitro. European Journal of Pharmacology. 1997b;319:R5–6. doi: 10.1016/s0014-2999(96)00964-8. [DOI] [PubMed] [Google Scholar]
- Khazipov R, Bregestovski P, Ben-Ari Y. Hippocampal inhibitory interneurons are functionally disconnected from excitatory inputs by anoxia. Journal of Neurophysiology. 1993;70:2251–2259. doi: 10.1152/jn.1993.70.6.2251. [DOI] [PubMed] [Google Scholar]
- Khazipov R, Congar P, Ben-Ari Y. Hippocampal CA1 lacunosum-moleculare interneurons: comparison of effects of anoxia on excitatory and inhibitory postsynaptic currents. Journal of Neurophysiology. 1995a;74:2138–2149. doi: 10.1152/jn.1995.74.5.2138. [DOI] [PubMed] [Google Scholar]
- Khazipov R, Congar P, Ben-Ari Y. Hippocampal CA1 lacunosum-moleculare interneurons: modulation of monosynaptic GABAergic IPSCs by presynaptic GABAB receptors. Journal of Neurophysiology. 1995b;74:2126–2137. doi: 10.1152/jn.1995.74.5.2126. [DOI] [PubMed] [Google Scholar]
- Korf J, Klein HC, Venema K, Postema F. Increases in striatal and hippocampal impedance and extracellular levels of amino acids by cardiac arrest in freely moving rats. Journal of Neurochemistry. 1988;50:1087–1096. doi: 10.1111/j.1471-4159.1988.tb10577.x. [DOI] [PubMed] [Google Scholar]
- Kral T, Luhmann HJ, Mittmann T, Heinemann U. Role of NMDA receptors and voltage-activated calcium channels in an in vitro model of cerebral ischemia. Brain Research. 1993;612:278–288. doi: 10.1016/0006-8993(93)91673-g. [DOI] [PubMed] [Google Scholar]
- Krnjevic K, Leblond J. Changes in membrane currents of hippocampal neurons evoked by brief anoxia. Journal of Neurophysiology. 1989;62:15–30. doi: 10.1152/jn.1989.62.1.15. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Hansen AJ. The effect of glutamate receptor blockade on anoxic depolarization and cortical spreading depression. Journal of Cerebral Blood Flow and Metabolism. 1992;12:223–229. doi: 10.1038/jcbfm.1992.32. [DOI] [PubMed] [Google Scholar]
- Leblanc MO, Bland BH. Developmental aspects of hippocampal electrical activity and motor behavior in the rat. Experimental Neurology. 1979;66:220–237. doi: 10.1016/0014-4886(79)90076-1. [DOI] [PubMed] [Google Scholar]
- Leblond J, Krnjevic K. Hypoxic changes in hippocampal neurons. Journal of Neurophysiology. 1989;62:1–14. doi: 10.1152/jn.1989.62.1.1. [DOI] [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7–14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- Leinekugel X, Khalilov I, Ben-Ari Y, Khazipov R. Giant depolarizing potentials: the septal pole of the hippocampus paces the activity of the developing intact septohippocampal complex in vitro. Journal of Neuroscience. 1998;18:6349–6357. doi: 10.1523/JNEUROSCI.18-16-06349.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leão AAP. Further observations on the spreading depression of activity in the cerebral cortex. Journal of Neurophysiology. 1947;10:409–414. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiological Reviews. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Luhmann HJ, Dzhala VI, Ben-Ari Y. Generation and propagation of 4-AP-induced epileptiform activity in neonatal intact limbic structures in vitro. European Journal of Neuroscience. 2000;12:2757–2768. doi: 10.1046/j.1460-9568.2000.00156.x. [DOI] [PubMed] [Google Scholar]
- Luhmann HJ, Kral T. Hypoxia-induced dysfunction in developing rat neocortex. Journal of Neurophysiology. 1997;78:1212–1221. doi: 10.1152/jn.1997.78.3.1212. [DOI] [PubMed] [Google Scholar]
