

Abstract
Macropatch recording was used to study release of acetylcholine in the frog neuromuscular junction evoked by either direct local depolarization or by an action potential.
The quantal content was established by directly counting the released quanta. The time course of release was obtained by constructing synaptic delay histograms.
Perfusion of the neuromuscular junction with methoctramine, a selective M2/M4 muscarinic antagonist, increased the quantal content and slowed the exponential decay of the synaptic delay histograms. Addition of the agonist muscarine reversed these effects.
Addition of acetylcholinesterase prolonged the decay of the delay histogram, and muscarine reversed this effect.
Methoctramine slowed the rise time of the postsynaptic current produced by axon stimulation without affecting either the excitatory nerve terminal current or the presynaptic Ca2+ current.
These results show that presynaptic M2 muscarinic receptors are involved in the process which terminates evoked ACh release.
In fast synapses, at 20 °C, neurotransmitter release starts about 0.5 ms after presynaptic depolarization, and stops within 3–5 ms. This implies that the release machinery is ‘primed’, and lacks only a triggering signal. It is accepted that the action potential provides this signal by opening Ca2+ channels (reviewed in Silinsky, 1985; Augustine et al. 1987; Neher, 1998), but the molecular mechanisms involved in initiation and termination of evoked release are unknown. However, if entry and removal of Ca2+ ions control the initiation and termination of the release process, it is expected that the time course of release will display dependence on the kinetics of entry and removal of Ca2+. But treatments known to affect Ca2+ entry or its removal, while affecting the amount of neurotransmitter released, do not alter the time course of release (Datyner & Gage, 1980; Andreu & Barrett, 1980; Parnas et al. 1986; Hochner et al. 1991). These findings suggest that Ca2+ entry and removal, although essential for release, do not control its time course (Parnas & Parnas, 1999).
Recent data suggest that the action potential, in addition to the opening of Ca2+ channels, controls initiation and termination of release by a mechanism which involves presynaptic inhibitory autoreceptors. Thus, it has been shown that the muscarinic acetylcholine receptor (mAChR), specifically the M2 subtype, known to mediate feedback inhibition of acetylcholine (ACh) release (Allen & Brown, 1993; Bellingham & Berger, 1996; Slutsky et al. 1999), interacts with the core proteins of the exocytotic machinery (SNARE proteins and synaptotagmin) in a voltage-dependent manner (Linial et al. 1997). This interaction is strong at resting potential and weakens as depolarization increases. It has also been shown that only when the receptor is occupied by ACh does it interact with syntaxin (Ilouz et al. 1999). Indeed, when exogenous acetylcholinesterase (AChE) was added, the interaction with syntaxin became much weaker, and the interaction lost its voltage dependence (Ilouz et al. 1999). Finally, it has been shown that the M2 receptor undergoes a voltage-dependent affinity shift. It displays high affinity at resting potential, and assumes a low-affinity state upon membrane depolarization (Ilouz et al. 1999).
It has also been shown that evoked release of ACh is enhanced by mAChR antagonists (hyoscine, scopolamine or atropine) in guinea-pig ileum (Morita et al. 1982; Peteris & Ogren, 1988), rat urinary bladder (D'Agostino et al. 1986), and both rat and mouse phrenic nerve (reviewed by Wessler, 1989) and by methoctramine (a specific M2/M4 antagonist) at the frog neuromuscular junction (Slutsky et al. 1999). These results support the notion that at rest, the release machinery is under tonic block produced by the tonic ACh concentration resulting in high occupancy of the muscarinic autoreceptor.
Based on the above biochemical and physiological data, the following hypothesis for voltage-dependent control of neurotransmitter release was suggested (Parnas et al. 2000). At rest (resting potential and resting concentration of transmitter in the synaptic cleft), the release machinery is maintained in a tonically blocked state. This is because at resting potential the receptor is in its high affinity state, such that even the low tonic concentration of transmitter in the synaptic cleft suffices to keep a large proportion of the autoreceptors occupied. The occupied autoreceptors associate tightly with the exocytotic machinery, thus maintaining the release machinery in a blocked state. Upon membrane depolarization, this block is removed and release is initiated. This is due to depolarization rapidly shifting the M2 receptor into its low affinity state, with concomitant dissociation of the transmitter. The unoccupied receptor detaches from the exocytotic machinery, the free machinery interacts with the Ca2+ that had entered during the depolarizing pulse, and transmitter release commences. Upon membrane repolarization, the autoreceptor rapidly regains its high affinity state, and again binds transmitter. The occupied receptor reassociates with the exocytotic machinery, and the block of release is reinstated, and hence, release terminates.
