

Abstract
L-type Ca2+ channels play an important role in vital cell functions such as muscle contraction and hormone secretion. Both a voltage-dependent and a Ca2+-dependent process inactivate these channels. Here we present evidence that inhibition of the mitochondrial Ca2+ import mechanism in rat (Sprague-Dawley) ventricular myocytes by ruthenium red (RR), by Ru360 or by carbonyl cyanide m-chlorophenylhydrazone (CCCP) decreases the magnitude of electrically evoked transient elevations of cytosolic Ca2+ concentration ([Ca2+]c). These agents were most effective at stimulus rates greater than 1 Hz.
RR and CCCP also caused a significant delay in the recovery from inactivation of L-type Ca2+ currents (ICa). This suggests that sequestration of cytosolic Ca2+, probably near the mouth of L-type Ca2+ channels, into mitochondria during cardiac contractile cycles, helps to remove the Ca2+-dependent inactivation of L-type Ca2+ channels.
We conclude that impairment of mitochondrial Ca2+ transport has no impact on either L-type Ca2+ currents or SR Ca2+ release at low stimulation frequencies (e.g. 0.1 Hz); however, it causes a depression of cytosolic Ca2+ transients attributable to an impaired recovery of L-type Ca2+ currents from inactivation at high stimulation frequencies (e.g. 3 Hz). The impairment of mitochondrial Ca2+ uptake and subsequent effects on Ca2+ transients at high frequencies at room temperature could be physiologically relevant since the normal heart rate of rat is around 5 Hz at body temperature. The role of mitochondria in clearing Ca2+ in the micro-domain near L-type Ca2+ channels could be impaired during high frequencies of heart beats such as in ventricular tachycardia, explaining, at least in part, the reduction of muscle contractility.
Activation of the cardiac L-type Ca2+ channels during an action potential leads to Ca2+-induced Ca2+ release from the sarcoplasmic reticulum (SR) (Fabiato, 1983; Beuckelmann & Wier, 1988; Bers, 1991; Eisner et al. 1998). These channels are subsequently inactivated by elevation of cytosolic Ca2+ concentration ([Ca2+]c) as well as by membrane depolarization (Chad & Eckert, 1984; Kass & Sanguinetti, 1984; Lee et al. 1985; Imredy & Yue, 1994). Therefore, [Ca2+]c in the vicinity of the channels critically controls their activity during excitation- contraction (EC) coupling.
The major factor contributing to channel inactivation in intact heart cells is Ca2+ released from the SR, with a small contribution from Ca2+ permeating through the channel itself (Satomi et al. 1996; Sun et al. 1997). Although the source of Ca2+ that inactivates L-type Ca2+ channels is well characterized, less is known about the mechanisms that clear cytosolic Ca2+ near the channel. In situ, mitochondria are located in tight apposition with endoplasmic reticulum (ER)/SR and can preferentially sequester the Ca2+ released from ER/SR (Rizzuto et al. 1993; Hajnóczky et al. 1995; Sharma et al. 2000). Furthermore, inhibitors of mitochondrial Ca2+ uptake severely compromise clearance of cytosolic Ca2+ after an imposed Ca2+ elevation in neurons (Thayer & Miller, 1990; Babcock & Hille, 1998). A similar role of mitochondria during the heartbeat is less likely, since the SR Ca2+ pump is the major system that removes Ca2+ from the cytosol (Bassani et al. 1994). However, recent work has revealed a preferential coupling of Ca2+ transport in rat ventricular myocytes from SR to mitochondria, because of their structural proximity (Sharma et al. 2000). Therefore, mitochondria might influence the local [Ca2+]c near L-type Ca2+ channels and thus modify their inactivation. To test this hypothesis, we monitored L-type Ca2+ currents (ICa) in cardiac myocytes and compared their rates of recovery from inactivation in the presence and absence of inhibitors of mitochondrial Ca 2+ uptake.
METHODS
Experiments upon rats were performed according to protocols approved by the Division of Laboratory Animal Medicine, University of Rochester, in compliance with state law, federal statute and NIH policy.
Cells and solutions
The method for myocyte isolation from adult rats has been described previously (Sharma et al. 2000). Briefly, isolated myocytes were prepared from the ventricles of adult male Sprague-Dawley rats (250–300 g, 5–6 months old). The rats were anaesthetized with sodium pentobarbital (60 mg (kg body weight)−1, i.p.). Rats were checked for deep anaesthesia by testing for tail and ear pinch reflexes. Only after making sure that the rat was under deep anaesthesia, the heart, along with a 5 mm section of the aortic arch, was quickly excised. The heart was mounted on a plastic cannula inserted into the aorta and perfused with Ca2+-free Joklik's tissue culture medium (Gibco, Grand Island, NY, USA) for 5 min to cleanse the heart of blood. The perfusion solution was then changed to Joklik's medium containing 50 μm CaCl2, 0.5 mg ml−1 collagenase (Worthington, Lakewood, NJ, USA, type II) and 0.1 % BSA (Sigma Chemical Co., St Louis, MO, USA). This enzyme solution was recirculated through the heart for approximately 30 min. The ventricles collected in separate flasks were shaken vigorously and filtered through 200 μm nylon mesh to obtain dissociated single cells. The isolated myocytes were kept in standard solution that contained (mm): NaCl 140, KCl 5, CaCl2 2, MgCl2 2, Hepes 10 and glucose 11, pH 7.4 at 37 °C with NaOH.
