

Abstract
We investigated the effect of moderate systemic hypoxia on the arterial, venous and interstital concentration of adenosine and adenine nucleotides in the neurally and vascularly isolated, constant-flow perfused gracilis muscles of anaesthetized dogs.
Systemic hypoxia reduced arterial PO2 from 129 to 28 mmHg, venous PO2 from 63 to 23 mmHg, arterial pH from 7.43 to 7.36 and venous pH from 7.38 to 7.32. Neither arterial nor venous PCO2 were changed. Arterial perfusion pressure remained at 109 ± 8 mmHg for the first 5 min of hypoxia, then increased to 131 ± 11 mmHg by 9 min, and then decreased again throughout the rest of the hypoxic period.
Arterial adenosine (427 ± 98 nm) did not change during hypoxia, but venous adenosine increased from 350 ± 52 to 518 ± 107 nm. Interstitial adenosine concentration did not increase (339 ± 154 nm in normoxia and 262 ± 97 nm in hypoxia). Neither arterial nor venous nor interstitial concentrations of adenine nucleotides changed significantly in hypoxia.
Interstitial adenosine, AMP, ADP and ATP increased from 194 ± 40, 351 ± 19, 52 ± 7 and 113 ± 36 to 764 ± 140, 793 ± 119, 403 ± 67 and 574 ± 122 nm, respectively, during 2 Hz muscle contractions.
Adenosine, AMP, ADP and ATP infused into the arterial blood did not elevate the interstitial concentration until the arterial concentration exceeded 10 μm.
We conclude that the increased adenosine in skeletal muscle during systemic hypoxia is formed by the vascular tissue or the blood cells, and that adenosine is formed intracellularly by these tissues. On the other hand, adenosine formation takes place extracellularly in the interstitial space during muscle contractions.
Acute systemic hypoxia increases adenosine output from skeletal muscle (Mo & Ballard, 1997). In rats, the adenosine released in hypoxia is suggested to act mainly on A1 receptors in the skeletal muscle (Bryan & Marshall, 1999a), through a largely NO-dependent mechanism (Bryan & Marshall, 1999b), to induce vasodilatation. It is suggested that adenosine's role is to alleviate the vasoconstrictor effects of several neuronal and hormonal factors released at the onset of systemic hypoxia, thus limiting the initial increase in arterial blood pressure, and to maintain the blood supply to the skeletal muscle in the face of a decreased arterial pressure in sustained hypoxia (Marshall, 1995). Indeed, it is thought that the skeletal muscle vasodilatation, to which adenosine makes a major contribution, is largely responsible for the net decrease in arterial blood pressure that occurs in the latter part of sustained hypoxia.
We have previously reported that the adenosine release in systemic hypoxia results from intracellular pH depression, caused by cellular uptake of the increased levels of circulating lactate (Mo & Ballard, 1997). However, little is known about the cellular origins of the adenosine or its distribution within the muscle: skeletal muscle, vascular smooth muscle and endothelial cells are all capable of producing adenosine, if suitably stimulated (Camici et al. 1985; Belloni et al. 1986; Shryock et al. 1988), and it is very well known that red blood cells and platelets can release adenosine precursors such as ADP or ATP. There have been many previous measurements of tissue or plasma adenosine concentrations (Dobson et al. 1971; Steffen et al. 1983; Ballard et al. 1987), but these cannot distinguish whether the adenosine is restricted to the blood and vascular tissues, or distributed throughout the interstitial space. However, there is a growing body of indirect evidence (Marshall, 2000) which suggests that the adenosine in systemic hypoxia may be released from the vascular endothelium. Based on reports that adenosine's actions in hypoxia can be blocked by the A1-selective inhibitor, DPCPX (Bryan & Marshall, 1999a), but adenosine acts on A2a receptors during muscle contractions (Poucher, 1996), it is suggested that the endothelial adenosine receptors (Balcells et al. 1992) are of the A1 type (Marshall, 2000). If this is so, then adenosine may remain entirely in the vascular compartment during hypoxia. This contrasts with the situation in muscle contractions, where the interstitial adenosine concentration is raised (Hellsten et al. 1998), and the skeletal muscle or interstitial cells are suggested to be the principal source of the adenosine. In the present study, we directly determined the concentrations of adenosine in the interstitial space (using microdialysis) and in the blood in order to distinguish in which compartment(s) the adenosine originated. Adenosine that originates from skeletal muscle cells must necessarily increase the interstitial concentration in order for it to diffuse into the blood, whereas adenosine arising from vascular tissues may not elevate the interstitial concentration (Costa et al. 1999).
There is also uncertainty as to whether adenosine itself or one of its precursors is released in sytemic hypoxia. Studies on a skeletal muscle homogenate in our own laboratory (Cheng et al. 2000) have shown that the activity of cytosolic 5′-nucleotidase is much lower than that of ecto-5′-nucleotidase, and that most adenosine is formed extracellularly. There are a number of possible sources for adenine nucleotide release in systemic hypoxia. It has long been known that sympathetic nerves co-release ATP, and sympathetic vasoconstrictor tone on the skeletal muscles is increased by peripheral chemoreceptor stimulation (Marshall, 1994). However, elevation of the blood pressure in the early part of the hypoxic response would presumably be detected by baroreceptors and lead to some withdrawal of sympathetic tone to the skeletal muscles, and so, the net effect on sympathetic vasoconstrictor tone remains uncertain. On the other hand, the intracellular pH depression resulting from lactate uptake in systemic hypoxia would stimulate Na+-H+ exchange, which is responsible for about 80 % of the pH correction in skeletal muscle after an acid load (Aickin & Thomas, 1977). It has been proposed that the resulting increase in intracellular Na+ stimulates the Na+-K+ pump, which consumes more ATP, and might lead to an increase in the release of its breakdown products (Phillis et al. 1999).
Therefore, the purpose of this study was to determine in which compartment adenosine originated, by measuring the interstitial, arterial and venous concentrations of adenosine before, during and after a period of acute systemic hypoxia. At the same time, its precursors, AMP, ADP and ATP, were also determined. The studies were performed in a denervated preparation, in order to separate the direct responses of the muscle and its vasculature from those arising indirectly, as a reflex response to chemoreceptor or baroreceptor stimulation. In some experiments, interstitial adenosine and adenine nucleotide concentrations were determined during muscle contractions in order to confirm that adenosine does appear in the interstitial space in this preparation in that condition. Finally, adenosine and adenine nucleotides were infused, in order to obtain a measure of the extent to which these substances can pass between the vascular and interstitial compartments.