To date, however, there has been no direct demonstration that the inhibitory autoreceptor plays a role in control of the time course of release. We demonstrate here that the presynaptic M2 receptor at the frog neuromuscular junction is indeed involved in control of the time course of release. Furthermore, we check one of the consequences of the hypothesis outlined above (Parnas et al. 2000). Specifically, the hypothesis predicts that after evoked release has commenced and the membrane potential has repolarized, retardation of rebinding of ACh to the M2 receptor should delay its reassociation with the exocytotic machinery. As a result, the decay phase of the time course of release should be prolonged, and both the quantal content and the rate of asynchronous (spontaneous) release should increase. In this paper, we present direct experimental support for this contention by showing that addition of either methoctramine or exogenous acetylcholinesterase (AChE) prolongs the decay of the time course of release, and increases both the quantal content and the rate of asynchronous release.
METHODS
Preparation and solutions
Frogs (Rana ridibunda) were killed by stunning and double pithing in accordance with institutional guidelines and Israel animal protection law. The cutaneous pectoris nerve-muscle preparation was isolated and it was held by small insect pins in a chamber with a Sylgard bottom (1.5 cm × 4 cm × 0.4 cm). For local depolarization (see below), and for construction of delay histograms, the temperature was controlled at 7 ± 1 °C by circulating (Gilson Minipuls 3) the fluid through a cooling device. In the experiments where release was evoked by an action potential, the temperature was kept at 20 ± 1 °C. The standard bathing solution contained (mm): NaCl, 116; KCl, 2; MgCl2, 1; CaCl2, 1; Tris, 5. The pH was adjusted to 7.4 by adding NaOH. For direct local depolarization of a small region of the terminal to different levels, 0.2 μm tetrodotoxin (TTX) was added to block sodium excitability (Dudel, 1981, 1993).
Recording and stimulation
Two modes of stimulation were employed. For local depolarization of a restricted region of the terminal and a concomitant recording of single quanta events, we used the macropatch technique (Dudel, 1981, 1993). To visualize the endplate, a long-distance objective lens (× 40) with a 1.8 mm working distance was used, requiring horizontal positioning of the macropatch electrode (Ravin et al. 1999). Macropatch electrodes with a long shaft were pulled on a DMZ-Universal puller (Zeitz-Instruments, Munich). The tip (∼8 μm) was slightly bent to allow positioning of the macropatch electrode over a branch of the endplate in the small space between the objective and the preparation. Pulse duration was 0.7 ms, pulse amplitude varied from −0.3 to −1.0 μA, stimulation frequency was 3 Hz.
For action potential-evoked release the nerve was stimulated with a suction electrode. The pulse duration was 0.2 ms, and the pulse amplitude was ∼20 % above threshold. The stimulation frequency was 1 Hz. To prevent contraction, 2 μmd-tubocurarine (dTC) was added. The nerve terminal current (ENTC) and the postsynaptic current (EPSC) were recorded with a macropatch electrode (Dudel, 1993).
Ca2+ current measurements
Ca2+ currents were measured with a macropatch electrode from a small region just below the electrode rim. dTC (2 μm) was added to avoid muscle contraction. In order to ensure that Ca2+ currents were measured as close as possible to a release site, the electrode was carefully guided along the terminal until ENTC and EPSC were detected (Fig. 1A). The dTC concentration was then increased to 10 μm to completely block the EPSC that would otherwise partially mask the measured presynaptic Ca2+ currents. Presynaptic Ca2+ currents were measured as described by Mallart & Brigant (1982). Briefly, 20 μm TTX was added to the macropatch electrode to inhibit sodium excitability below the electrode (Fig. 1B, continuous line). This allowed propagation of an action potential with a reduced or abolished inward sodium current in the region below, and in the vicinity of, the electrode. The potassium channel blockers tetraethylammonium chloride (TEA; 1 mm) and 3,4-diaminopyridine (3,4-DAP; 100 μm) were added to the bath solution to block the potassium current, leaving only Ca2+ and capacitative currents (Fig. 1B, dotted line). Addition of cadmium (50 μm) (Fig. 1B, dashed line) and subsequent subtraction revealed the Ca2+ current in isolation (Fig. 1C). The first outward current of the triphasic wave reflects the inward Ca2+ current produced by the invading action potential at the region proximal to the electrode (seen as an outward current at the site of recording). The inward current reflects Ca2+ current below the electrode, and the late outward current reflects the retrograde spread of the inward Ca2+ current produced at a region distal to the recording site.
Figure 1. Measurement of presynaptic Ca2+ currents at a release site on the frog nerve terminal.
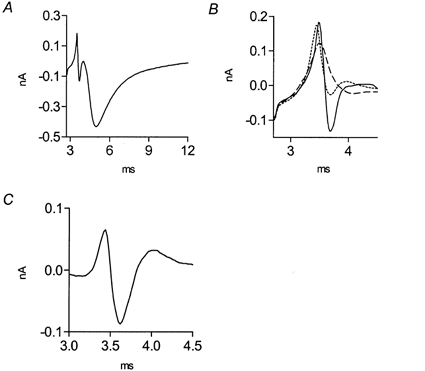
A, recording (average of 256 traces) of ENTCs and EPSCs at 1 Hz, 20 °C, in the presence of 2 μm dTC. B, enlarged ENTC from A after complete block of EPSC by 10 μm dTC: in the presence of 20 μm TTX in the electrode (continuous line), after addition of 1 mm TEA and 100 μm 3,4-DAP (dotted line), and after subsequent addition of 50 μm cadmium (dashed line). C, Ca2+ current obtained after subtraction of currents (dashed line subtracted from dotted line of B).
