

Abstract
Gradients of ion channels across the left ventricular (LV) wall have been well characterized and it has been shown that disruption of such gradients leads to altered rates of repolarization across the wall, which is associated with the generation of arrhythmias.
We have hypothesized that a transmural gradient of INa is present and have directly measured this current in adult rat myocytes isolated from both the epicardial and endocardial layers of the left ventricle. Currents were also recorded in right ventricular (RV) myocytes for comparison.
Peak inward INa currents, at −30 mV, were −49.7 ± 2.5 pA pF−1 (n = 22), −32.9 ± 3.2 pA pF−1 (n = 16) and −49.7 ± 3.7 pA pF−1 (n = 24) for RV, LV epicardial and LV endocardial myocytes, respectively. No differences in the voltage dependence of inactivation, the voltage dependence of steady-state inactivation, or reactivation were reported.
Our results demonstrate that a gradient of sodium current density is present across the LV wall of adult rat hearts.
Gradients of ion channels across the LV wall are known to play key roles in normal cardiac function (Antzelevitch et al. 1999) as well as having the potential to generate arrhythmias (Kimura et al. 1990; Geller et al. 1993; Wolk et al. 1999; Yu et al. 1999). It has been well established in several species that the Ca2+-independent transient outward K+ current (Ito) is greater in the LV sub-epicardial layer compared to the endocardium (Litovsky & Antzelevitch, 1988, 1989; Liu et al. 1993). Additionally, delayed rectifier K+ currents (IK) are also reported to be heterogeneous with significantly less current in myocytes of the mid-myocardium (Liu et al. 1993). Together, these differences combine to establish a heterogeneous pattern of repolarization across the LV wall forming the basis for an upright T-wave (Shimizu & Antzelevitch, 1997). Several disease states such as myocardial ischaemia and reperfusion as well as long-QT syndromes (LQTS) are known to disrupt the relative rates of repolarization, thus increasing the incidence of life-threatening arrhythmias (Vincent, 2000).
Although there are several K+ channel mutations associated with LQTS, novel long-QT mutations (LQT3) in cardiac sodium channels have also been identified (Kambouris et al. 1998). Such mutations induce persistent inward sodium current (INa,P) and delay repolarization (Kambouris et al. 1998). Interestingly, recent indirect evidence suggests that a transmural gradient of INa may exist (Sicouri & Antzelevitch, 1995; Sicouri et al. 1996; Cook et al. 1997; Sakmann et al. 2000; Zygmunt et al. 2001), providing further insight into an additional means by which arrhythmias may be initiated. Such a gradient may also be of importance to ischaemia-reperfusion injury as we have previously shown that hydrogen peroxide, endogenously generated during reperfusion, selectively alters the rate of INa inactivation (similar to known sodium channel gene mutations underlying LQT3), dramatically prolonging action potential duration (Ward & Giles, 1997). Together, these studies indicate that in addition to K+ currents, INa may also contribute to heterogeneous rates of repolarization across the LV wall. In the present study, we characterized a transmural gradient of INa in myocytes isolated from the epicardial and endocardial regions of adult rat left ventricle.
METHODS
Cell isolation
LV epicardial and endocardial as well as RV myocytes were isolated from adult Sprague-Dawley rats (200–225 g) using the methods of Ward & Giles (1997) with modifications as indicated. Rats were decapitated (under sodium pentobarbital anaesthesia; 50 mg kg−1i.p.) using protocols approved by the Queen's University Animal Care Committee in accordance with Canadian Council on Animal Care guidelines. For isolation of epicardial and endocardial cell populations the LV free wall was removed following enzymatic digestion and, by fine dissection, either the extreme endocardial or epicardial layer was removed and minced in 10 ml of Tyrode solution (mm: NaCl 140; KCl 5.4; Na2HPO4 1; Hepes 5; glucose 10; MgCl2 1; pH adjusted to 7.4 with 1 m NaOH and continuously gassed with 100 % O2) containing collagenase (0.5 mg ml−1; Yakult, Japan), protease (0.1 mg ml−1; Sigma), bovine serum albumin (3 mg ml−1, Sigma) and CaCl2 (50 μm). The tissue was gently agitated in a shaking water bath for 10–30 min at 36 °C until single dissociated cells were observed. Once myocytes were present, aliquots were removed at 5 min intervals over 30 min and stored in 4 ml of modified KB solution containing (mm): potassium glutamate 100; potassium aspartate 10; KCl 25; glucose 20; KH2PO4 10; Hepes 5; MgSO4 2; taurine 20; creatine 5; and EGTA 0.5; with 0.1 % BSA, pH adjusted to 7.2 with 1 m KOH. RV myocyte cell suspensions were considered to be a homogeneous cell population as we have been unable to demonstrate differences in the ionic currents between the epicardial and endocardial layers of this tissue.