Measurement of cytosolic and mitochondrial [Ca2+]
The method for recording of [Ca2+]c from single cardiac myocytes has been previously published (Sharma et al. 1996). Briefly, isolated myocytes were loaded with 3 μm fura-2 AM for 15 min at room temperature in standard Tyrode solution. After loading, the cells were incubated in dye-free solution for over 45 min to allow the conversion of dye to its Ca2+-sensitive, free acid form. The coverslip with dye-loaded cells was mounted in a tissue chamber on the stage of a Nikon Diaphot inverted microscope equipped for epifluorescence (Deltascan 1, Photon Technology International, Princeton, NJ, USA). To evoke Ca2+ transients, myocytes were electrically stimulated at various frequencies with an S8 Grass stimulator. The cell was sequentially excited at 340 and 380 nm wavelength light using two excitation monochromators at a switching frequency of 100 Hz controlled by an optical chopper. The emission fluorescence was collected at 510 nm. The results are then presented as the ratio (R) of fluorescence excited at 340 nm (F340) to that excited at 380 nm (F380). The amplitude of each individual Ca2+ transient is defined as the difference between the systolic (peak) fluorescence ratio values and diastolic (baseline) fluorescence ratio values. To minimize the variation, amplitudes of six consecutive transients recorded at the steady state were averaged to produce the representative Ca2+ transient amplitude at a particular frequency of stimulation.
The details of mitochondrial calcium concentration ([Ca2+]m) and [Ca2+]c measurements in chemically skinned myocytes with rhod-2 and fura-2, respectively, have been described in detail before (Sharma et al. 2000). Rhod-2 has been used to measure changes of [Ca2+]m in living cells (Hajnóczky et al. 1995; Jou et al. 1996). One of the requirements for using this technique to measure [Ca2+]m is that the recorded fluorescence signals for Ca2+ must originate solely from mitochondria. To achieve this, intact myocytes were first loaded with the dye by incubating the cells in standard solution containing rhod-2 AM (2 μm, Molecular Probes, Eugene, OR, USA) for 40–50 min at room temperature. Since rhod-2 AM consists of a cationic rhodamine molecule, it accumulates preferentially inside the mitochondria due to their negative membrane potential. After loading, the cells were kept in rhod-2 AM-free standard solution for at least 1 h to allow conversion of the dye to its Ca2+-sensitive, free acid form. A droplet of the suspension of cells was then transferred to the laminin-coated Lab-Tek perfusion chamber (VWR, Rochester, NY, USA). To remove residual rhod-2 in the cytosol, the plasma membrane was skinned by exposing the myocytes for 20 s with a solution containing (mm): KCl 120, NaCl 10, glucose 10, MgCl2 2, Na2ATP 5, Na2CrP 15, succinate 5, EGTA 0.1, Hepes 10 and saponin 0.2 mg ml−1, pH 7.20 (Neary et al. 1996). The myocytes were then perfused with the above buffer containing 100 nm free Ca2+ but without saponin to wash away rhod-2 that leaked out from cytosol after the plasma membrane was permeabilized. The free Ca2+ concentrations were calculated according to a computer program developed by Fabiato & Fabiato (1979). The cells were then exposed to the excitation light of 555 nm wavelength and the emission fluorescence was collected at 590 nm (Sharma et al. 2000). Since rhod-2 is not a ratiometric dye, its fluorescence intensity was not calibrated to obtain absolute values of [Ca2+]m. All fluorescence signals (F) are expressed relative to the control value (F0).
For measurements of [Ca2+]c in chemically skinned myocytes, 10 μm pentapotassium salt of fura-2 was added to the incubating solution. Similar to the measurements of [Ca2+]c in intact myocytes, the results were presented as ratio values (R =F340/F380). In some cases, in situ calibration was done to convert the ratio values into absolute values of [Ca2+]c. Permeabilized myocytes were perfused with calibration solution containing
where R is the measured ratio between F340 and F380 and Rmin and Rmax are the ratios obtained in solutions containing 0 Ca2+ and 0.1 mm Ca2+, respectively. Sf2 is the 380 nm excitation signal in 0 Ca2+, and Sb2 is the 380 nm excitation signal at 0.1 mm Ca2+ in calibrating buffer. Kd is the dissociation constant for fura-2-Ca2+ and taken to be 224 nm (Grynkiewicz et al. 1985).
Electrophysiological techniques
Inward Ca2+ currents were recorded from rat myocytes. To record membrane currents, the patch-clamp technique was used in the whole-cell configuration (Hamill et al. 1981). Pipettes were double-pulled from hard glass (KIMAX-51, Kimble Glass, Toledo, OH, USA) using a horizontal Brown-Fleming puller. Seventy to ninety per cent of the series resistance was compensated.
Membrane currents in response to voltage-step depolarizations applied from the holding potential were measured with an EPC-7 amplifier (List Electronics, Darmstadt, Germany) and sampled by a microcomputer. Analog signals were digitized with a Labmaster interface (Axon Instruments, Foster City, CA, USA) at a 12 bit resolution. Currents were amplified and filtered with an active four-pole, low-pass Bessel filter with a corner frequency of no more than half the sampling frequency. To measure activation of Ca2+ currents, command pulses of 50–100 ms duration and variable amplitude were delivered. Linear currents were subtracted with the P/-6 procedure. Data were analysed by a combination of pCLAMP 6.0 (Axon Instruments) and in-house software. The fitting of numerical formulae to the experimental data employed a non-linear least squares algorithm. The pipette solution contained (mm): caesium aspartate 100, CsCl 20, TEACl 20, Mg-ATP 5, MgCl2 1, EGTA 0.05, Hepes 5, pH 7.2. Previous work has shown that mitochondrial Ca2+ transport capabilities are not impaired in the presence of high Cs+ solutions (Herrington et al. 1996; Zhou et el. 1998; see also Results). The standard bath solution contained (mm): NaCl 140, KCl 5, CaCl2 1, MgCl2 2, glucose 10 and Hepes 10, pH 7.5.
All experiments were carried out at room temperature (23 °C).