METHODS
Animal preparation
All experimental protocols were approved by the University of Hong Kong Committee on the Use of Live Animals in Teaching and Research. Twenty-four mongrel dogs, weighing 12–24 kg, were used. The dogs were anaesthetized by an injection of ketamine and xylazine (5 and 2 mg kg−1, respectively) to the longissimus lumborum muscle, followed by intravenous infusion of sodium pentobarbital (8 mg kg−1). The anaesthesia was subsequently maintained by intravenous infusion of sodium pentobarbital (average dose 4.0 mg kg−1 h−1). Bicarbonate (28 mm) in 0.9 % saline was infused as required (usually 1 ml min−1) as the vehicle for drug delivery and to replace any volume losses caused by evaporation or surgical procedures.
The trachea was cannulated. Positive pressure ventilation with a mixture of room air and oxygen was started at a stroke rate of 17 min−1, and an initial stroke volume of 17 ml kg−1. Arterial PO2 was determined using a Strathkelvin Instruments model 781 oxygen meter (S. I., Glasgow, UK), and PCO2 using an Orion model 9502BN carbon dioxide electrode attached to an Orion model 710A meter (Orion Research Inc., Beverley, MA, USA). The stroke volume and/or the content of oxygen in the inspired air were adjusted to keep arterial PO2 at 129 ± 5 mmHg (mean ±s.e.m.., n = 18) and PCO2 at 29 ± 2 mmHg. The arterial pH was measured using an AgCl pH electrode (model 5993-11, Cole-Parmer, Vernon Hills, IL, USA) attached to an Orion model EA 920 ion analyser, and maintained at 7.43 ± 0.02 by infusion of sodium bicarbonate (50–300 mm), if required. Oesophageal temperature was kept at 36.9 ± 0.2 °C by heating coils under the operating table.
The gracilis muscle was vascularly isolated as previously described (Ballard, 1991). One hour after completion of the surgery, the dog was given heparin sodium (500 i.u. kg−1, followed by 100 i.u. kg−1 h−1) and connected to an extracorporeal perfusion circuit as previously described (Mo & Ballard, 1997). In brief, the gracilis muscle was perfused with blood from the contralateral femoral artery. Venous blood was returned to the dog via the jugular vein. Two pressure transducers (model P23XL, Gould) were connected to the perfusion circuit and the carotid artery to measure the perfusion and systemic arterial pressures, respectively. Signals were amplified using Gould 13-4615-50 transducer amplifiers, and recorded on a Gould RS 3400 recorder. The brachial artery was connected to a pressure reservoir to control the arterial pressure. The femoral, sciatic and obturator nerves were sectioned. We let the muscle blood flow stabilize for 30 min, then measured the free flow, and set the constant flow to about 150 % of the free flow.
Analytical procedures
Plasma extraction
Plasma (600 μl) mixed with 6 μl (5 × 10−5m) methyl-adenosine, the internal standard, was extracted with 75 μl perchloric acid (1.5 m) and 1 mm EDTA. The supernatant (500 μl) was withdrawn after refrigerated centrifugation for 5 min at 10 000 r.p.m. (model 16R, Hettich Zentrifugen, Tuttlingen, Germany) and neutralized with 29 μl 4 m KOH. After ice cooling for 10 min, the sample was again centrifuged at 10 000 r.p.m. and 4 °C for 5 min. The pH of the supernatant was finally adjusted to 6–7 prior to high performance liquid chromatography (HPLC).
HPLC
Adenosine, AMP, ADP, ATP and methyl-adenosine were determined by ion-pair reverse-phase HPLC, using a model 600 chromatography workstation (Bio-Rad, Hercules, CA, USA). Samples were chromatographed on a 25 cm × 4.6 mm column, packed with 3 μm particles of LC18-T (Supelco, Bellefonte, PA, USA). The mobile phase consisted of solution A (0.1 m KH2PO4 and 0.004 m tetrabutyl-ammonium chloride, pH 6) and solution B (20 % methanol in solution A, pH 5.5). AMP, adenosine, methyl-adenosine and ADP were eluted with a solvent B gradient from 3 to 40 %, run from 3 to 28 min, and the peak of ATP appeared in the isocratic phase of 45 % solvent B, run from 32 to 45 min. The solvent flow rate was 1 ml min−1. Peaks were detected by their UV absorbance at 254 nm and 280 nm (UV2000, Thermo Separation Products, Riviera Beach, FL, USA). The peaks of adenosine, AMP, methyl-adenosine, ADP and ATP in the HPLC chromatogram were identified by comparison of their retention times to the retention time of the standard and the ‘spiked’ sample, in which a certain ratio of standard and plasma sample were mixed. The mean retention times (n = 144) of AMP, adenosine, methyl-adenosine, ADP and ATP were 9.61 ± 0.04, 11.00 ± 0.06, 19.75 ± 0.12, 23.62 ± 0.13 and 38.49 ± 0.15 min, respectively. Therefore, the peaks of all the tested compounds were clearly separated. The sensitivities for detection of the peaks of adenosine, AMP, ADP and ATP were all about 10 nm, which was equivalent to about 30 nm in undiluted microdialysate or about 15 nm in undiluted plasma extract.
Recovery from sample processing
The recovery rate from sample processing was determined by the extraction of plasma, to which a known concentration (1, 5 or 10 μm) of a mixture of adenosine, methyl-adenosine, AMP, ADP and ATP had been added. The recovery of each test compound was calculated from the concentration remaining in the sample after processing (100 %× (final concentration after processing – endogenous concentration)/known concentration before processing). Results are shown in Table 1. The mean recovery of methyl-adenosine was 72 ± 2 %. The recoveries of adenosine and AMP were higher than that of the internal standard, and those of ADP and ATP were lower (column 1). Concentrations given in this paper have been corrected for the recovery of the internal standard (methyl adenosine), and should therefore be compared to the recovery values in column 2 of Table 1.
Table 1.
Recovery of adenosine, AMP, ADP, ATP and methyl-adenosine after sample extraction
| Recovery (%) | Adjusted recovery (%) | |
|---|---|---|
| Adenosine | 94 ± 5 | 114 ± 5 |
| AMP | 81 ± 2 | 103 ± 3 |
| Methyl-adenosine | 72 ± 2 | 100 |
| ADP | 47 ± 2 | 66 ± 3 |
| ATP | 53 ± 6 | 79 ± 8 |
The mean recoveries were calculated following extraction of a 1, 5 or 10 μm mixture of adenosine, AMP, ADP, ATP and the internal standard, methyl-adenosine, added to plasma from seven dogs (n = 66). Column 1 and column 2 are the recoveries without or with, respectively, adjustment for the recovery of internal standard.