Electrophysiological methods
Pipette resistances ranged from 1.5 to 2.5 MΩ when filled with internal solution. Junction potentials were corrected for. Whole-cell currents and action potentials were recorded with an Axopatch-1D amplifier (Axon Instruments) using pCLAMP 8.0 data acquisition software (Axon Instruments). Membrane potential and whole-cell currents were filtered at 2 kHz and sampled between 5 and 20 kHz depending on the experiment type.
Action potential waveforms were recorded using a standard internal pipette solution containing (mm): KCl 20; potassium aspartate 110; EGTA 10; Hepes 10; MgCl2 1; K2ATP 5; CaCl2 1; NaCl 10; pH adjusted to 7.2 using 1 m KOH. Tyrode solution with 1 mm CaCl2 added was used for superfusion. Action potentials were elicited by 5 ms, 800 pA current injection at a stimulation frequency of 1 Hz.
Ito was recorded using solutions identical to those for action potential recordings. From a holding potential of −80 mV, whole-cell currents were elicited by 500 ms square-wave voltage steps to command potentials between −120 and +60 mV, in 10 mV increments, at a rate of 0.5 Hz. This protocol was repeated with a 100 ms prepulse to −40 mV immediately prior to the test potential to voltage-inactivate Ito. The currents derived by subtracting these protocols represent the isolated Ito.
Whole-cell INa was recorded using a pipette solution containing (mm): CsF 120; Hepes 10; MgCl2 2; EGTA 10; CaCl2 1; NaCl 10; pH adjusted to 7.4 with 1 m KOH. To further isolate INa the superfusion solution contained (mm): NaCl 10; Hepes 10; KCl 5; MgCl2 1; CaCl2 1; CoCl2 5; CsCl 5; glucose 10; choline chloride 110; pH adjusted to 7.4 with 1 m NaOH. To ensure adequate voltage control a minimum of 80 % series resistance compensation was applied. INa was elicited using 50 ms rectangular voltage steps from a holding potential of −90 mV to command potentials ranging from −100 to 20 mV in 5 mV steps applied at a frequency of 1 Hz.
For some experiments the steady-state voltage dependence of INa was examined by applying a 10 s conditioning step to membrane potentials between −140 and −20 mV, in 5 mV increments, followed by a test pulse to −30 mV to activate INa. The time dependence of INa reactivation was also examined using a typical two-pulse protocol. The conditioning pulse (P1) consisted of a 20 ms square-wave depolarization to −30 mV from a holding potential of −100 mV. The test pulse (P2) was identical and was applied between 3 and 150 ms, in 3 ms increments, following the conditioning pulse. Reactivation data were expressed as the ratio of P2/P1 and fitted to the Boltzmann equation.
Whole-cell currents were statistically analysed using Student's unpaired t test or analysis of variance where appropriate. Differences were considered statistically significant when P < 0.05.