Quantal content, time course of release and rate of asynchronous release
At 7 °C, quanta appeared after the stimulus artefact and could easily be discerned. The total number of quanta (measured during 10 ms following the depolarizing pulse at 3 Hz) divided by the number of applied pulses yields the quantal content. Current traces were digitized using the NeuroData Instrument Corp. (Neuro-Corder DR-484) A/D converter at 50 kHz and transferred to a Pentium II computer (450 MHz) using the Labview (AT-MIO-16F-5, NI-DAQ 4.9.0 driver software) interface. Synaptic delay histograms, reflecting the time course of transmitter release (Katz & Miledi, 1965), were determined as described earlier (Ravin et al. 1999). Briefly, the delays from the beginning of the depolarizing pulse to the beginning of the quantal events were measured, and delay histograms were constructed (bin size was 0.1 ms). The delay histograms, from which asynchronous release was subtracted, were smoothed by nine-point averaging (GraphPad Prizm). Asynchronous release was measured starting 10 ms after the depolarizing pulse until the following pulse (320 ms).
The decay phases of the various delay histograms (y(t)) were fitted by either one or more exponentials, according to the following equation:
| (1) |
where τd1 and τd2 are the time constants of decay of the corresponding exponents and b is the constant achieved at t→∞.
We first tried to fit the decay phases of the various histograms by two exponentials. In so doing, we found either that τd1 was equal to τd2 (within standard deviation limits), or that, irrespective of the treatment (control, methoctramine or AChE), τd1 and τd2 differed by a factor of 50–100, so that the ‘slow’ time constant hardly contributed over the time range of the experimental measurements (up to 10 ms). The ‘fast’ time constants were found to be virtually the same as those found when the data were fitted by a single exponent. Accordingly, we fitted the decay phase with a single exponent.
The parameters, a, b and τd of the various single exponents were evaluated employing a standard least-squares-sum fit technique provided by the GraphPad Prizm software (same results were obtained using MATLAB). Goodness of fit was evaluated from the value of R2 (R2 > 0.9 indicated a significant fit).
Statistical evaluation
Significance was checked by Student's paired (the same experiment) and unpaired (different experiments) two-tail t tests. Results are given as mean ±s.d. throughout.
AChE preparation
AChE, in its water-soluble globular G2 dimeric form, was purified from Torpedo californica electric organ by affinity chromatography, after solubilization with phosphatidylinositol-specific phospholipase C (Futerman et al. 1985). Its specific activity was ca 3000 units (mg protein)−1, one unit corresponding to hydrolysis of 1 μmol min−1 of acetylthiocholine, assayed according to Ellman et al. (1961). When required, the AChE was inhibited by incubation with phospholine iodide (Storz Division of Wyeth/Ayerst Canada, Inc., Montreal, Canada) as follows. A stock solution of 100 mm phospholine in 0.9 % NaCl was added, to a final concentration of 1 mm, to a solution of 1.34 mg ml−1 AChE in buffered Ringer (pH 7.4). After 30 min at room temperature, > 99.5 % inhibition was achieved. Both the native and the inhibited AChE solutions were stored at 4 °C until used.
RESULTS
Methoctramine and exogenous AChE prolong the decay of the time course of ACh release
In the frog, feedback inhibition of ACh release is mediated by the M2 presynaptic muscarinic receptor (Slutsky et al. 1999). Hence, reduction of binding of ACh to the M2 receptor, following the onset of release, can be achieved by adding methoctramine, a selective M2/M4 antagonist. In so doing, it should be recalled that within the period of transmitter release (a few milliseconds after depolarization), the average concentration of ACh in the synaptic cleft rises briefly above its resting level. This transient elevation may displace the added antagonist from the M2 receptor. We therefore expected that rather high levels of methoctramine would be needed to compete with the elevated ACh; hence, we used 5 μm.
Figure 2 shows data obtained before and after addition of 5 μm methoctramine. The quantal content increased from 0.2 (control) to 0.5 after addition of methoctramine (Fig. 2A), and the time course of release was prolonged (Fig. 2B). The time constant of decay (τd) of the delay histogram increased from 0.55 ± 0.1 ms to 1.0 ± 0.1 ms (Fig. 2C). It can also be seen that the amplitude of the unitary current was reduced by ∼50 % (Fig. 2A, inset), indicating also a postsynaptic effect.
Figure 2. Methoctramine prolongs the time course and increases the quantal content of ACh release.