RESULTS
First, we established the experimental conditions that allow a selective blockade of mitochondrial Ca2+ uptake in rat ventricular myocytes. For this we used ruthenium red (RR), a blocker of the mitochondrial uniporter in several cell types including heart cells (McCormack & England, 1983; Hansford, 1987; Trollinger et al. 2000). Alternatively, we used Ru360, a selective inhibitor of mitochondrial uniporter in single cardiac myocytes (Matlib et al. 1998), or CCCP. In CCCP experiments, oligomycin was added to remove the voltage gradient for Ca2+ influx through mitochondrial uniporter without ATP depletion (Nieminen et al. 1990; Budd & Nicholls, 1996). Figure 1A and B illustrates that RR (1–10 μm) blocks the mitochondrial Ca2+ uptake, confirming previous results (Sharma et al. 2000). In permeabilized myocytes, caffeine (10 mm) caused a rapid increase (4.10 ± 0.37-fold, n = 14) in peak rhod-2 fluorescence (Fig. 1A). RR (4 μm) significantly blocked the caffeine response. The increase in the rhod-2 fluorescence signal in response to caffeine was only 0.06 ± 0.01-fold (Fig. 1B, n = 14), consistent with results from a previous report in digitonin-permeabilized HeLa cells (Rizzuto et al. 1993). To test the effects of RR on Ca2+ release from SR, we measured [Ca2+]c in skinned cardiac myocytes using the fluorescent indicator fura-2. Under control conditions (Fig. 1C), caffeine produced an immediate transient increase in fluorescence. A similar response was obtained with 4 μm RR (Fig. 1D). Peak [Ca2+]c levels were 412 ± 17 nm (n = 14) in control conditions and 459 ± 13 nm (n = 5) in the presence of RR. Likewise, the resting [Ca2+]c levels were 87 ± 4 nm (n = 14) and 92 ± 4 nm (n = 5), respectively. These results agree with previous work indicating that 4 μm RR has no significant effect on caffeine-induced contractures or on Ca2+-induced Ca2+ release in heart (Zhu & Nosek, 1992; Sharma et al. 2000). Finally, we tested the effects of RR on the caffeine-induced [Ca2+]c response in intact cells. Caffeine (20 mm) produced a robust increase in [Ca2+]c (Fig. 1E). After that, the cell was incubated with normal solution containing RR (10 μm) for 12 min and tested again. Myocytes were pretreated with RR or Ru360 for a period of 12 min as these chemicals permeate the intact plasma membrane slowly. Matlib et al. (1998) have shown that it takes at least 10 min for externally applied Ru360 to achieve a maximum concentration in isolated cardiac myocytes. RR had no effect on caffeine-induced Ca2+ signals in the intact myocyte (Fig. 1F). The ratio between the peak amplitude in response to caffeine of the RR-treated cells to that of the untreated control cells averaged 1.05 ± 0.17 (n = 7).
Figure 1. The effect of RR on [Ca2+]m and [Ca2+]c in skinned and intact cardiac myocytes during caffeine application.
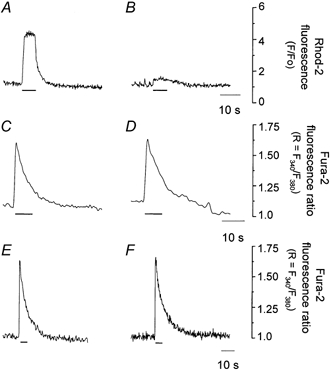
Mitochondrial rhod-2 signals from a skinned cell in response to caffeine (10 mm): A, control; B, after incubation in RR (4 μm). Fluorescence data are expressed relative to resting fluorescence (F/Fo). Cytosolic fura-2 ratiometric signals (R = F340/F380) from a skinned cell in response to caffeine (10 mm): C, control; D, after incubation in RR (4 μm). Cytosolic fura-2 ratiometric signals from an intact cell in response to caffeine (20 mm): E, control; F, in the presence of RR (10 μm). Horizontal bars indicate time of caffeine application.
After establishing that RR does not interfere with Ca2+ release from SR by caffeine under the present experimental conditions, we investigated its effect on Ca2+ transients produced by electrical stimulation. An intact cardiac myocyte was electrically stimulated at 0.1 Hz and its cytosolic Ca2+ transients were recorded when the steady state was achieved (Fig. 2A). The stimulation was paused and the cell was exposed to 10 μm RR for at least 12 min. The stimulation was then resumed and the Ca2+ transients were recorded again. The electrical stimulation protocol used ensured a steady state level in the amplitude of Ca2+ transients as we found experimentally. The stimulation was paused to allow a complete and fast exchange of the extracellular solutions. These transients in the steady state were identical to those recorded in the control solution (Fig. 2B). Similar experiments were performed in additional myocytes using higher frequencies of stimulation. Figure 2C illustrates Ca2+ transients recorded at a frequency of 3.0 Hz under control conditions. RR significantly decreased the amplitude of individual Ca2+ transients at this frequency of stimulation (Fig. 2D). Data from several cells were summarized by plotting the ratio of the mean amplitude of evoked Ca2+ transients recorded in the presence of RR to that recorded in its absence, as a function of the frequency of stimulation (Fig. 2E). The results indicate that as the frequency of stimulation increased, the ratio decreased, suggesting that blocking the mitochondrial uniporter reduces Ca2+ release by SR more effectively at high frequencies of stimulation.
Figure 2. The effect of RR on the cytosolic Ca2+ transients measured with fura-2 at different frequencies of stimulation.
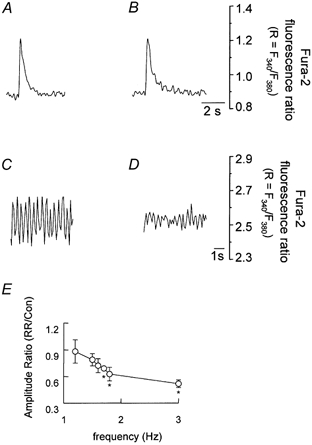
Ca2+ transients from an intact cardiomyocyte electrically stimulated at 0.1 Hz: A, control; B, after incubation in RR (10 μm). Ca2+ transients from an intact cardiomyocyte electrically stimulated at 3.0 Hz: C, control; D, after incubation in RR (10 μm). E, the relationship between action of RR (ordinate) and frequency of stimulation (abscissa). The amplitude ratio is the mean amplitude of evoked [Ca2+]c response observed in the presence of RR divided by that recorded in its absence. Points with error bars are the average (±s.e.m.) of 3–8 experiments. *P < 0.05.