RESULTS
To demonstrate the successful isolation of LV epicardial and endocardial myocytes, Ito was measured in the two cell populations, since it is well documented that Ito is significantly greater in LV epicardial myocytes (Litovsky & Antzelevitch, 1988, 1989; Liu et al. 1993). At +50 mV we report peak Ito as 24.7 ± 3.1 pA pF−1 (n = 10) and 2.8 ± 0.8 pA pF−1 (n = 10) for epicardial (Fig. 1D) and endocardial (Fig. 1F) myocytes, respectively. Ito was also recorded from RV cells (Fig. 1B) for comparison and was found to be 32.3 ± 4.6 pA pF−1 (n = 8). Representative action potential waveforms of LV epicardial, LV endocardial and RV myocytes are also shown in Fig. 1. As expected, LV epicardial and RV-derived myocytes characteristically had shorter action potential durations compared to LV endocardial myocytes. At 90 % repolarization these values were 20 ± 2 ms (n = 7), 22 ± 5 ms (n = 5) and 73 ± 12 ms (n = 8), respectively.
Figure 1. Representative action potential waveforms and Ca2+-independent transient outward K+ current (Ito) traces in cardiac myocytes.

Action potentials from RV, LV epicardial and LV endocardial myocytes were elicited as outlined in Methods and are shown in A, C and E, respectively. Representative Ito traces for RV (B), LV epicardial (D) and LV endocardial myocytes (F) were recorded at command potentials of −40, 0 and +40 mV. Currents were normalized for cell capacitance.
INa was measured using voltage clamp to determine whether there are differences in INa across the LV wall and compared to RV myocytes. RV peak inward (at −30 mV) INa density was −49.7 ± 2.5 pA pF−1 (n = 22; Fig. 2A). A similar INa density of −49.7 ± 3.7 pA pF−1 (n = 24; Fig. 2B) was recorded in LV endocardial myocytes. In contrast, INa was significantly lower in LV epicardial myocytes compared to endocardial myocytes, with a peak inward current of −32.9 ± 3.2 pA pF−1 at −30 mV (n = 16, P < 0.05; Fig. 2C). INa current-voltage relationships for all three cell populations are illustrated in Fig. 2D. The voltage dependence of the inactivation kinetics was assessed by fitting the data to a single exponential equation and is illustrated in Fig. 3. No differences in calculated time constant (tau) values were found between the three populations of myocytes over the voltage ranges examined.
Figure 2. INa densities recorded from myocardial cell populations.
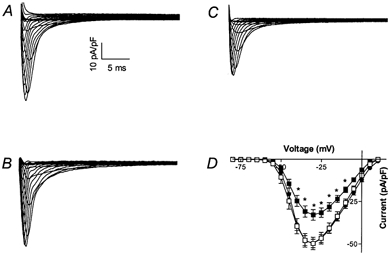
Representative families of RV (A), LV endocardial (B) and LV epicardial (C) sodium currents were elicited using 50 ms rectangular voltage steps from a holding potential of −90 mV to command potentials ranging from −100 to +20 mV in 5 mV steps. D, current-voltage relationship of INa density recorded from LV epicardial (▪; n = 16), LV endocardial (□; n = 22) and RV (•; n = 24) myocytes. Values are all normalized to cell capacitance and expressed as means ±s.e.m.* Significantly different (P < 0.05) compared to endocardial myocytes at the same membrane potential.
Figure 3. Voltage dependence of inactivation kinetics for RV (•), LV epicardial (▪) and LV endocardial (□) myocytes.

Tau values were calculated by fitting INa traces to a single exponential decay equation and are plotted against command potential. Data points represent the mean ±s.e.m. for n = 10 myocytes.
Steady-state inactivation was assessed to confirm that the LV transmural differences in current density were not due to an altered voltage dependence of inactivation (Fig. 4A). For each individual myocyte, steady-state voltage-dependent inactivation data were fitted to the Boltzmann equation to determine the membrane potential resulting in 50 % inactivation (V11/2sol;2). LV endocardial and epicardial cells revealed V11/2sol;2 values of −68 ± 1 mV (n = 10) and −68 ± 1 mV (n = 7), respectively, indicating that the voltage dependence of inactivation of these channels was identical. A V11/2sol;2 value of −70 ± 1 mV (n = 15) was determined for RV myocytes.