Microdialysis
Procedures
Three microdialysis probes (LM10, Bioanalytical Systems, West Lafayette, IN, USA) were implanted through a surgical needle into a gracilis muscle in a position longitudinally aligned with the myofibrils. The probes were perfused with a solution of similar composition to the interstitial fluid (mm: NaCl 142, KCl 4, CaCl2, 24, MgSO4 0.5, NaH2PO4 1.2, glucose 5.6, pH 7.3–7.4) at a perfusion rate of 2 μl min−1. The probe was perfused for more than 90 min prior to sampling. The dialysate was collected in an ice-cooled vial for HPLC analysis.
The probe dialysate was protein free, and did not require any treatment prior to HPLC analysis. Adenosine, AMP, ADP and ATP were separated by HPLC, using 40 μl dialysate mixed with 80 μl distilled water. Recovered concentrations were corrected for the dilution factor.
Determination of microdialysis probe recovery for adenosine and adenine nucleotides
Two series of experiment were performed: in the first series of experiments, involving four dogs, a mixture of adenosine and the adenine nucleotides at known concentrations of 0, 50, 100, 500 or 1000 nm were perfused through the microdialysis probe, and their concentrations then determined in the dialysate. In the second series of experiments, involving a further five dogs, each compound (adenosine, AMP or ADP) was perfused separately to determine the recovery rate. The perfusion period was 20 min and the dialysate was collected for two successive 10 min periods. As the test compounds passed through the probe, concentrations of the substances changed according to the concentration gradient between the probe and the interstitial space; the interstitial concentrations were assumed to remain constant in steady-state conditions. Figure 1 shows the regression lines for dialysate concentration versus perfusate (probe) concentrations. In all cases, the relationship was linear, indicating that the dialysate concentration was directly proportional to the concentration gradient: the correlation factors (r2) for adenosine, AMP, ADP and ATP were 0.759, 0.758, 0.863 and 0.886, respectively. The slopes of the regression lines were equivalent to the recovery factors for the probe, which were 60, 62, 52, and 59 % for adenosine, AMP, ADP and ATP, respectively. In this paper, all concentrations have been adjusted for the probe recovery.
Figure 1. Determination of the microdialysis probe recovery for adenosine and the adenine nucleotides.
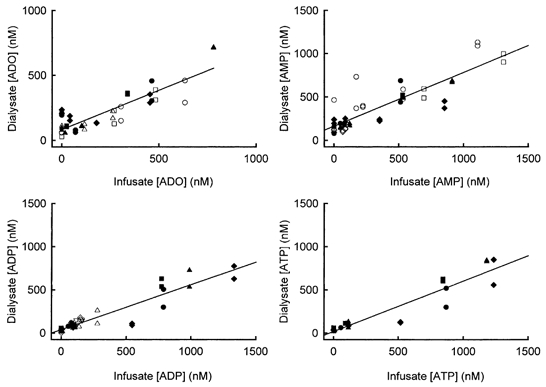
Each point represents one sample at one perfusate concentration: the filled symbols are the experiments (n = 4) in which a mixture of adenosine and adenine nucleotides was infused, and the open symbols are the experiments (n = 5) in which the compounds were infused separately. Recovery was equal to the slope of the line.
Experimental procedures
Systemic hypoxia
Two pre-control samples of microdialysate were collected for successive 10 min periods during normoxia. Blood samples were collected from the arterial and venous sections of the perfusion circuit for analysis of adenosine, adenine nucleotides, pH, PO2 and PCO2, at the beginning, and the 10th and 20th minutes of the dialysate collection. Systemic hypoxia was then induced by supplementing the inspired air with N2. The level of hypoxia was adjusted to be moderate (arterial PO2 around 30 mmHg) within the first 5 min, as confirmed by the blood gases. Blood samples were taken at this point. The microdialysate was then collected for two successive 7 min periods in hypoxia. At the end of each dialysate collection, blood samples were also withdrawn. After a total of 20 min of hypoxia, the animal was returned to ventilation with air supplemented with oxygen. Three post-controls of blood and dialysate sample, each for 10 min, were taken after the restoration of normoxia.
Infusion of adenosine and adenine nucleotides to the muscle
In order to quantify the movement of substances from the blood to the interstitial space, mixtures of adenosine, AMP, ADP and ATP, at concentrations of 1 × 10−5, 1 × 10−4 or 1 × 10−3m, were infused via the femoral artery to the gracilis muscle at an infusion rate approximately one-tenth of the plasma flow. The microdialysate was collected for two successive 10 min periods, and the arterial and venous samples were collected at the 10th and 20th minutes of the infusion.
Muscle contractions
Muscle contractions were induced by stimulation of the peripheral end of the obturator nerve using 2 Hz, 6 V and 1 ms pulses (model S48, Grass). Blood samples were collected from the muscle at rest, at the end of 10 min of contractions and at 10 min intervals throughout the 30 min post-control for pH and blood gas analysis. Microdialysis samples were collected for adenosine and adenine nucleotide analysis for 10 min each, two in the pre-control period, one throughout the muscle contractions, and three in the post-control period. Then, in two out of six experiments, after the blood gas and pressure values had returned to pre-control values, the entire pre-control, stimulation and post-control sampling was repeated, except that only two post-controls were taken in the second experiment.
When all experiments had been completed, animals were killed with an intravenous infusion of saturated potassium chloride.
Statistical analysis
Values are the means ±s.e.m..; n is the number of tests given at the beginning of each section. Student's paired t test was used for comparison between the control and treatment data.
RESULTS
Twenty-four dogs were tested. The resting blood flow to the gracilis muscles was 5.0 ± 0.9 ml min−1 (100 g)−1. The constant flow rate was set to 7.6 ± 1.1 ml min−1 (100 g)−1.
Systemic hypoxia
Systemic hypoxia was induced 13 times in 11 dogs. Since all the controls at 10 and 20 min were consistent, only the 20 min control values are shown.
Arterial and venous blood gases and pH
The arterial and venous PO2, PCO2 and pH are shown in Table 2. The arterial PO2 in normoxia was set between 100 and 150 mmHg, and moderate hypoxia was induced by reducing it to about 30 mmHg (P < 0.001 for all hypoxic PO2). Systemic hypoxia did not alter the arterial or venous PCO2. The arterial and venous pH did not significantly change at 5 or 12 min of hypoxia. However, at the end of hypoxia, arterial and venous pH significantly decreased (P < 0.05) and had not recovered after 30 min of post-control.
Table 2.