Experiments with RR were further substantiated by using Ru360, a compound that inhibits mitochondrial Ca2+ uptake without inhibiting Ca2+ release from SR (Matlib et al. 1998; Zhou et al. 1998). After obtaining Ca2+ transients at frequencies of 0.1 and 3 Hz, the stimulation was paused and the cell was exposed to 10 μm Ru360 for a period of 30 min. The stimulation was then resumed and Ca2+ transients were recorded again. Figure 3 shows that there was no difference in the amplitude of Ca2+ transients in control and Ru360-treated cells when the cell was stimulated at a frequency of 0.1 Hz (Fig. 3A, control; and Fig. 3B, with Ru360). However, the amplitude of Ca2+ transients was significantly reduced in a Ru360-treated cell when it was stimulated at a frequency of 3.0 Hz (Fig. 3C, control; and Fig. 3D, with Ru360). Incubation of cardiac cells with 10 μm Ru360 reduced the amplitude of Ca2+ transients to 68 ± 13 % (n = 5, P < 0.001) compared to the amplitude of Ca2+ transients in untreated cells at 3 Hz of stimulation.
Figure 3. The effect of Ru360 on the cytosolic Ca2+ transients measured with fura-2 at different frequencies of stimulation.
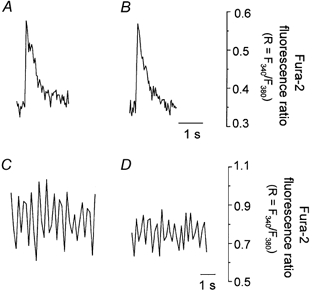
Ca2+ transients from an intact cardiac cell electrically stimulated at 0.1 Hz: A, control; B, after incubation in Ru360 (10 μM). Ca2+ transients from an intact cardiac cell electrically stimulated at 3.0 Hz: C, control; D, after incubation in Ru360 (10 μm).
The effect of mitochondrial Ca2+ uniporter blockade upon Ca2+ transients was further verified by treating the cardiac cells with CCCP (5 μm) and oligomycin (5 μm), to remove the voltage gradient for Ca2+ influx through Ca2+ uniporter without ATP depletion. As shown in Fig. 4, the transients obtained at the frequency of 0.1 Hz were similar in control (Fig. 4A) and CCCP-treated cells (Fig. 4B). However, the amplitude of Ca2+ transients in CCCP- and oligomycin-treated cells was significantly reduced upon stimulation of the cell at a frequency of 3.0 Hz (Fig. 4C, control; and Fig. 4D, CCCP treated). Incubation of cardiac cells with CCCP and oligomycin reduced the amplitude of Ca2+ transients to 60 ± 12 % (n = 5, P < 0.001), compared to the amplitude of Ca2+ transients in untreated cells at 3 Hz stimulation.
Figure 4. The effect of CCCP on the cytosolic Ca2+ transients measured with fura-2 at different frequencies of stimulation.
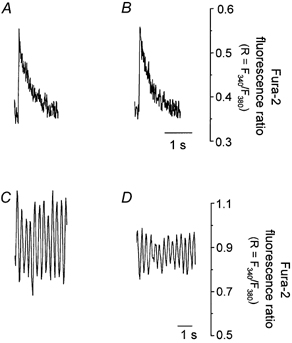
Ca2+ transients from an intact cardiac cell electrically stimulated at 0.1 Hz: A, control; B, after incubation in CCCP (5 μm along with 5 μm oligomycin). Ca2+ transients from an intact cardiac cell electrically stimulated at 3.0 Hz: C, control; D, after incubation in CCCP (5 μm along with 5 μm oligomycin).
It is possible that blocking mitochondrial Ca2+ uptake decreases the clearance of cytosolic Ca2+ near the L-type Ca2+ channels, especially at high frequencies of stimulation. This would in turn increase the inactivation of the L-type Ca2+ channels leading to a decrease in Ca2+ transients. To test this hypothesis, we recorded ICa with the voltage-clamp technique (Hamill et al. 1981). Again, the first set of experiments was carried out in skinned rhod-2-loaded myocytes to show that the intracellular electrode solution does not affect the ability of mitochondria to accumulate Ca2+ and RR and CCCP do not affect ICa directly. Figure 5 shows that in rhod-2-loaded chemically skinned cardiac cells, caffeine-induced increases in the mitochondrial Ca2+ were similar in a cell suspended in either normal Na+- and K+-containing buffer (Fig. 5A) or patch pipette solution containing 120 mm Cs+ and 20 mm TEACl (Fig. 5B, 98 ± 9 %, n = 3, P > 0.05). These data show that the electrode solution does not alter the ability of SR to release Ca2+ and mitochondria to sequester Ca2+.
Figure 5. The effect of internal electrode solution upon caffeine-induced [Ca2+]m increase.
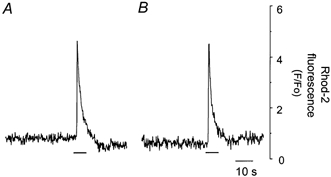
Rhod-2-loaded and chemically skinned cardiac cells were suspended in either normal Hepes buffered solution (A), or internal electrode solution (B). Horizontal bars indicate time of caffeine application (10 mm). The caffeine-induced [Ca2+]m increase was similar in cardiac cells suspended in control or internal electrode solution.