Figure 4. Voltage dependence of sodium current steady-state inactivation and recovery from fast inactivation.
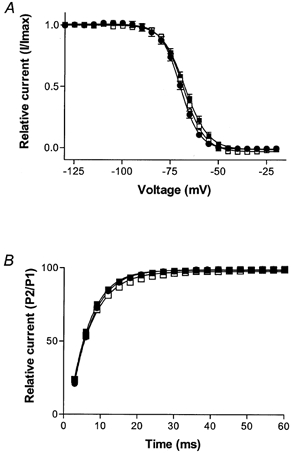
A, plot of steady-state inactivation of INa is recorded as voltage versus relative peak current from LV epicardial (▪; n = 7), LV endocardial (□; n = 10) and RV (•; n = 15) myocytes. Data were normalized to peak inward current and expressed as the mean ±s.e.m.B, reactivation data were recorded using a paired-pulse protocol and expressed as the P2/P1 ratio for LV epicardial (▪; n = 13), LV endocardial (□; n = 11) and RV (•; n = 12) myocytes (means ±s.e.m.).
The time course of recovery from fast inactivation was assessed to demonstrate that this parameter was similar for each of the three cell populations (Fig. 4B). P2/P1 data were acquired for individual myocytes at incrementing time intervals and were fitted to the Boltzmann equation to determine the time at which 50 % of INa had recovered from fast inactivation (RT50). LV endocardial and epicardial myocytes had derived RT50 values of 6.0 ± 0.3 ms (n = 8) and 5.8 ± 0.2 ms (n = 9), respectively. For RV myocytes, this value was 6.2 ± 0.2 ms (n = 9). Statistical analysis determined that these data were not significantly different from each other.
DISCUSSION
In the present study we have demonstrated that Na+ current density is significantly lower in LV epicardial myocytes compared to those isolated from either the LV endocardium or RV and that the activation, inactivation and re-activation kinetics of these currents are similar for the three cell populations. These findings are of significance as they identify a novel LV transmural current gradient and suggest that this gradient is not attributed to differential distribution of Na+ channel isoforms.
While there have been no reports on a transmural INa gradient, previous studies have examined the possibility of a transmural gradient of INa,P. Sakmann et al. (2000) have reported that although INa,P is detectable in guinea-pig LV epicardial and endocardial layers there are no differences in current density. Upon closer examination of the macroscopic recordings our results are in agreement in that we also did not detect any difference of INa,P across the ventricular wall. However, this may be attributed to the fact that we did not observe INa,P in any of our recordings, suggesting either that this current is not present in rat ventricular myocytes or that differences in recording conditions have masked this current. The former is unlikely since we have previously demonstrated that in RV myocytes INa,P can be enhanced/activated by exposing the myocyte to the reactive oxygen species hydrogen peroxide, yet this current was not present under control conditions (Ward & Giles, 1997). Clearly, additional studies are required to further explore these discrepancies.
The presence of a INa gradient is of potential importance to several disease states. Specifically, LQT3 is known to be associated with mutations in the SCN5A gene encoding INa (Bennett et al. 1995; Wang et al. 1996; Abriel et al. 2000). LQT3 mutations prolong the QT interval by inducing a small persistent inward current caused by the reopening of Na+ channels (Bennett et al. 1995; Dumaine et al. 1996). Our previous studies have reported similar single channel events relevant to ischaemic heart disease. We have shown that hydrogen peroxide, a reactive oxygen species known to be produced during ischaemia and subsequent reperfusion, also induces a small persistent inward current that we now attribute to INa,P (Ward & Giles, 1997). What remains unknown is whether INa,P is a distinct ionic current or whether it is a modified conductance state of INa. The latter is more likely as it has been shown that phosphorylation of the inactivation gate of INa results in a INa,P-like current (Qu et al. 1996; Franceschetti et al. 2000) and several drugs and toxins such as BDF9148 and anthopleura toxin also modulate the inactivation kinetics of INa to induce INa,P (Ward & Giles, 1997; Yuill et al. 2000).