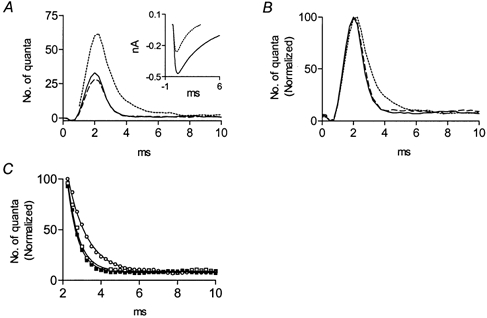
A, delay histograms (1000 pulses) in control (continuous line), 10 min after addition of 5 μm methoctramine (dotted line), and after subsequent addition of 30 μm muscarine (dashed line); 30 μm pirenzepine was present throughout. The control quantal content was 0.2; increased to 0.5 after application of methoctramine, and decreased to 0.18 after addition of muscarine. Inset, average amplitude of the unitary current (n = 100, without any selection) in control (continuous line), and 10 min after application of methoctramine (dotted line). B, peak normalization of the histograms presented in A. C, exponential fits of the decay phases of the histograms shown in B. Symbols correspond to experimental data and continuous lines correspond to the exponential fits. The time constant of decay in the control (▪) was 0.55 ± 0.1 ms (R2= 0.99); this increased to 1.0 ± 0.1 ms (R2= 0.99) 10 min after addition of methoctramine (□), and reversed to 0.6 ± 0.1 ms (R2= 0.99) after subsequent addition of muscarine (○).
We would expect that addition of an agonist should counterbalance the effect of an antagonist. We chose muscarine as the agonist because it is non-hydrolysable and has no postsynaptic effects at the frog neuromuscular junction (Slutsky et al. 1999). Figure 2 shows that 30 μm muscarine, in the presence of 30 μm pirenzepine (to block the M1 receptor that enhances release; Slutsky et al. 1999) abolished the presynaptic effects of methoctramine. The value of τd, which was 1.0 ± 0.1 ms in the presence of methoctramine, returned to the control value (0.6 ± 0.1 ms) after adding muscarine. As expected (Slutsky et al. 1999), the methoctramine-mediated enhancement of release was also abolished (Fig. 2A).
In five similar experiments, addition of methoctramine increased the quantal content and the rate of asynchronous release by (2.6 ± 0.3)-fold and (6.8 ± 1.2)-fold (P < 0.001), respectively. The unitary current declined (0.5 ± 0.1)-fold (P < 0.001) and τd was prolonged (1.9 ± 0.2)-fold (P < 0.0001), and reverted to (1.0 ± 0.2)-fold (P > 0.8) following subsequent addition of muscarine.
While we were encouraged by the observation that methoctramine prolonged the time course of release, we nevertheless sought another means to retard binding of ACh. This was because cholinergic antagonists can interact with multiple targets (as seen in Fig. 2A, inset), and are only specific for a given target over a limited concentration range. It is thus very difficult to select a concentration of antagonist sufficiently low to minimize postsynaptic effects, yet large enough to retard binding to the M2 receptors in the face of an elevated level of ACh, prevailing during the period of release.
An alternative way to retard binding of ACh should be by directly reducing its concentration in the synaptic cleft. This was achieved by perfusion of the potent synaptic enzyme, AChE.
Figure 3A shows that, as expected, the quantal content increased from 0.35 (control) to 0.56, 5 min following perfusion with 22 μg ml−1 AChE. Concomitantly, the decay of the time course of release was prolonged (Fig. 3B). τd increased from 0.7 ± 0.1 ms to 1.2 ± 0.2 ms (Fig. 3C). As for methoctramine, 30 μm muscarine, in the presence of 30 μm pirenzepine, reversed the prolongation of release caused by the exogenous AChE (Fig. 3B), with τd decreasing to 0.7 ± 0.1 ms (Fig. 3C). In addition, muscarine inhibited release (Fig. 3A).
Figure 3. AChE prolongs the time course and increases the quantal content of ACh release.
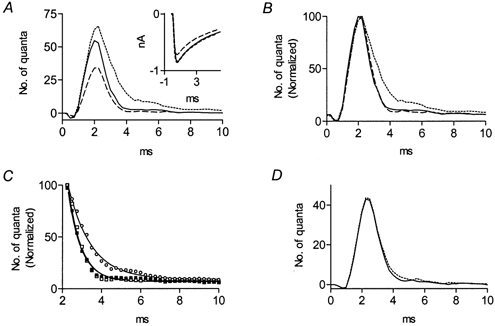
A, delay histograms (1000 pulses) in control (continuous line), 5 min after addition of 22 μg ml−1 AChE (dotted line), and after subsequent addition of 30 μm muscarine (dashed line); 30 μm pirenzepine was present throughout. In the control, quantal content was 0.35; quantal content increased to 0.56 after application of AChE, and decreased to 0.21 after subsequent addition of muscarine. Inset, average amplitude of the unitary current (n = 100) in control (continuous line), and after application of 22 μg ml−1 (dotted line) and 88 μg ml−1 (dashed line) AChE. B, peak normalization of the histograms presented in A. C, exponential fits of the decay phases of the histograms shown in B. Symbols correspond to experimental data and continuous lines correspond to the exponential fits. The time constant of decay in the control (▪) was 0.7 ± 0.1 ms (R2= 0.99); it increased to 1.2 ± 0.2 ms after addition of AChE (□), and decreased to 0.7 ± 0.1 ms (R2= 0.99) after subsequent addition of muscarine (○). D, catalytically inactive AChE had no effect on release. Delay histogram (2000 pulses) in control (continuous line) and 5 min after application of 22 μg ml−1 inactivated AChE (dotted line). In the control and after treatment with inactivated enzyme, quantal content was 0.17; the time constant of decay was 0.65 ± 0.15 ms (R2= 0.99) in the control and 0.7 ± 0.1 ms (R2= 0.99) following addition of inactive AChE.