- Martin RL, Lloyd HG, Cowan AI. The early events of oxygen and glucose deprivation: setting the scene for neuronal death? Trends in Neurosciences. 1994;17:251–257. doi: 10.1016/0166-2236(94)90008-6. [DOI] [PubMed] [Google Scholar]
- Müller Somjen GG. Na+ and K+ concentrations, extra- and intracellular voltages, and the effect of TTX in hypoxic rat hippocampal slices. Journal of Neurophysiology. 83:735–745. doi: 10.1152/jn.2000.83.2.735. [DOI] [PubMed] [Google Scholar]
- Romijn HJ, Hofman MA, Gramsbergen A. At what age is the developing cerebral cortex of the rat comparable to that of the full-term newborn human baby? Early Human Development. 1991;26:61–67. doi: 10.1016/0378-3782(91)90044-4. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Gahwiler, Thompson SM. Presynaptic inhibition of excitatory synaptic transmission by muscarinic and metabotropic glutamate receptor activation in the hippocampus: are Ca2+ channels involved? Neuropharmacology. 1995;34:1549–1557. doi: 10.1016/0028-3908(95)00119-q. [DOI] [PubMed] [Google Scholar]
- SickSolowRoberts ELJ. Extracellular potassium ion activity and electrophysiology in the hippocampal slice: paradoxical recovery of synaptic transmission during anoxia. Brain Research. 1987;418:227–234. doi: 10.1016/0006-8993(87)90090-4. [DOI] [PubMed] [Google Scholar]
- Somjen GG, Aitken PG, Balestrino M, Crain BJ, Czeh G, Kawasaki K, Nadler JV, Neill KH, Urban L. Extracellular ions, hypoxic irreversible loss of function and delayed postischemic neuron degeneration studied in vitro. Acta Physiologica Scandinavica. 1989;(suppl. 582):58. [PubMed] [Google Scholar]
- Tanaka E, Yamamoto S, Kudo Y, Mihara S, Higashi H. Mechanisms underlying the rapid depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. Journal of Neurophysiology. 1997;78:891–902. doi: 10.1152/jn.1997.78.2.891. [DOI] [PubMed] [Google Scholar]
- Whittington MA, Traub RD, Jefferys JG. Synchronized oscillations in interneuron networks driven by metabotropic glutamate receptor activation. Nature. 1995;373:612–615. doi: 10.1038/373612a0. [DOI] [PubMed] [Google Scholar]
- Whittington MA, Traub RD, Kopell N, Ermentrout B, Buhl EH. Inhibition-based rhythms: experimental and mathematical observations on network dynamics. International Journal of Psychophysiology. 2000;38:315–336. doi: 10.1016/s0167-8760(00)00173-2. [DOI] [PubMed] [Google Scholar]
- Xie Y, Zacharias E, Hoff P, Tegtmeier F. Ion channel involvement in anoxic depolarization induced by cardiac arrest in rat brain. Journal of Cerebral Blood Flow and Metabolism. 1995;15:587–594. doi: 10.1038/jcbfm.1995.72. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Tanaka E, Higashi H. Mediation by intracellular calcium-dependent signals of hypoxic hyperpolarization in rat hippocampal CA1 neurons in vitro. Journal of Neurophysiology. 1997;77:386–392. doi: 10.1152/jn.1997.77.1.386. [DOI] [PubMed] [Google Scholar]
- Zeng YC, Domenici MR, Frank C, Sagratella S, Scotti De Carolis A. Effects of adenosinergic drugs on hypoxia-induced electrophysiological changes in rat hippocampal slices. Life Sciences. 1992;51:1073–1082. doi: 10.1016/0024-3205(92)90507-l. [DOI] [PubMed] [Google Scholar]
- Zhu PJ, Krnjevic K. Anoxia selectively depresses excitatory synaptic transmission in hippocampal slices. Neuroscience Letters. 1994;166:27–30. doi: 10.1016/0304-3940(94)90832-x. [DOI] [PubMed] [Google Scholar]
- Zhu PJ, Krnjevic K. Persistent block of CA1 synaptic function by prolonged hypoxia. Neuroscience. 1999;90:759–770. doi: 10.1016/s0306-4522(98)00495-3. [DOI] [PubMed] [Google Scholar]