Arterial and venous pH and blood gases before, during and after systemic hypoxia
| pH | PO2 | PCO2 | ||||
|---|---|---|---|---|---|---|
| Arterial | Venous | Arterial | Venous | Arterial | Venous | |
| Pre-control | 7.43 ± 0.02 | 7.38 ± 0.03 | 129 ± 7 | 63 ± 6 | 29 ± 2 | 34 ± 2 |
| Hypoxia | ||||||
| 5 min | 7.46 ± 0.03 | 7.38 ± 0.03 | 30 ± 2** | 36 ± 4** | 28 ± 2 | 34 ± 2 |
| 12 min | 7.41 ± 0.04 | 7.36 ± 0.04 | 28 ± 2** | 27 ± 3** | 28 ± 2 | 34 ± 2 |
| 19 min | 7.36 ± 0.04* | 7.32 ± 0.04* | 28 ± 3** | 23 ± 2** | 29 ± 2 | 35 ± 2 |
| Post-control | ||||||
| 10 min | 7.32 ± 0.03** | 7.28 ± 0.03** | 120 ± 16 | 52 ± 4 | 29 ± 2 | 35 ± 2 |
| 20 min | 7.34 ± 0.02** | 7.29 ± 0.03** | 155 ± 26 | 58 ± 5 | 29 ± 2 | 34 ± 2 |
| 30 min | 7.37 ± 0.02* | 7.31 ± 0.02** | 135 ± 5 | 58 ± 6 | 29 ± 2 | 34 ± 2 |
Values are the means ±s.e.m. of 13 tests in 11 dogs.
P < 0.05
P < 0.001 in a paired t test compared with the normoxic pre-control.
Haemodynamic responses
Haemodynamic data are the means of 12 dogs. A typical pressure tracing is shown in Fig. 2. Arterial perfusion pressure (108 ± 8 mmHg) was not significantly changed for 5 min after the onset of hypoxia, but then gradually increased to 131 ± 11 mmHg at 9 ± 1.8 min (P < 0.004). The responses in the later part of the hypoxia were more variable: in some dogs, the pressure decreased only slightly throughout the remainder of the hypoxic period, but in others, it decreased to, or even below, control by the end of hypoxia. The average values were 117 ± 10 mmHg at 12 min and 115 ± 12 mmHg at 19 min. The post-control was 111 ± 12 mmHg at 10 min and 103 ± 9 mmHg at 20 min, which was maintained through the remainder of the recovery period.
Figure 2. Typical experimental recordings of the response to 19 min of hypoxia.
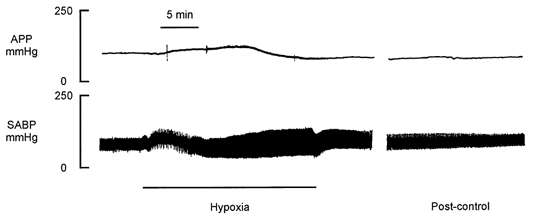
The upper panel shows the arterial perfusion pressure (APP) and the lower panel shows the systemic arterial blood pressure (SABP).
Systemic arterial blood pressure was controlled by a pressure reservoir connected to a brachial artery. In the first 5 min of systemic hypoxia, the mean systemic pressure increased more rapidly than the pressure system could respond, and a net increase of about 30 mmHg was seen. Shortly thereafter, the systemic arterial pressure was adjusted to a level not significantly different from the pre-control where it was maintained throughout the rest of the hypoxic period and the recovery. Approximately 100 ml of blood was withdrawn into the reservoir in the first 10 min of hypoxia. However, during the second half of the hypoxic period, blood had to be transferred from the reservoir to the animal in order to maintain the blood pressure, indicating that the blood pressure would have fallen below control if it had not been regulated. The average volume transferred back into the arterial system was about 60 ml. Since the arterial reservoir had been primed with about 200 ml of a saline-dextran mixture, a significant net transfer of fluid from the reservoir to the arterial system would reduce the haematocrit: the haematocrit did not change significantly during hypoxia, but it decreased from 47 ± 2 to 43 ± 3 % (P < 0.05) in the first 10 min of post-control.
Plasma and interstitial adenosine and adenine nucleotides concentration
The plasma and interstitial values are shown in Fig. 3 and Fig. 4, respectively, and are the data from 9–11 dogs. Arterial adenosine concentration was 427 ± 98 nm. Values were not significantly changed during hypoxia (535 ± 127, 559 ± 115 and 442 ± 105 nm at 5, 12 and 19 min) and back to normoxia in three post-controls (466 ± 102, 461 ± 126 and 475 ± 131 nm at 10, 20 and 30 min). Systemic hypoxia significantly increased venous adenosine concentration (Fig. 3, P < 0.01 for the 5th minute of hypoxia, and P < 0.05 for the 12th and 19th minutes of hypoxia). Despite the increased venous adenosine, there was no significant difference in the interstitial adenosine concentration between hypoxia and normoxia (Fig. 4).
Figure 3. Venous adenosine and adenine nucleotides concentrations determined at 10 min intervals in normoxia and 5–7 min intervals in hypoxia.
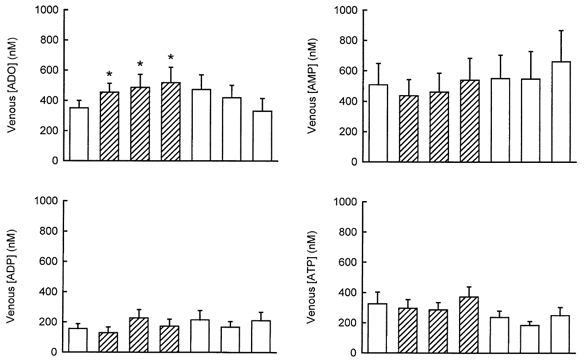
*P < 0.05 in a paired t test compared with the pre-control. Values are the means ±s.e.m.. of 10–11 tests in 11 dogs. □, normoxia;  hypoxia.
hypoxia.
Figure 4. Interstitial adenosine and adenine nucleotides concentrations in normoxia or systemic hypoxia.
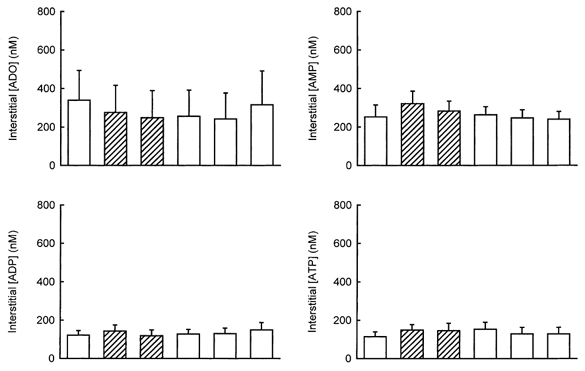
Samples were collected for 10 min each during normoxia and for 7 min each during hypoxia. Hypoxic sample collection was started after the fifth minute of hypoxia. The values are the means ±s.e.m.. of 10–11 tests in 11 dogs. □, normoxia; 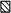 , hypoxia. There were no significant differences between any of the hypoxic samples and their pre-controls in a paired t test.