Modulation of repolarization across the LV wall is well described to be an initiator of arrhythmic events (Antzelevitch et al. 1999). The direct evidence presented in this study of a transmural difference of INa density between LV epicardial and endocardial myocytes suggests that INa is also important with respect to arrhythmia initiation. However, additional studies are required to further reveal the intricate relationship between INa and INa,P and the role of these currents as modulators of repolarization across the LV wall.
Acknowledgments
This research was supported by an operating grant from the Heart and Stroke Foundation of Ontario (no. NA4173) to C.A.W. and equipment grants from the J. P. Bickell Foundation, Canadian Foundation for Innovation and Ontario Innovation Trust. C.A.W. is a Research Scholar of the Heart and Stroke Foundation of Canada.
References
- Abriel H, Wehrens XH, Benhorin J, Kerem B, Kass RS. Molecular pharmacology of the sodium channel mutation D1790G linked to the long-QT syndrome. Circulation. 2000;102:921–925. doi: 10.1161/01.cir.102.8.921. [DOI] [PubMed] [Google Scholar]
- Antzelevitch C, Yan GX, Shimizu W. Transmural dispersion of repolarization and arrhythmogenicity: the Brugada syndrome versus the long QT syndrome. Journal of Electrocardiology. 1999;32(suppl.):158–165. doi: 10.1016/s0022-0736(99)90074-2. [DOI] [PubMed] [Google Scholar]
- Bennett PB, Yazawa K, Makita N, George ALJ. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- Cook SJ, Chamunorwa JP, Lancaster MK, O'Neill SC. Regional differences in the regulation of intracellular sodium and in action potential configuration in rabbit left ventricle. Pflügers Archiv. 1997;433:515–522. doi: 10.1007/s004240050307. [DOI] [PubMed] [Google Scholar]
- Dumaine R, Wang Q, Keating MT, Hartmann HA, Schwartz PJ, Brown AM, Kirsch GE. Multiple mechanisms of Na+ channel-linked long-QT syndrome. Circulation Research. 1996;78:916–924. doi: 10.1161/01.res.78.5.916. [DOI] [PubMed] [Google Scholar]
- Franceschetti S, Taverna S, Sancini G, Panzica F, Lombardi R, Avanzini G. Protein kinase C-dependent modulation of Na+ currents increases the excitability of rat neocortical pyramidal neurons. Journal of Physiology. 2000;528:291–304. doi: 10.1111/j.1469-7793.2000.00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller JC, Rosen MR. Persistent T-wave changes after alteration of the ventricular activation sequence. New insights into cellular mechanisms of ‘cardiac memory’. Circulation. 1993;88:1811–1819. doi: 10.1161/01.cir.88.4.1811. [DOI] [PubMed] [Google Scholar]
- Kambouris NG, Nuss HB, Johns DC, Tomaselli GF, Marban E, Balser JR. Phenotypic characterization of a novel long-QT syndrome mutation (R1623Q) in the cardiac sodium channel. Circulation. 1998;97:640–644. doi: 10.1161/01.cir.97.7.640. [DOI] [PubMed] [Google Scholar]
- Kimura S, Bassett AL, Furukawa T, Cuevas J, Myerburg RJ. Electrophysiological properties and responses to simulated ischemia in cat ventricular myocytes of endocardial and epicardial origin. Circulation Research. 1990;66:469–477. doi: 10.1161/01.res.66.2.469. [DOI] [PubMed] [Google Scholar]
- Litovsky SH, Antzelevitch C. Rate dependence of action potential duration and refractoriness in canine ventricular endocardium differs from that of epicardium: role of the transient outward current. Journal of the American College of Cardiology. 1989;14:1053–1066. doi: 10.1016/0735-1097(89)90490-7. [DOI] [PubMed] [Google Scholar]