AchE, 22 μg ml−1, did not affect the amplitude of the unitary current, but when 88 μg ml−1 AChE was superfused, the amplitude of the unitary current was reduced by 13 % (Fig. 3A, inset). These results indicate that the ACh-mediated presynaptic and postsynaptic processes exhibit different sensitivity to the added AChE.
Similar results to those in Fig. 3 (with 22 μg ml−1 AChE) were obtained in eight experiments. The quantal content increased by a factor of 1.5 ± 0.2 (P < 0.001). The frequency of asynchronous release increased (4.2 ± 1.3)-fold (P < 0.001), and the amplitude of the unitary current was not affected (P > 0.7). The τd was prolonged (1.8 ± 0.3)-fold (P < 0.001) following addition of AChE, and decreased back to 1.1 ± 0.3 (P > 0.6) upon subsequent addition of muscarine. With 88 μg ml−1 AChE the average unitary current declined (0.86 ± 0.03)-fold (n = 4, P < 0.001).
Catalytically inactive AChE had no effect
To establish that the effects produced by the exogenous AChE were indeed due to increased hydrolysis of ACh, we repeated the experiments of Fig. 3A, but utilizing AChE which had been irreversibly inactivated by the organophosphate inhibitor, phospholine (Maglothin & Wilson, 1974). The catalytically inactive AChE had no effect on the release parameters (Fig. 3D). Similar results were obtained in six such experiments. The ratios of the τd values, the quantal contents, the rates of asynchronous release and the amplitudes of the unitary current, before and after treatment with the inactive enzyme, were, respectively, 0.9 ± 0.3, 1.1 ± 0.3, 1.0 ± 0.2 and 1.0 ± 0.1 (n = 6, P > 0.6).
Methoctramine, by competing with ACh for binding to the M2 receptor, and AChE, by reducing the actual concentration of ACh, both produced similar effects on the time course of release. It is, nevertheless, possible that the prolongation of release by AChE is associated with the products of hydrolysis of ACh: acetate and choline. Accumulation of acetate could reduce the pH of the bathing solution, which could, in turn, affect the time course of ACh release. Increasing the concentration of the buffer (Tris) from 5 to 15 mm, had no effect. Thus, this possibility could be excluded. With regard to choline, it might be expected that choline would accumulate due to the faster hydrolysis of ACh, and hence, might affect the time course of ACh release. Figure 4 shows that choline affected ACh release differently from AChE. Choline (50 μm) did not prolong the time course of release (Fig. 4B), but reduced, as reported previously, both the quantal content (Glavinovic, 1987) by a factor of 0.6 (Fig. 4A), and the amplitude of the unitary current (Sterz et al. 1986; Glavinovic, 1987) by a factor of 0.87 (Fig. 4A, inset). It also reduced the rate of asynchronous release (by a factor of 0.6, not shown).
Figure 4. Choline (50 μm) does not affect the time course, but decreases both the quantal content of ACh release and the amplitude of the unitary current.
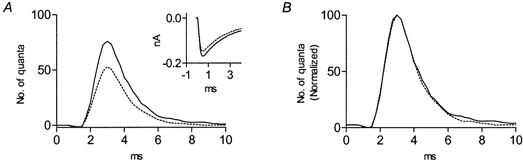
A, delay histograms obtained in control (continuous line) and after addition of 50 μm choline (dotted line). The quantal content for the control was 0.37 and it was reduced to 0.23 in the presence of choline. Inset, average amplitude of the unitary current (n = 100) in control (continuous line) and after application of 50 μm choline (dotted line). B, peak normalization of the delay histograms shown in A.
On average, choline (50 μm) did not affect the time course of ACh release (n = 6, P > 0.8), but reduced the quantal content, the rate of asynchronous release and the amplitude of the unitary current by factors of 0.6 ± 0.2, 0.5 ± 0.1 and 0.84 ± 0.05, respectively (n = 6, P < 0.001).