, hypoxia. There were no significant differences between any of the hypoxic samples and their pre-controls in a paired t test.
Arterial AMP, ADP and ATP concentrations were 486 ± 114, 425 ± 115 and 1251 ± 509 nm, respectively, in normoxia, and 468 ± 131, 402 ± 128 and 1184 ± 511 nm, respectively, in hypoxia (no significant difference). Neither the venous (Fig. 3) nor the interstitial (Fig. 4) concentrations of any of the nucleotides were changed during systemic hypoxia.
The relationship of arterial to interstitial adenosine, AMP, ADP and ATP
In order to estimate what degree of elevation of the arterial concentration of adenosine, AMP, ADP and ATP would be required to increase the interstitial concentration, mixtures of all the compounds at concentrations of 10, 100 or 1000 μm were infused in five dogs. The individual values of arterial and interstitial concentration are plotted against each other in Fig. 5. There was no significant movement of adenosine, AMP, ADP or ATP from the vascular to the interstitial space until the arterial concentration was more than 50 times higher than the interstitial: all four compounds appeared to be elevated in the interstitial space when the arterial concentration exceeded about 10 μm.
Figure 5. Relationship between arterial concentration of infused adenosine or adenine nucleotides, and their interstitial concentrations in five dogs.
Each point represents one sample in one experiment.
Muscle contractions
Eight tests were performed in six dogs
Arterial and venous blood gases and pH
Control arterial PO2 was 127 ± 19 mmHg, PCO2 was 36 ± 5 mmHg and pH was 7.44 ± 0.03; none of the values changed significantly during the contraction and post-control periods. Venous PO2 decreased from 66 ± 8 to 19 ± 2 mmHg (P < 0.001) during muscle contractions, but had already recovered to 63 ± 1 mmHg by the tenth minute of post-control. Venous PCO2 increased from 38 ± 4 to 69 ± 6 mmHg (P < 0.0005), and venous pH decreased from 7.42 ± 0.03 to 7.16 ± 0.03 (P < 0.00005) during muscle contractions; both remained significantly different from pre-control at 10 and 20 min of recovery, but had returned to values not significantly different from control after 30 min, when PCO2 was 48 ± 7 mmHg and pH was 7.31 ± 0.03.
Haemodynamic and force responses
Arterial perfusion pressure decreased from 111 ± 17 to 39 ± 2 mmHg (P < 0.001) during the first minute of contractions, and remained at 36 ± 2 mmHg until the end of the contraction period. Perfusion pressure remained depressed at 10 and 20 min post-control, but had recovered to 78 ± 5 mmHg (not significantly different from pre-control) by the 30th minute of recovery. Contractile force reached its maximum value of 40.0 ± 8.5 g (g wet weight of muscle)−1 after 0.5 ± 0.03 min, and thereafter declined, reaching 8.6 ± 1.1 g (g wet weight of muscle)−1 at the end of the 10 min contraction period.
Interstitial adenosine, AMP, ADP and ATP concentrations
Interstitial adenosine, AMP, ADP and ATP concentrations during pre-control, contractions and recovery are shown in Fig. 6. All four compounds were significantly increased (P < 0.005) in contracting muscles; adenosine remained elevated (P < 0.05) in the first post-control sample, but all three adenine nucleotides had returned to values not significantly different from the pre-control within the first 10 min of recovery.
Figure 6. Interstitial adenosine and adenine nucleotides concentrations at rest or during muscle contractions.
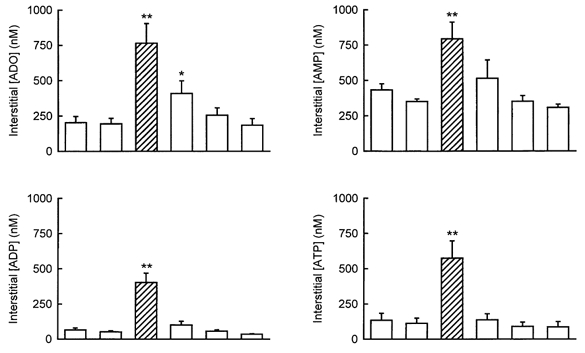
Samples were collected for 10 min each. Values are the means ±s.e.m.. of eight tests in six dogs. □, rest;  , contractions. Asterisks indicate values that were significantly different from pre-control in a paired t test (**P < 0.005; *P < 0.05).
, contractions. Asterisks indicate values that were significantly different from pre-control in a paired t test (**P < 0.005; *P < 0.05).
DISCUSSION
The main findings of these experiments were that: (1) the interstitial adenosine did not increase in response to systemic hypoxia; (2) consistent with our previous finding (Mo & Ballard, 1997), the venous adenosine was significantly increased 5 min after the onset of hypoxia, and remained elevated until the end of 20 min hypoxia; (3) hypoxia did not increase the precursors of adenosine, AMP, ADP and ATP in the interstitial fluid or the venous blood; and (4) the interstitial adenosine and adenine nucleotides were all increased during muscle contractions.
Our values of resting interstitial adenosine and adenine nucleotides are in good agreement with those reported by others (Hellsten et al. 1998; MacLean et al. 1998; Dela & Stallknecht, 1999). The measurements of interstitial adenosine and adenine nucleotides during muscle contractions were performed principally to ensure that our methodology was capable of detecting any changes in these substances, should they occur: the values we obtained are similar to, although slightly lower than, those reported by others (Hellsten et al. 1998). This difference most likely arises from the different types of muscle contraction employed by the two groups: we used isometric contractions whereas Hellsten's group used dynamic contractions, which would result in greater movement of the dialysis probe, and might produce more trauma to the muscle cells. The finding that all three adenine nucleotides, as well as adenosine, are increased in the interstitial space during muscle contractions lends further support to the concept that adenosine is formed extracellularly, from extracellular AMP, by ecto-5′-nucleotidase (Cheng et al. 2000).