We then studied the effects of RR and CCCP on ICa.ICa was recorded during 100 ms step depolarizations at a frequency of 1 Hz during pulses from −35 mV to +95 mV in 10 mV steps from the holding potential of −40 mV. Figure 6A shows records of ICa obtained under control conditions during step depolarizations to −15 mV (○) and to +5 mV (•). The time course of decay of ICa was faster when the peak current was larger, as expected by a Ca2+-dependent inactivation process. This was further confirmed by the experiments showing that the inactivation of Ca2+ channels was greatly reduced when Ba2+ was used as charge carrier. In four separate experiments, the ratio between the Ba2+ current at the end of a 45 ms depolarizing pulse to +5 mV, relative to its peak amplitude, averaged 0.99 ± 0.01 (n = 4), indicating very little inactivation. This is consistent with previous reports describing that entry of Ba2+ through calcium channels fails to trigger the release of Ca2+ from SR to induce Ca2+ channel inactivation (Nabauer et al. 1989) even though Ba2+ might have some effects on Ca2+ transport mechanisms in mitochondria or SR (Akerman et al. 1977). In the presence of RR or CCCP (plus oligomycin, 5 μm), we found that the relationship between the time course of decay of ICa and the peak amplitude of the current was similar to that in control conditions (Fig. 6B and C). The decay phase of the currents at each potential was fitted to a single exponential and the corresponding time constant was plotted as a function of the peak amplitude of ICa. Figure 6D (control), E (RR) and F (CCCP) summarizes data from these experiments (n = 6). As the current amplitude increased (x-axis), the time constant of decay decreased (y-axis) with slopes (ms (pA pF−1)−1) of −1.98 (○), −1.73 (▿) and −1.63 (□), indicating no major differences in the rate of inactivation of ICa by these compounds, at all voltages tested. Since channel inactivation is dependent on [Ca2+]c, these results suggest that Ca2+ release from SR is not affected under these experimental conditions. Furthermore, they also suggest that RR or CCCP does not have a direct action on the L-type Ca2+ channel itself. In agreement with this conclusion, no changes in the amplitude of ICa were observed when the currents were recorded at step potentials to +5 mV in cells treated with RR or CCCP and stimulated at a frequency of 0.1 Hz. Thus, peak ICa averaged (pA pF−1) −4.39 ± 0.56 (n = 7) in control experiments, −4.94 ± 0.51 (n = 14) in RR-incubated cells, and −4.45 ± 0.72 (n = 9) in CCCP-treated cells.
Figure 6. Blockade of mitochondrial uniporter and ICa.
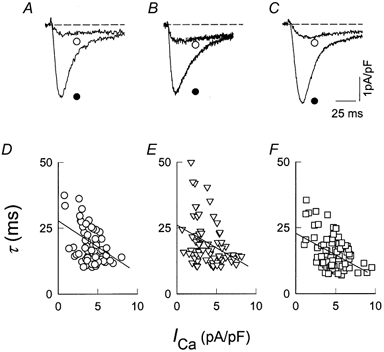
ICa was recorded in control conditions (A), in the presence of RR (10 μm) (B), and in the presence of CCCP (5 μm) plus oligomycin (5 μm) (C). Currents were activated by stepping the membrane potential from a holding potential of −40 mV to −15 mV (○) and to +5 mV (•). D–F, the relation between the time constant of decay and the amplitude of ICa under control conditions (○, n = 14), in RR- (▿, n = 18) and in CCCP-incubated (□, n = 28) cells. Straight lines have slopes (ms (pA pF−1)−1) of −1.98 in D, −1.73 in E and −1.63 in F.
We then studied the effect of RR and CCCP on the recovery from inactivation of ICa, elicited with pulse frequencies higher than 1 Hz. Figure 7A shows superimposed records of ICa from a control experiment. To evaluate the recovery from inactivation, currents were elicited by the double-pulse protocol shown in Fig. 7C in which the interval separating two similar pulses was progressively increased from 100 to 1000 ms (Brehm et al. 1980). The first pulse elicited a maximal ICa that decayed during the pulse, as shown previously. The amplitude of ICa during the second pulse was critically dependent on the duration of the interval elapsed since the end of the first pulse. When the interval was short, the current was small and decayed more slowly. In the presence of RR, currents showed a decreased recovery from inactivation at every given interval as shown in Fig. 7B, especially after short intervals. A similar effect was observed with CCCP (with oligomycin added). Figure 7D illustrates the recovery of ICa as a function of the interval between pulses, in control experiments (○), and in the presence of RR (•) or CCCP (▪). The recovery from inactivation was delayed by approximately 100 ms after mitochondrial Ca2+ influx was impaired in either way. Using the same data, the fraction of inactivated channels was plotted in a semilogarithmic scale against pulse interval (Fig. 7E). The graph indicates that recovery from inactivation followed a single exponential with time constants of 300 ms (○, control), or 540 ms (•, RR) and 654 ms (▪, CCCP). Therefore, inhibition of mitochondrial Ca2+ uptake lengthened the time constant by as much as 50 %. The fact that only the recovery from inactivation is altered by inhibition of mitochondrial Ca2+ uptake while the time constants of the Ca2+ current itself are unaffected suggests that clearance of focal Ca2+ by mitochondria proceeds more slowly than the activation and inactivation of the channels during one single pulse.
Figure 7. The effect of mitochondrial uniporter inhibition on the recovery of ICa from inactivation.
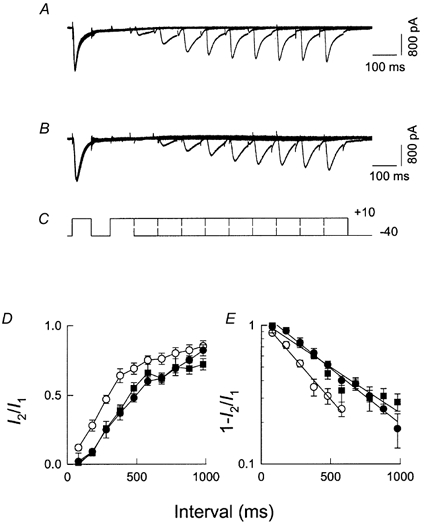
ICa was produced by pairs of identical pulses delivered at intervals varying from 100 to 1000 ms. The holding potential was −40 mV and the test potential was +10 mV. Families of ICa produced by each paired pulse were superimposed to show the time-dependent recovery from inactivation. A, control; B, a separate experiment in a cell incubated in RR (10 μm); C, the pulse protocol. D, the amplitude of ICa during the second pulse (I2) relative to its value during the first pulse (I1), as a function of the time interval between pulses, under control conditions (○), in cells incubated with RR (10 μm) (•), and in cells incubated in (10 μm) CCCP (▪). Each point is the mean (±s.e.m.) of at least four determinations. E, fraction of inactivated channels plotted semi-logarithmically against pulse interval from the same experiments as in D. The time constants for recovery from inactivation are 300 ms (○, control), 540 ms (•, RR) and 654 ms (▪, CCCP).