Methoctramine affects the time course of action potential-evoked release
We tested whether methoctramine also prolongs the time course of ACh release evoked by the natural stimulus, the action potential, under physiological conditions of high quantal content. Under such conditions the time course of release must be inferred, as was done by others (Schneggenburger & Neher, 2000), from the postsynaptic current (EPSC). Figure 5A shows the action potential-evoked nerve terminal current (ENTC) and the postsynaptic current (EPSC) in the control (continuous line) and after addition of 5 μm methoctramine (dotted line). Methoctramine did not affect the ENTC (Fig. 5A and B, enlarged), but prolonged the rise time of the EPSC (reflecting the time course of transmitter release; Aumann & Parnas, 1991), τrise, defined as t[90 % of peak EPSC]−t[10 % of peak EPSC] (Franke et al. 1991), from 0.39 to 0.56 ms (see normalized currents in Fig. 5C). Methoctramine also decreased the amplitude of the EPSC (Fig. 5A) by a factor of 0.7. This reduction can be accounted for by the postsynaptic effect causing a reduction of the unitary current (see Fig. 2A inset).
Figure 5. Methoctramine slows the rise time of the action potential-evoked postsynaptic current.
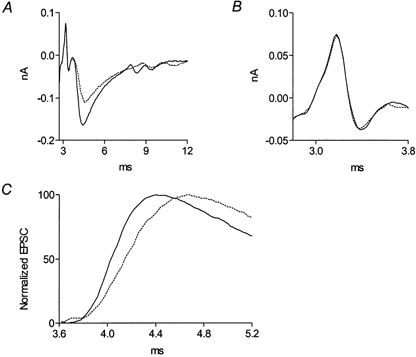
A, recording (average of 256 traces) of ENTCs and EPSCs in the control (continuous line) and after addition of 5 μm methoctramine (dotted line) at 1 Hz and 20 °C. B, enlarged ENTC of A. C, normalization of the EPSCs presented in A, taking the peak current as 100 %. τrise in the control was 0.39 ms, and increased 1.4-fold to 0.56 ms after addition of methoctramine.
On average, methoctramine increased τrise of the EPSC (1.3 ± 0.1)-fold (n = 4, P < 0.001), decreased the amplitude of the EPSC (0.7 ± 0.05)-fold (n = 4, P < 0.001), but did not affect the ENTC (n = 4, P > 0.7).
Methoctramine does not affect presynaptic Ca2+ currents
The prolongation of the time course of ACh release by methoctramine (Fig. 2 and Fig. 5) could have resulted from a change in the profile of the presynaptic Ca2+ current. To check this possibility, we measured Ca2+ currents from the nerve terminal before and after application of methoctramine. Figure 6 shows that 5 μm methoctramine (dotted line) did not affect the profile of the presynaptic Ca2+ current measured near a release site. Similar results were obtained in six experiments (P > 0.8).
Figure 6. Methoctramine does not affect the profile of the presynaptic Ca2+ currents.
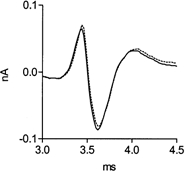
Control, continuous line; following addition of 5 μm methoctramine, dotted line (same experiment as in Fig. 1).
Taken together, the data presented in Figs 1–6 suggest that under physiological conditions the M2 receptor is involved in the mechanisms which terminate ACh release following membrane repolarization.
DISCUSSION
In this study, experiments were designed to test the possibility that presynaptic M2 receptors play a role in determining the time course of ACh release, and in particular, of its termination. We show that both methoctramine, which retards binding of ACh to the M2 receptor, and exogenous AChE, which lowers the concentration of ACh in the synaptic cleft, prolong the decay of the time course of release.
With respect to the action of methoctramine, it might be argued that both the decrease in the amplitude of the unitary current and the prolongation of the decay of the time course of release result from a postsynaptic effect of methoctramine. For example methoctramine might serve as an open channel blocker. This possibility seems unlikely since prolongation of the time course of release was demonstrated in two ways: the decay of the synaptic delay histogram was prolonged, and the rise time of the EPSC was slowed. Synaptic delay histograms have no bearing on postsynaptic processes and reflect only the presynaptic process of release. With respect to the postsynaptic current, had methoctramine acted as an open channel blocker, it should have affected the decay phase of the postsynaptic current, but not its rising phase, in a complex manner (Dudel et al. 1999), as seen here. Also, the fact that two different agents, methoctramine and AChE, which act by different mechanisms, produced the same effect, slowing the decay phase of evoked release, and that in both cases muscarine cancelled the prolongation, makes it less likely that the effect is unspecific. Rather, it seems reasonable to deduce from the data that retardation of binding of ACh to the presynaptic M2 receptor is somehow involved in the prolongation of the time course of ACh release.
It should be asked, at this point, whether addition of M2 receptor agonists, or of AChE inhibitors, might be expected to shorten the time course of release. The answer is negative. This is because, at resting potential, the condition prevailing at the time of release, the M2 receptor is in its high affinity state (Ilouz et al. 1999); hence, the normal ACh concentration in the synaptic cleft is sufficient to ensure binding of ACh to the receptor.