The arterial adenosine concentration did not change in systemic hypoxia, but the venous adenosine concentration progressively increased throughout the period of hypoxia, and returned to control over about 30 min following the return to normoxic conditions, indicating a net adenosine output from the muscle. This is in agreement with our previous measurements (Mo & Ballard, 1997) and with studies utilizing adenosine receptor antagonists (Neylon & Marshall, 1991), which showed a significant contribution of adenosine to the increase in muscle conductance during systemic hypoxia. However, the interstitial adenosine concentration did not increase in hypoxia, which suggests that adenosine was unlikely to have originated from the skeletal muscle or interstitial cells. If skeletal muscle or parenchymal cells had been the source of the increased venous adenosine in hypoxia, then a large concentration gradient from the interstitial to the vascular space would be expected, since the endothelium takes up as much as two-thirds of the total amount in transit (Wangler et al. 1989). The increased intravascular adenosine did not diffuse into the interstitial space. The concentration gradient from the blood to the interstitial space was only around 250 nm, whereas the infusion study showed that adenosine would not begin to appear in the interstitial space until a concentration gradient of more than 10 μm had been achieved. Presumably, the avid endothelial uptake of adenosine (Wangler et al. 1989) prevented it from crossing the vascular wall. On the basis of this finding, we assume that either the vascular tissues or the blood cells must have been the source of the released adenosine. Since the arterial adenosine was not elevated during systemic hypoxia, it is unlikely that the blood cells or platelets were the source of the increased adenosine. Costa et al. (1999) have also reported that platelets were not the source of the increased venous adenosine during forearm ischaemia. Our previous studies of the dog model showed that the hypoxic adenosine release resulted from cellular uptake of lactate, since an inhibitor of lactate uptake abolished the adenosine release (Mo & Ballard, 1997). This is in accordance with our current finding, since the lactate is delivered to the muscle in the arterial blood, and it is logical to suppose that it would mainly enter vascular endothelial and blood cells. The elevated venous adenosine failed to recirculate to the arterial side of the circulation; this is presumably because of its very rapid uptake by blood and endothelial cells (Catravas, 1984). It is thought that a single pass through the pulmonary circulation can reduce the blood adenosine concentration back to basal levels, because of the very large surface area of capillary endothelium available in this organ.
We found that none of the adenine nucleotides were significantly increased in the interstitium or the venous blood in response to systemic hypoxia (Fig. 3 and Fig. 4). This is similar to the situation in the heart, where both adenosine and adenine nucleotides are released in well-oxygenated conditions, but only adenosine release increases in hypoxia (Borst & Schrader, 1991; Raatikainen, 1994). There are two possible explanations for the lack of nucleotide output in hypoxia: either the adenosine that appeared in the venous blood during hypoxia was formed intracellularly, by cells in the vascular wall or the blood, and transported to the extracellular compartment, or else the activities of the ecto-enzymes, ATPase, ADPase and ecto-5′-nucleotidase were so high that adenine nucleotides were rapidly converted to adenosine, and thus, adenine nucleotides did not accumulate in the blood. Studies of the 5′-nucleotidase from skeletal muscle homogenates show that the predominant form is the ecto-5′-nucleotidase (Camici et al. 1985; Cheng et al. 2000), which accounts for around 85 % of the adenosine formation. In those studies, the cytosolic 5′-nucleotidase was reported to be either absent (Camici et al. 1985), or of such low activity that it could not contribute to adenosine formation (Cheng et al. 2000). However, in a whole-muscle homogenate, the properties of small mass of tissue, such as the vascular endothelium, may not be well represented. Immunostaining of the heart confirms that an AMP-specific cytosolic 5′-nucleotidase is present in the cytoplasm of endothelium and red blood cells (Darvish et al. 1993), and cytosolic adenosine formation by endothelium or red blood cells therefore seems possible. Estimates of the proportion of adenosine formed extracellularly by cultured endothelial cells yield widely varying results (Deussen et al. 1993; Catravas & Watkins, 1995), which is not altogether surprising since the expression of 5′-nucleotidase declines rapidly in culture (Hayes et al. 1979; Chesterman et al. 1983). The maximal velocities for ecto-ATPase, ADPase and AMPase are all very high, with a rank order ATPase > ADPase > AMPase (Gordon et al. 1986). However, the enzymes for adenosine removal are intracellular, and thus, the removal of adenosine is limited by the rate at which it can be taken up into cells. Therefore, when adenosine is formed extracellularly from ATP, adenosine accumulates to a greater extent than any of the adenine nucleotides (e.g. as in the muscle contraction experiments of this study). We can confirm that this relationship is also true for the intravascular compartment in our preparation, since the arterial infusion of identical concentrations of adenosine and adenine nucleotides raised the venous adenosine to 77 ± 12 μm, but the venous AMP, ADP and ATP concentrations to only 13 ± 4, 17 ± 8 and 10 ± 5 μm, respectively. Similarly, whilst interstitial adenosine and all the adenine nucleotides were elevated during muscle contractions, only the interstitial adenosine remained elevated during the first 10 min of post-control. However, we could not detect even a small elevation of adenine nucleotides in hypoxia, and we conclude, therefore, that only a small proportion, if any, of the increased adenosine in can have been formed extracellularly.
The haemodynamic responses to hypoxia in our study are different from those reported in other studies (Neylon & Marshall, 1991): in our study, the blood pressure was controlled by a reservoir system, and the muscle was denervated, which eliminated any reflex effects arising from the baro- or chemoreceptors. Therefore, any haemodynamic effects can be attributed either to metabolites produced in the muscle itself or else to metabolites or hormones delivered in the arterial blood. We saw no change in the vascular resistance during the first 5 min of hypoxia, followed by an increase of about 20 % in the vascular resistance between 5 and 9 min, and then by a steady decrease in vascular resistance throughout the remainder of the hypoxic period. As we know from the HPLC results that the concentration of adenosine, a vasodilator metabolite, is already significantly elevated in the venous blood within the first 5 min of hypoxia, this suggests that both vasodilator and vasoconstrictor metabolites are competing for the control of the skeletal muscle vasculature throughout the hypoxic period. Based on the 5 min delay before vasoconstriction began (whereas blood pressure increased immediately after the onset of hypoxia), we would tentatively suggest that this is mediated by a metabolite or hormone delivered in the arterial blood. However, the switch from vasoconstriction to vasodilatation in the test muscle occurred at the same time as the blood began to move out of the arterial reservoir into the artery (signifying that blood pressure would decrease if not maintained by the reservoir system): therefore, we assume this to be caused by vasodilator metabolite synthesis in both our test muscle and the rest of the body. In other studies of systemic hypoxia, in which the nerves are left intact and the blood pressure is allowed to change freely, a broadly similar pattern of blood pressure changes are seen: the blood pressure usually increases initially, and then decreases to below the control level; in small mammals such as the rat, the initial increase in blood pressure is much less pronounced, and the switch to a net decrease in pressure occurs much earlier (Neylon & Marshall, 1991). In those studies, then, there is a reduction in the perfusion pressure to the muscle. This is accompanied by a substantial increase in the vascular conductance, which maintains blood flow at around the control level, despite the reduced perfusion pressure. Around half of the increased vascular conductance can be attributed to adenosine in those studies (Neylon & Marshall, 1991). The differences between the responses of these two preparations suggest that the nerves normally contribute to the muscle's haemodynamic response to hypoxia: this might be achieved either through the release of further amounts of adenosine or adenine nucleotides (or other substances) from the nerves, or else if the nerves mediate a part of the vasodilator response to adenosine. Another group, have reported an increase in interstitial adenosine in human skeletal muscle during hypoxia (MacLean et al. 1998): this difference might arise from the presence of an intact nerve in the human subjects, or else simply from a species difference. We think it unlikely that a species difference accounts for the finding in humans, as others have shown that muscle contractions raise interstitial adenosine in humans, but ischaemia does not (Costa et al. 1999). Therefore, it is more likely that there is a neuronal contribution to interstitial adenosine formation in the human study. In an intact-nerve preparation, ATP could be released into the interstitial space from the purinergic nerves (Johnson et al. 1999), or from the sympathetic nerves, either as the co-transmitter for catecholamines or from the post-synaptic membrane (Coney & Marshall, 1999a,b); if it were released, then it would almost certainly be converted to adenosine by the ectoenzymes lining the interstitial space. Experiments are currently underway in our laboratory to investigate the role of the nerve in mediating in adenosine formation in systemic hypoxia.