- Litovsky SH, Antzelevitch C. Transient outward current prominent in canine ventricular epicardium but not endocardium. Circulation Research. 1998;62:116–126. doi: 10.1161/01.res.62.1.116. [DOI] [PubMed] [Google Scholar]
- Liu DW, Gintant GA, Antzelevitch C. Ionic bases for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circulation Research. 1993;72:671–687. doi: 10.1161/01.res.72.3.671. [DOI] [PubMed] [Google Scholar]
- Qu Y, Rogers JC, Tanada TN, Catterall WA, Scheuer T. Phosphorylation of S1505 in the cardiac Na+ channel inactivation gate is required for modulation by protein kinase C. Journal of General Physiology. 1996;108:375–379. doi: 10.1085/jgp.108.5.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakmann FASS, Spindler AJ, Bryant SM, Linz KW, Noble D. Distribution of a persistent sodium current across the ventricular wall of guinea pigs. Circulation Research. 2000;87:910–914. doi: 10.1161/01.res.87.10.910. [DOI] [PubMed] [Google Scholar]
- Shimizu W, Antzelevitch C. Sodium current block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade de pointe in LQT2 and LQT3 models of the long AT syndrome. Circulation. 1997;96:2038–2047. doi: 10.1161/01.cir.96.6.2038. [DOI] [PubMed] [Google Scholar]
- Sicouri S, Antzelevitch C. Electrophysiologic characteristics of M cells in the canine left ventricular free wall. Journal of Cardiovascular Electrophysiology. 1995;6:591–603. doi: 10.1111/j.1540-8167.1995.tb00435.x. [DOI] [PubMed] [Google Scholar]
- Sicouri S, Quist M, Antzelevitch C. Evidence for the presence of M cells in the guinea pig ventricle. Journal of Cardiovascular Electrophysiology. 1996;7:503–511. doi: 10.1111/j.1540-8167.1996.tb00557.x. [DOI] [PubMed] [Google Scholar]
- Vincent GM. Long QT syndrome. Cardiology Clinics. 2000;18:309–325. doi: 10.1016/s0733-8651(05)70143-0. [DOI] [PubMed] [Google Scholar]
- Wang DW, Yazawa K, George ALJ, Bennett PB. Characterization of human cardiac Na+ channel mutations in the congenital long QT syndrome. Proceedings of the National Academy of Sciences of the USA. 1996;93:13200–13205. doi: 10.1073/pnas.93.23.13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CA, Giles WR. Ionic mechanism of the effects of hydrogen peroxide in rat ventricular myocytes. Journal of Physiology. 1997;500:631–642. doi: 10.1113/jphysiol.1997.sp022048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolk R, Cobbe SM, Hicks MN, Kane KA. Functional, structural, and dynamic basis of electrical heterogeneity in healthy and diseased cardiac muscle: implications for arrhythmogenesis and anti-arrhythmic drug therapy. Pharmacology and Therapeutics. 1999;84:207–231. doi: 10.1016/s0163-7258(99)00033-9. [DOI] [PubMed] [Google Scholar]
- Yu H, McKinnon D, Dixon JE, Gao J, Wymore R, Cohen IS, Danilo PJ, Shvilkin A, Anyukhovsky EP, Sosunov EA, Hara M, Rosen MR. Transient outward current, Ito1, is altered in cardiac memory. Circulation. 1999;99:1898–1905. doi: 10.1161/01.cir.99.14.1898. [DOI] [PubMed] [Google Scholar]
- Yuill KH, Convery MK, Dooley PC, Doggrell SA, Hancox JC. Effects of BDF9198 on action potentials and ionic currents from guinea-pig isolated ventricular myocytes. British Journal of Pharmacology. 2000;130:1753–1766. doi: 10.1038/sj.bjp.0703476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt AC, Eddlestone GT, Thomas GP, Nesterenko VV, Antzelevitch C. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. American Journal of Physiology. 2001;281:H689–697. doi: 10.1152/ajpheart.2001.281.2.H689. [DOI] [PubMed] [Google Scholar]