What could be the mechanism by which activation of the M2 receptor modulates the decay of the time course of release? One possibility would be that addition of methoctramine, or of exogenous AChE, relieves the Ca2+ channels from a tonic block caused by the resting concentration of ACh, and as a result, more Ca2+ enters upon membrane depolarization. This possibility can be excluded because Fig. 6 shows that the prolongation of release was not accompanied by an effect of methoctramine on the amplitude or profile of the Ca2+ currents. The results here are thus compatible with the notion that both methoctramine and AChE modulate, in some way, the vesicle fusion machinery. The question is, how? One possibility would be that such a modulation occurs downstream of Ca2+ influx. For example the M2 receptor may be involved in modulation of the rate constants of Ca2+ binding to the Ca2+ sensor, or alternatively, it could affect the fusion kinetics after Ca2+ had already been bound.
Our results cannot rule out the above-mentioned or other possible mechanisms. However, the results we present are compatible with the theory presented in the Introduction. According to this theory, release terminates, upon membrane repolarization, due to binding of ACh to the M2 receptor and a consequent association of the bound receptor with the vesicle fusion machinery. Retardation of binding of ACh to the M2 receptor slows this association and, as a result, prolongs the decay of the time course of release.
Finally, we wish to emphasize that the hypothesis suggested and tested in the present study, concerning the role of presynaptic autoreceptors in determining the time course of release, may not be limited to the cholinergic system. The mechanism we propose may serve as a general mechanism for control of neurotransmitter release at fast releasing synapses, where the type of the autoreceptor will obviously be specific for the particular transmitter.
Acknowledgments
This work was supported by an SFB 391 grant from DFG, Germany. I. Parnas is grateful for ongoing support from the Goldie Anna Fund. I. Parnas is the Greenfield Professor of Neurobiology and I. Silman is the Bernstein-Mason Professor of Neurochemistry. We are grateful to Lilly Toker for preparing the AChE.
References
- Allen TG, Brown DA. M2 muscarinic receptor-mediated inhibition of the Ca2+ current in rat magnocellular cholinergic basal forebrain neurones. Journal of Physiology. 1993;466:173–189. [PMC free article] [PubMed] [Google Scholar]
- Andreu R, Barrett EF. Calcium dependence of evoked transmitter release at very low quantal contents at the frog neuromuscular junction. Journal of Physiology. 1980;308:79–97. doi: 10.1113/jphysiol.1980.sp013463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ, Charlton MP, Smith SJ. Calcium action in synaptic transmitter release. Annual Review of Neuroscience. 1987;10:633–693. doi: 10.1146/annurev.ne.10.030187.003221. [DOI] [PubMed] [Google Scholar]
- Aumann Y, Parnas H. Evaluation of the time course of neurotransmitter release from the measured PSC and MPSC. Bulletin of Mathematical Biology. 1991;53:537–555. doi: 10.1007/BF02458628. [DOI] [PubMed] [Google Scholar]
- Bellingham MC, Berger AJ. Presynaptic depression of excitatory synaptic inputs to rat hypoglossal motoneurons by muscarinic M2 receptors. Journal of Neurophysiology. 1996;76:3758–3770. doi: 10.1152/jn.1996.76.6.3758. [DOI] [PubMed] [Google Scholar]
- D'Agostino G, Kilbinger H, Chiari MC, Grana F. Presynaptic inhibitory muscarinic receptors modulating [3H]acetylcholine release in the rat urinary bladder. Journal of Pharmacology and Experimental Therapeutics. 1986;239:522–528. [PubMed] [Google Scholar]
- Datyner NB, Gage PW. Phasic secretion of acetylcholine at a mammalian neuromuscular junction. Journal of Physiology. 1980;303:299–314. doi: 10.1113/jphysiol.1980.sp013286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudel J. The effect of reduced calcium on quantal unit current and release at the crayfish neuromuscular junction. Pflügers Archiv. 1981;391:35–40. doi: 10.1007/BF00580691. [DOI] [PubMed] [Google Scholar]
- Dudel J. Inhibition of Ca2+ inflow at nerve terminals of frog muscle blocks facilitation while phasic transmitter release is still considerable. Pflügers Archiv. 1993;415:566–574. doi: 10.1007/BF02583507. [DOI] [PubMed] [Google Scholar]
- Dudel J, Schramm M, Franke C, Ratner E, Parnas H. Block of quantal end-plate currents of mouse muscle by physostigmine and procaine. Journal of Neurophysiolology. 1999;81:2386–2397. doi: 10.1152/jn.1999.81.5.2386. [DOI] [PubMed] [Google Scholar]