The muscle vasodilatation induced by systemic hypoxia or infused adenosine was found to be largely NO dependent in the rat (Bryan & Marshall, 1999b), which led to the suggestion that the actions of adenosine in systemic hypoxia were mediated by the endothelium. However, neuronal NO-synthase (NOS) is also found in the skeletal muscle, mainly in nerve cells and in skeletal muscle fibres at the motor endplate (Grozdanovic & Baumgarten, 1999), and at lower levels in vascular smooth muscle (Segal et al. 1999). Neuronal NOS is suggested to be responsible for the NO-modulation of α-adrenergic vasoconstriction in muscle (Thomas et al. 1998). The finding that interstitial adenosine is not elevated in hypoxia rules out any contribution of the neuronal NOS in skeletal muscle fibres to the hypoxic response, but it is hard to be certain whether or not adenosine could permeate the vascular wall sufficiently to activate nNOS in vascular smooth muscle or perivascular nerves. However, on the basis of the adenosine infusion results in this study, we consider it unlikely; thus, adenosine most probably does act at an endothelial site in systemic hypoxia.
Acknowledgments
This work was funded by the Research Grants Council of Hong Kong. We are grateful to Mr Yip Man Keung for technical assistance.
References
- Aickin CC, Thomas RC. An investigation of the ionic mechanism of intracellular pH regulation in mouse soleus muscle fibres. Journal of Physiology. 1977;273:295–316. doi: 10.1113/jphysiol.1977.sp012095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balcells E, Suarez J, Rubio R. Functional role of intravascular coronary endothelial adenosine receptors. European Journal of Pharmacology. 1992;210:1–9. doi: 10.1016/0014-2999(92)90644-j. [DOI] [PubMed] [Google Scholar]
- Ballard HJ. The role of pH in the release of adenosine from skeletal muscle in anaesthetised dogs. Journal of Physiology. 1991;433:95–108. doi: 10.1113/jphysiol.1991.sp018416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard HJ, Cotterrell D, Karim F. Appearance of adenosine in venous blood from the contracting gracilis muscle and its role in vasodilatation in the dog. Journal of Physiology. 1987;387:401–413. doi: 10.1113/jphysiol.1987.sp016580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belloni FL, Phair RD, Sparks HV. The role of adenosine in prolonged vasodilation following flow-restricted exercise of canine skeletal muscle. Circulation Research. 1986;44:756–766. doi: 10.1161/01.res.44.6.759. [DOI] [PubMed] [Google Scholar]
- Borst MM, Schrader J. Adenine nucleotide release from isolated perfused guinea pig hearts and extracellular formation of adenosine. Circulation Research. 1991;68:797–806. doi: 10.1161/01.res.68.3.797. [DOI] [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Adenosine receptor subtypes and vasodilatation in rat skeletal muscle during systemic hypoxia: a role for A1 receptors. Journal of Physiology. 1999a;514:151–162. doi: 10.1111/j.1469-7793.1999.151af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Cellular mechanisms by which adenosine induces vasodilatation in rat skeletal muscle: significance for systemic hypoxia. Journal of Physiology. 1999b;514:163–175. doi: 10.1111/j.1469-7793.1999.163af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camici M, Fini C, Ipata PL. Isolation and kinetic properties of 5′-nucleotidase from guinea-pig skeletal muscle. Biochimica et Biophysica Acta. 1985;840:6–12. doi: 10.1016/0304-4165(85)90155-2. [DOI] [PubMed] [Google Scholar]
- Catravas JD. Removal of adenosine from the rabbit pulmonary circulation, in vivo and in vitro. Circulation Research. 1984;54:603–611. doi: 10.1161/01.res.54.5.603. [DOI] [PubMed] [Google Scholar]
- Catravas JD, Watkins CA. Plasmalemmal metabolic activities in cultured calf pulmonary arterial endothelial cells. Research Communications in Chemical Pathology and Pharmacology. 1995;50:163–179. [PubMed] [Google Scholar]
- Cheng B, Essackjee HC, Ballard HJ. Evidence for control of adenosine metabolism in rat oxidative skeletal muscle by changes in pH. Journal of Physiology. 2000;522:467–477. doi: 10.1111/j.1469-7793.2000.t01-1-00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesterman CN, Ager A, Gordon JL. Regulation of prostaglandin production and ectoenzyme activities in cultured aortic endothelial cells. Journal of Cell Physiology. 1983;116:45–50. doi: 10.1002/jcp.1041160108. [DOI] [PubMed] [Google Scholar]
- Coney AM, Marshall JM. Modulations by adenosine of vasoconstriction evoked in rat skeletal muscle by different patterns of sympathetic nerve stimulation. Journal of Physiology. 1999a;515.P:142P. [Google Scholar]
- Coney AM, Marshall JM. Systemic hypoxia diferenctially modulates vasoconstriction of rat skeletal muscle evoked by different patterns of sympathetic nerve stimulation. Journal of Physiology. 1999b;518.P:177P. [Google Scholar]
- Costa F, Sulur P, Angel M, Cavalcante J, Haile V, Christman B, Biaggioni I. Intravascular source of adenosine during forearm ischemia in humans: implications for reactive hyperemia. Hypertension. 1999;33:1453–1457. doi: 10.1161/01.hyp.33.6.1453. [DOI] [PubMed] [Google Scholar]
- Darvish A, Postlethwaite JJ, Metting PJ. Immunogold localization of adenosine 5′-monophosphate-specific cytosolic 5′-nucleotidase in dog heart. Hypertension. 1993;21:906–910. doi: 10.1161/01.hyp.21.6.906. [DOI] [PubMed] [Google Scholar]