DISCUSSION
Ultrastructural studies (Sharma et al. 2000) have shown that Ca2+ release units in cardiomyocytes are preferentially located in close proximity to mitochondria. The nearest distance between them is in the 37–270 nm range. At such a distance, mitochondria are exposed to a transient Ca2+ elevation in the tens of micromolar range during Ca2+ release from SR/ER (Smith & Augustine, 1988; Neher, 1998). Since Ca2+ influx through the uniporter is half-maximally activated when free Ca2+ reaches values in that range (Gunter et al. 1994), mitochondria are expected to take up Ca2+ during Ca2+ transients associated with action potentials. This focal Ca2+ uptake by mitochondria in intact cardiomyocytes is supported by recent experimental evidence showing that mitochondria are depolarized by spontaneous Ca2+ release from SR (Ca2+ sparks) (Duchen et al. 1998).
At full velocity, mitochondria are capable of lowering free cytosolic Ca2+ at a rate of 4.6 μm s−1 in chromaffin cells, where mitochondria account for 6 % of the cell volume (Babcock et al. 1997). These numbers are expected to be higher in rat cardiomyocytes where the volume occupied by mitochondria is 35 % (Sommer & Johnson, 1979). The distance between mitochondria and the centre of T-tubules averages 145 nm and the distance between nearest SR T tubule junctions and mitochondria averages 37 nm (Sharma et al. 2000). This relatively short distance makes it feasible for the mitochondria to reduce [Ca2+]c, mostly due to Ca2+ release from SR, near the L-type Ca2+ channel, and thereby reduces Ca2+-dependent inactivation. The notion that mitochondrial buffering of Ca2+ close to the mouth of the channel could regulate channel inactivation has also been observed in other preparations (Budd & Nicholls, 1996; Hoth et al. 1997). This spatial characteristic of mitochondrial Ca2+ signalling, although complex, could be quite critical in determining the functional diversities that exist intrinsically in different cell types (Duchen, 1999).
Recently, it has been described that the blockade of the mitochondrial Ca2+ uptake mechanism in chromaffin cells causes a reduction in the amplitude of Ca2+ currents during depolarizing steps through L and other types of Ca2+ channels (Hernandez-Guijo et al. 2001). This reduction is due to inactivation of the channels by a Ca2+-dependent mechanism. The difference between these results and ours can be explained considering the fact that in chromaffin cells, mitochondria play a major role buffering the bulk cytosolic Ca2+ concentration. Thus, CCCP causes a distinct increase of cytosolic Ca2+ in non-stimulated cells (Babcock et al. 1997; Hernandez-Guijo et al. 2001). This increase in the levels of Ca2+ leads to inactivation of the channels (Hernandez-Guijo et al. 2001). This role of mitochondria holds for chromaffin cells and neurons but not for muscle cells with a highly elaborated sarcoplasmic reticulum.
Our results are consistent with the picture that at high frequencies, mitochondria play a significant role in buffering [Ca2+]c near L-type channels leading to a lesser degree of channel inactivation. From this perspective, mitochondria could be extremely important in ensuring the ability of the heart to contract effectively during rate increases. When the uniporter is blocked, the Ca2+ buffering capacity of mitochondria is compromised and so is EC coupling, because of the decrease in the recovery from inactivation of the L-type channel. It is important to point out that the effect of RR on Ca2+ transients might conceivably be explained by a frequency-dependent blocking effect of RR on calcium-induced calcium release (CICR) directly. This is, however, unlikely because if RR is blocking CICR at higher frequencies, then the local [Ca2+]c should decrease near the L-type channel and this would in turn inactivate the L-type channel less. This is opposite to what we observe in our voltage clamp experiments.
The use of RR as a specific blocker for the mitochondrial Ca2+ uniporter in intact cells is far from ideal (Duchen, 1992). Its effectiveness depends on the cell types used, time of exposure and concentration. Fortunately, several lines of evidence have shown that the use of RR in cardiac muscle cells is less problematic. In the perfused rat heart, RR (3.2 μm for 5 min) inhibits the positive inotropic agent-induced activation of pyruvate dehydrogenase, a Ca2+-dependent intramitochondrial enzyme, consistent with the idea that RR blocks the mitochondrial Ca2+ influx (McCormack & England, 1983). Similarly, in isolated cardiac myocytes, RR (12 μm for 13 min) prevents activation of pyruvate dehydrogenase by high KCl solution (Hansford, 1987). In addition, RR does not attenuate the KCl-induced increase in [Ca2+]c, consistent with the idea that RR does not have an effect on the voltage-gated Ca2+ channels in the plasma membrane under this condition. It has been shown that RR at a concentration of 5 μm completely inhibits rat heart myocyte contraction at a high stimulation rate of 6 Hz (Griffiths, 2000). However, this study also reported that RR at low concentrations (0.5–1 μm) inhibited mitochondrial Ca2+ uptake in this preparation. The author concluded that the inhibition of [Ca2+]m uptake was secondary to a reduction in [Ca2+]c as a result of decreased Ca2+ release from SR, a conclusion supported by the ability of RR to inhibit cell shortening. However, in our studies as shown in Fig. 1C–F and 2A and B, RR up to 10 μm concentration had no inhibitory effect upon the capability of SR to release Ca2+ in response to caffeine or electrical stimulation at 0.1 Hz. Our experimental results are similar to that reported in a recent study showing that RR (10 μm for 20 min) inhibits electrically evoked mitochondrial rhod-2 fluorescence transients (1 Hz) but not cytosolic fluo-3 in rabbit cardiac myocytes (Trollinger et al. 2000). Our experimental results appear to be consistent with these studies showing the specificity of RR. In addition, similar results on Ca2+ transients were obtained with 10 μm Ru360, a concentration that was used to inhibit mitochondrial Ca2+ uptake without causing side effects on the other Ca2+ transport mechanisms in intact cardiac myocytes (Matlib et al. 1998).