- Ellman GL, Courtney KD, Andres VJ, Featherstone RM. A new and rapid determination of acetylcholinesterase activity. Biochemical Pharmacology. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Franke C, Hatt H, Parnas H, Dudel J. Kinetic constants of the acetylcholine (ACh) receptor reaction deduced from the rise in open probability after steps in ACh concentration. Biophysical Journal. 1991;60:1008–1016. doi: 10.1016/S0006-3495(91)82138-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman AH, Low MG, Ackermann KE, Sherman WR, Silman I. Identification of covalently bound inositol in the hydrophobic membrane-anchoring domain of Torpedo acetylcholinesterase. Biochemical and Biophysical Research Communications. 1985;129:312–317. doi: 10.1016/0006-291x(85)91439-1. [DOI] [PubMed] [Google Scholar]
- Glavinovic MI. Synaptic depression in frog neuromuscular junction. Journal of Neurophysiology. 1987;58:230–246. doi: 10.1152/jn.1987.58.1.230. [DOI] [PubMed] [Google Scholar]
- Hochner B, Parnas H, Parnas I. Effects of intra-axonal injection of Ca2+ buffers on evoked release and on facilitation in the crayfish neuromuscular junction. Neuroscience Letters. 1991;125:215–218. doi: 10.1016/0304-3940(91)90032-o. [DOI] [PubMed] [Google Scholar]
- Ilouz N, Branski L, Pranis J, Parnas H, Linial M. Depolarization affects the binding properties of muscarinic acetylcholine receptors and their interaction with proteins of the exocytic apparatus. Journal of Biological Chemistry. 1999;274:29519–29528. doi: 10.1074/jbc.274.41.29519. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. The effect of reduced calcium on quantal unit current and release at the crayfish neuromuscular junction. Proceedings of the Royal Society B. 1965;161:483–495. doi: 10.1098/rspb.1965.0016. [DOI] [PubMed] [Google Scholar]
- Linial M, Ilouz N, Parnas H. Voltage-dependent interaction between the muscarinic ACh receptor and proteins of the exocytic machinery. Journal of Physiology. 1997;504:251–258. doi: 10.1111/j.1469-7793.1997.251be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglothin JA, Wilson IB. Equilibrium constants for the phosphorylation of acetylcholinesterase by some diethyl phosphorothiolates. Biochemistry. 1974;13:3520–3527. doi: 10.1021/bi00714a017. [DOI] [PubMed] [Google Scholar]
- Mallart A, Brigant JL. Electrical activity at motor nerve terminals of the mouse. Journal de Physiologie. 1982;78:407–411. [PubMed] [Google Scholar]
- Morita K, North RA, Tokimasa T. Muscarinic presynaptic inhibition of synaptic transmission in myenteric plexus of guinea-pig ileum. Journal of Physiology. 1982;333:141–149. doi: 10.1113/jphysiol.1982.sp014444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Parnas I, Parnas H. Perspectives: Different mechanisms control the amount and time course of neurotransmitter release. Journal of Physiology. 1999;517:629. doi: 10.1111/j.1469-7793.1999.0629s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnas H, Dudel J, Parnas I. Neurotransmitter release and its facilitation in crayfish. VII. Another voltage dependent process beside CA entry controls the time course of phasic release. Pflügers Archiv. 1986;406:121–130. doi: 10.1007/BF00586672. [DOI] [PubMed] [Google Scholar]
- Parnas H, Segel L, Dudel J, Parnas I. Autoreceptors, membrane potential and regulation of transmitter release. Trends in Neurosciences. 2000;23:60–68. doi: 10.1016/s0166-2236(99)01498-8. [DOI] [PubMed] [Google Scholar]
- Peteris A, Ogren VR. Interaction of forskolin with effect of atropine on [3H] acetylcholine secretion in guinea-pig ileum myenteric plexus. Journal of Physiology. 1988;395:441–453. doi: 10.1113/jphysiol.1988.sp016928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravin R, Parnas H, Spira ME, Parnas I. Partial uncoupling of neurotransmitter release from [Ca2+]i by membrane hyperpolarization. Journal of Neurophysiology. 1999;81:3044–3053. doi: 10.1152/jn.1999.81.6.3044. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- Silinsky EM. The biophysical pharmacology of calcium-dependent acetylcholine secretion. Pharmacological Reviews. 1985;37:81–132. [PubMed] [Google Scholar]
- Slutsky I, Parnas H, Parnas I. Presynaptic effects of muscarine on ACh release at the frog neuromuscular junction. Journal of Physiology. 1999;514:769–782. doi: 10.1111/j.1469-7793.1999.769ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterz R, Peper K, Simon J, Ebert JP, Edge M, Pagala M, Bradley RJ. Agonist and blocking effects of choline at the neuromuscular junction. Brain Research. 1986;385:99–114. doi: 10.1016/0006-8993(86)91551-9. [DOI] [PubMed] [Google Scholar]
- Wessler I. Control of transmitter release from the motor nerve by presynaptic nicotinic and muscarinic autoreceptors. Trends in Pharmacological Sciences. 1989;10:110–114. doi: 10.1016/0165-6147(89)90208-3. [DOI] [PubMed] [Google Scholar]