- Dela F, Stallknecht B. No role of interstitial adenosine in insulin-mediated vasodilation. Acta Physiologica Scandinavica. 1999;167:37–42. doi: 10.1046/j.1365-201x.1999.00583.x. [DOI] [PubMed] [Google Scholar]
- Deussen A, Bading B, Kelm M, Schrader J. Formation and salvage of adenosine by macrovascular endothelial cells. American Journal of Physiology. 1993;264:H692–H700. doi: 10.1152/ajpheart.1993.264.3.H692. [DOI] [PubMed] [Google Scholar]
- Dobson JG, Jr, Rubio R, Berne RM. Role of adenosine nucleotides, adenosine and inorganic phosphate in the regulation of skeletal muscle blood flow. Circulation Research. 1971;24:375–384. doi: 10.1161/01.res.29.4.375. [DOI] [PubMed] [Google Scholar]
- Gordon EL, Pearson JD, Slakey LL. The hydrolysis of extracellular adenine nucleotides by cultured endothelial cells from pig aorta. Feed-forward inhibition of adenosine production at the cell surface. Journal of Biological Chemistry. 1986;261:15496–15507. [PubMed] [Google Scholar]
- Grozdanovic Z, Baumgarten HG. Nitric oxide synthase in skeletal muscle fibers: a signalling component of the dystrophin-glycoprotein complex. Histology and Histopathology. 1999;14:243–256. doi: 10.14670/HH-14.243. [DOI] [PubMed] [Google Scholar]
- Hayes LW, Goguen CA, Stevens AL, Magargal WW, Slakey LL. Enzyme activities in endothelial cells and smooth muscle cells from swine aorta. Proceedings of the National Academy of Sciences of the USA. 1979;76:2532–2535. doi: 10.1073/pnas.76.6.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten Y, MacLean D, Radegran G, Saltin B, Bangsbo J. Adenosine concentrations in the interstitium of resting and contracting human skeletal muscle. Circulation. 1998;98:6–8. doi: 10.1161/01.cir.98.1.6. [DOI] [PubMed] [Google Scholar]
- Johnson CD, Coney AM, Marshall JM. Responses evoked in tail and hindlimb circulations of the rat by natural patterns of sympathetic nerve activity: Contributions from noradrenaline and ATP. Journal of Physiology. 1999;515.P:144P. [Google Scholar]
- MacLean DA, Sinoway LI, Leuenberger U. Systemic hypoxia elevates skeletal muscle interstitial adenosine levels in humans. Circulation. 1998;98:1990–1992. doi: 10.1161/01.cir.98.19.1990. [DOI] [PubMed] [Google Scholar]
- Marshall JM. Peripheral chemoreceptors and cardiovascular regulation. Physiological Reviews. 1994;74:543–594. doi: 10.1152/physrev.1994.74.3.543. [DOI] [PubMed] [Google Scholar]
- Marshall JM. Skeletal muscle vasculature and systemic hypoxia. News in Physiological Sciences. 1995;10:274–280. [Google Scholar]
- Marshall JM. Adenosine and muscle vasodilatation in acute systemic hypoxia. Acta Physiologica Scandinavica. 2000;168:561–573. doi: 10.1046/j.1365-201x.2000.00709.x. [DOI] [PubMed] [Google Scholar]
- Mo FM, Ballard HJ. Intracellular lactate controls adenosine output from dog gracilis muscle during moderate systemic hypoxia. American Journal Physiology. 1997;272:H318–H324. doi: 10.1152/ajpheart.1997.272.1.H318. [DOI] [PubMed] [Google Scholar]
- Neylon M, Marshall JM. The role of adenosine in the respiratory and cardiovascular response to systemic hypoxia in the rat. Journal of Physiology. 1991;440:529–545. doi: 10.1113/jphysiol.1991.sp018723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillis JW, Song D, O'Regan MH. The role of adenosine in rat coronary flow regulation during respiratory and metabolic acidosis. European Journal of Pharmacology. 1999;356:199–206. doi: 10.1016/s0014-2999(98)00512-3. [DOI] [PubMed] [Google Scholar]
- Poucher SM. The role of the A2a adenosine receptor subtype in functional hyperaemia in the hindlimb of anaesthetised cats. Journal of Physiology. 1996;492:495–503. doi: 10.1113/jphysiol.1996.sp021324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raatikainen MJ, Peuhkurinen KJ, Hassinen IE. Contribution of endothelium and cardiomyocytes to hypoxia-induced adenosine release. Journal of Molecular and Cellular Cardiology. 1994;26:1069–1080. doi: 10.1006/jmcc.1994.1127. [DOI] [PubMed] [Google Scholar]
- Segal SS, Brett SE, Sessa WC. Codistribution of NOS and caveolin throughout peripheral vasculature and skeletal muscle of hamsters. American Journal of Physiology. 1999;277:H1167–1177. doi: 10.1152/ajpheart.1999.277.3.H1167. [DOI] [PubMed] [Google Scholar]
- Shryock JC, Rubio R, Berne RM. Release of adenosine from pig aortic endothelial cells during hypoxia and metabolic inhibition. American Journal of Physiology. 1988;254:H223–2299. doi: 10.1152/ajpheart.1988.254.2.H223. [DOI] [PubMed] [Google Scholar]
- Steffen RP, McKenzie JE, Bockman EL, Haddy FJ. Changes in dog gracilis muscle adenosine during exercise and acetate infusion. American Journal of Physiology. 1983;244:H387–395. doi: 10.1152/ajpheart.1983.244.3.H387. [DOI] [PubMed] [Google Scholar]
- Thomas GD, Sander M, Lau KS, Huang PL, Stull JT, Victor RG. Impaired metabolic modulation of alpha adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proceedings of the National Academy of Sciences U.S.A. 1998;95:15090–15095. doi: 10.1073/pnas.95.25.15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangler RD, Gorman MW, Wang CY, Dewitt DF, Chan IS, Bassingthwaighte JB, Sparks HV. Transcapillary adenosine transport and interstitial adenosine concentration in guinea pig hearts. American Journal of Physiology. 1989;257:H89–106. doi: 10.1152/ajpheart.1989.257.1.H89. [DOI] [PMC free article] [PubMed] [Google Scholar]