Although it is expected that cardiac activity would lead to an increase in mitochondrial Ca2+, this does not necessarily imply the presence of Ca2+ oscillations inside the mitochondria on a beat-to-beat basis. It remains unclear whether intramitochondrial Ca2+ can follow the cytosolic oscillations of Ca2+ during cardiac activity, and there is evidence for and against this possibility (Hüser et al. 2000; Trollinger et al. 2000). The presence of oscillations would depend not only on the Ca2+ influx but also on the kinetics of the Ca2+ efflux mechanism in heart mitochondria.
Changes in the physical architecture between L-type channels and SR Ca2+ release units have been shown to play a critical role in the reduction of contractile force under pathological conditions such as hypertrophy and failing heart (Gómez et al. 1997). The role of mitochondria in EC coupling that we propose here depends on their apposition to this refined architecture. It is conceivable that this structural integrity might also be altered in these pathological conditions so that the ability of mitochondria to regulate Ca2+ inactivation is compromised contributing to severe contractile dysfunction or heart failure.
Acknowledgments
This work was supported by NIH grants HL-33333 and HL-56782, and American Heart Association/New York State Affiliate Grant-in-aid 0050839T. K.C.Y. is a pre-doctoral fellow of the American Heart Association/New York State Affiliate grant 0010078T. We thank D. Dimanno for technical assistance and Drs T. Begenisich, R. Dirksen, D. Giovannucci and R. Gross and everyone in Dr Sheu's laboratory for helpful discussion of the manuscript.
References
- Akerman KE, Wikstrom MK, Saris NE. Effect of inhibitors on the sigmoidicity of the calcium ion transport kinetics in rat liver mitochondria. Biochimica et Biophysica Acta. 1977;464:287–294. doi: 10.1016/0005-2736(77)90004-9. [DOI] [PubMed] [Google Scholar]
- Babcock DF, Herrington J, Goodwin PC, Park YB, Hille B. Mitochondrial participation in the intracellular Ca2+ network. Journal of Cell Biology. 1997;136:833–844. doi: 10.1083/jcb.136.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock DF, Hille B. Mitochondrial oversight of cellular Ca2+ signaling. Current Opinion in Neurobiology. 1998;8:398–404. doi: 10.1016/s0959-4388(98)80067-6. [DOI] [PubMed] [Google Scholar]
- Bassani RA, Bassani JW, Bers DM. Relaxation in ferret ventricular myocytes: unusual interplay among calcium transport systems. Journal of Physiology. 1994;476:295–308. doi: 10.1113/jphysiol.1994.sp020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Dordrecht, The Netherlands: Kluwer Academic Press; 1991. p. 258. [Google Scholar]
- Beuckelmann DJ, Wier WG. Mechanism of release of calcium from sarcoplasmic reticulum of guinea-pig cardiac cells. Journal of Physiology. 1988;405:233–255. doi: 10.1113/jphysiol.1988.sp017331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm P, Eckert R, Tillotson D. Calcium-mediated inactivation of calcium current in Paramecium. Journal of Physiology. 1980;306:193–203. doi: 10.1113/jphysiol.1980.sp013391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. Journal of Neurochemistry. 1996;66:403–411. doi: 10.1046/j.1471-4159.1996.66010403.x. [DOI] [PubMed] [Google Scholar]
- Chad JE, Eckert R. Calcium domains associated with individual channels can account for anomalous voltage relations of Ca2+-dependent responses. Biophysical Journal. 1984;45:993–999. doi: 10.1016/S0006-3495(84)84244-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Ca2+-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochemical Journal. 1992;283:41–50. doi: 10.1042/bj2830041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Leyssens A, Crompton M. Transient mitochondrial depolarizations reflect focal sarcoplasmic reticular calcium release in single rat cardiomyocytes. Journal of Cell Biology. 1998;142:975–988. doi: 10.1083/jcb.142.4.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. Journal of Physiology. 1999;516:1–17. doi: 10.1111/j.1469-7793.1999.001aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner DA, Trafford AW, Diaz ME, Overend CL, O'neill SC. The control of Ca2+ release from the cardiac sarcoplasmic reticulum: regulation versus autoregulation. Cardiovascular Research. 1998;38:589–604. doi: 10.1016/s0008-6363(98)00062-5. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. American Journal of Physiology. 1983;245:1–14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Calculator programs for computing the compositions of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. Journal of Physiology. 1979;75:463–505. [PubMed] [Google Scholar]
- Gómez AM, Benitah JP, Henzel D, Vinet A, Lorente P, Delgado C. Modulation of electrical heterogeneity by compensated hypertrophy in rat left ventricle. American Journal of Physiology. 1997;272:H1078–1086. doi: 10.1152/ajpheart.1997.272.3.H1078. [DOI] [PubMed] [Google Scholar]
- Griffiths EJ. Use of ruthenium red as an inhibitor of mitochondrial Ca2+ uptake in single rat cardiomyocytes. FEBS Letters. 2000;486:257–260. doi: 10.1016/s0014-5793(00)02268-7. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gunter TE, Gunter KK, Sheu S-S, Gavin CE. Mitochondrial calcium transport: physiological and pathological relevance. American Journal of Physiology. 1994;267:C313–339. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- Hajnóczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hansford RG. Relation between cytosolic free Ca2+ concentration and the control of pyruvate dehydrogenase in isolated cardiac myocytes. Biochemical Journal. 1987;241:145–151. doi: 10.1042/bj2410145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernándej-Guijo JM, Maneu-Flores VE, Ruiz-Nuño A, Villarroya M, García AG, Gandía L. Calcium-dependent inhibition of L, N, and P/Q Ca2+ channels in chromaffin cells: role of mitochondria. Journal of Neuroscience. 2001;21:2553–2560. doi: 10.1523/JNEUROSCI.21-08-02553.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington J, Park YB, Babcock DF, Hille B. Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron. 1996;16:219–228. doi: 10.1016/s0896-6273(00)80038-0. [DOI] [PubMed] [Google Scholar]
- Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. Journal of Cell Biology. 1997;137:633–648. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüser J, Blatter LA, Sheu S-S. Mitochondrial calcium in heart cells: beat-to-beat oscillations or slow integration of cytosolic transients? Journal of Bioenergetics and Biomembranes. 2000;32:27–33. doi: 10.1023/a:1005556227425. [DOI] [PubMed] [Google Scholar]
- Imredy JP, Yue DT. Mechanism of Ca2+-sensitive inactivation of L-type Ca2+ channels. Neuron. 1994;12:1301–1318. doi: 10.1016/0896-6273(94)90446-4. [DOI] [PubMed] [Google Scholar]
- Jou M-J, Peng T-I, Sheu S-S. Histamine induces oscillations of mitochondrial free Ca2+ concentration in single rat brain astrocytes. Journal of Physiology. 1996;497:299–308. doi: 10.1113/jphysiol.1996.sp021769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass RS, Sanguinetti MC. Inactivation of calcium channel current in the calf cardiac Purkinje fiber. Evidence for voltage- and calcium-mediated mechanisms. Journal of General Physiology. 1984;84:705–726. doi: 10.1085/jgp.84.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Marban E, Tsien RW. Inactivation of calcium channels in mammalian heart cells: joint dependence on membrane potential and intracellular calcium. Journal of Physiology. 1985;364:395–411. doi: 10.1113/jphysiol.1985.sp015752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JG, England PJ. Ruthenium red inhibits the activation of pyruvate dehydrogenase caused by positive inotropic agents in the perfused rat heart. Biochemical Journal. 1983;214:581–585. doi: 10.1042/bj2140581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlib MA, Zhou Z, Knight S, Ahmed S, Choi KM, Krause-Bauer J, Phillips R, Altschuld R, Katsube Y, Sperelakis N, Bers DM. Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. Journal of Biological Chemistry. 1998;273:10223–10231. doi: 10.1074/jbc.273.17.10223. [DOI] [PubMed] [Google Scholar]
- Nabauer M, Callewaert G, Cleemann L, Morad M. Regulation of calcium release is gated by calcium current, not gating charge, in cardiac myocytes. Science. 1989;244:800–803. doi: 10.1126/science.2543067. [DOI] [PubMed] [Google Scholar]
- Neary P, Steele DS, Orchard CH, Smith GL. Measurements of Ca2+ release from saponin-permeabilized single cardiac myocytes. Journal of Physiology. 1996;497:7P. [Google Scholar]
- Neher E. Vesicle pools and Ca2+ micro domains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Nieminen AL, Dawson TL, Gores GJ, Kawanishi T, Herman B, Lemasters JJ. Protection by acidotic pH and fructose against lethal injury to rat hepatocytes from mitochondrial inhibitors, ionophores and oxidant chemicals. Biochemical and Biophysical Research Communications. 1990;167:600–606. doi: 10.1016/0006-291x(90)92067-a. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Murgia M, Pozzan T. Micro domains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- Satomi A-A, Cleemann L, Morad M. Cross-signaling between L-type Ca2+ channels and ryanodine receptors in rat ventricular myocytes. Journal of General Physiology. 1996;108:435–454. doi: 10.1085/jgp.108.5.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma VK, Colecraft HM, Wang DX, Levey AL, Grigorenko EV, Yeh HH, Sheu S-S. Molecular and functional identification of m1 muscarinic acetylcholine receptors in rat ventricular myocytes. Circulation Research. 1996;79:86–93. doi: 10.1161/01.res.79.1.86. [DOI] [PubMed] [Google Scholar]
- Sharma VK, Ramesh V, Franzini-Armstrong C, Sheu S-S. Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. Journal of Bioenergetics and Biomembranes. 2000;32:97–104. doi: 10.1023/a:1005520714221. [DOI] [PubMed] [Google Scholar]
- Smith SJ, Augustine GJ. Calcium ions, active zones and synaptic transmitter release. Trends in Neurosciences. 1988;11:458–464. doi: 10.1016/0166-2236(88)90199-3. [DOI] [PubMed] [Google Scholar]
- Sommer JR, Johnson EA. The ultra structure of cardiac muscle. In: Berne RM, editor. Handbook of Physiology, section 2, The Cardiovascular System, The Heart. I. Washington, DC: American Physiological Society; 1979. pp. 113–186. [Google Scholar]
- Sun H, Leblanc N, Nattel S. Mechanisms of inactivation of L-type calcium channels in human atrial myocytes. American Journal of Physiology. 1997;272:H1625–1635. doi: 10.1152/ajpheart.1997.272.4.H1625. [DOI] [PubMed] [Google Scholar]
- Thayer SA, Miller RJ. Regulation of the intracellular free calcium concentration in single rat dorsal root ganglion neurons in vitro. Journal of Physiology. 1990;425:85–115. doi: 10.1113/jphysiol.1990.sp018094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trollinger DR, Cascio WE, Lemasters JJ. Mitochondrial calcium transients in adult rabbit cardiac myocytes: inhibition by ruthenium red and artifacts caused by lysosomal loading of Ca2+-indicating fluorophores. Biophysical Journal. 2000;79:39–50. doi: 10.1016/S0006-3495(00)76272-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Matlib MA, Bers DM. Cytosolic and mitochondrial Ca2+ signals in patch clamped mammalian ventricular myocytes. Journal of Physiology. 1998;507:379–403. doi: 10.1111/j.1469-7793.1998.379bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Nosek TM. Ruthenium red affects the contractile apparatus but not sarcoplasmic reticulum Ca2+ release of skinned papillary muscle. Pflügers Archiv. 1992;420:255–258. doi: 10.1007/BF00374455. [DOI] [PubMed] [Google Scholar]