

Abstract
The effects that muscarinic receptor stimulation have on the cAMP-dependent regulation of L-type Ca2+ currents were studied in isolated guinea-pig ventricular myocytes using the whole-cell configuration of the patch-clamp technique.
The muscarinic agonist ACh inhibited the Ca2+ current stimulated by the β-adrenergic agonist isoprenaline (Iso), and washout of ACh revealed a stimulatory response that appeared as a transient rebound increase in the amplitude of the Ca2+ current. The ACh-induced stimulatory effect was not observed in the absence of Iso.
ACh-induced rebound stimulation was also observed in the presence of H2 histamine receptor activation and cholera toxin treatment, which like β-adrenergic receptor activation enhance adenylyl cyclase (AC) activity in a stimulatory G protein (Gs)-dependent manner. ACh-induced rebound stimulation was not observed in the presence of forskolin, which enhances AC activity in a Gs-independent manner.
Pertussis toxin (PTX) treatment blocked both the stimulatory and inhibitory effects of ACh. Intracellular dialysis with QEHA, a peptide that binds free G protein βγ subunits, selectively antagonized the stimulatory effect, leaving an enhanced inhibitory effect.
Evidence for the expression of AC4, an isoform of AC that can be stimulated by Gβγ but only in the presence of Gαs, was obtained by Western blot analysis of guinea-pig ventricular myocyte membrane preparations.
These results suggest that muscarinic receptor stimulation facilitates as well as inhibits cAMP-dependent regulation of the Ca2+ current and that the net response is a balance between these two actions. We suggest that the stimulatory effect is due to a direct activation of AC4 by the βγ subunits of a PTX-sensitive G protein.
Parasympathetic stimulation exerts a significant influence over the electrical and mechanical activity of the heart. These effects are mediated by the neurotransmitter ACh, which acts via muscarinic receptors that are intrinsic to all cardiac myocytes. In general, ACh exerts inhibitory effects on heart rate and contractility, and these effects can be mediated by both direct and indirect signalling mechanisms. Direct signalling mechanisms involve coupling of the M2 muscarinic receptor to G protein-activated, inwardly rectifying K+ channels, which are expressed mainly in the atrial and pacemaker cells of the heart. Indirect signalling mechanisms involve M2 muscarinic receptor-mediated modulation of cAMP-dependent β-adrenergic responses throughout the heart. At one time, the dogma was that muscarinic receptor stimulation simply inhibits or antagonizes β-adrenergic responses via M2 muscarinic receptor-dependent activation of the inhibitory G protein Gi, which then directly inhibits adenylyl cyclase (AC) activity (Hartzell, 1988). However, there is clear evidence that muscarinic receptor activation not only inhibits β-adrenergic responses, it also facilitates β-adrenergic responses. The stimulatory effects appear most obviously as rebound increases in heart rate and contractility that can be observed immediately following the termination of vagal stimulation or cessation of exposure to ACh (Hollenberg et al. 1965; Levy, 1971; Burke & Calaresu, 1972; Gilmour & Zipes, 1985).
In isolated cardiac myocytes, ACh has been shown to produce both inhibition and rebound stimulation of L-type Ca2+, cAMP-regulated Cl−, and pacemaker (If) currents (Wang & Lipsius, 1995, 1996; Zakharov & Harvey, 1997; Song et al. 1998). The coexistence of both inhibitory and stimulatory effects of ACh in the same cell suggests that the net response to muscarinic stimulation represents a balance between these opposing actions. Furthermore, ACh-induced rebound stimulatory effects can explain physiological phenomena such as post-vagal tachycardia (Wang & Lipsius, 1996), and they are also believed to play a role in triggering certain types of arrhythmogenic mechanisms (Wang et al. 1997; Song et al. 1998).
As in the case of indirect inhibitory effects, ACh appears to exert its stimulatory actions by modulating the cAMP-dependent responses of cardiac myocytes (Linden, 1987; Wang & Lipsius, 1995; Zakharov & Harvey, 1997). In cat atrial myocytes, the stimulatory response has been explained by the nitric oxide synthase (NOS)-dependent generation of nitric oxide (NO) and subsequent stimulation of soluble guanylyl cyclase activity (Wang et al. 1998). The resulting production of cGMP then causes a decrease in cAMP breakdown by inhibiting type III phosphodiesterase (PDE III) activity. However, we have demonstrated previously that NO and cGMP do not contribute significantly to the stimulatory effect of ACh in ventricular myocytes (Zakharov & Harvey, 1997; Belevych & Harvey, 2000). Therefore, the main objective of the present study was to investigate the possible molecular mechanism responsible for ACh-induced stimulatory effects in ventricular myocytes. A preliminary report of some of these results has been presented in abstract form (Belevych et al. 2001).
METHODS
Cell isolation
Single ventricular myocytes were isolated from adult Hartley guinea-pigs using the modification of a method described previously (Zakharov & Harvey, 1997). Briefly, guinea-pigs were anaesthetized by i.p. injection of pentobarbital (150 mg kg−1), in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by National Institutes of Health and approved by the Institutional Animal Care and Use Committee at Case Western Reserve University. Following this procedure, hearts were quickly excised and the coronary arteries were perfused via the aorta with physiological salt solution (PSS) containing (mm): NaCl 140, KCl 5.4, MgCl2 2.5, CaCl2 1.5, glucose 11 and Hepes 5.5 (pH 7.4). Hearts were initially perfused with calcium-containing PSS for 5 min. The solution was then switched to nominally calcium-free PSS for a further 5 min, after which time enough collagenase A (Boehringer Mannheim) was added to achieve a final concentration of ∼0.5 mg ml−1. After 20–35 min of digestion at 36 °C, the ventricles were removed and placed in a Kraft-Brühe solution containing (mm): potassium glutamate 110, KH2PO4 10, KCl 25, MgSO4 2, taurine 20, creatine 5, EGTA 0.5, glucose 20 and Hepes 5 (pH 7.4). The tissue was then minced, and single myocytes were obtained by filtering it through 100 μm nylon mesh. Cells were stored in PSS at room temperature and used on the day of isolation only.
Electrophysiological recording methods
Whole-cell Ca2+ currents were recorded using either the conventional whole-cell patch-clamp technique (Hamill et al. 1981) or the perforated-patch variation of that technique employing amphotericin B (Rae et al. 1991). Microelectrodes were pulled from borosilicate glass capillary tubing (Corning 7052, Garner Glass) and had resistances between 1 and 2 MΩ when filled with intracellular solution. For conventional whole-cell experiments, the intracellular solution used contained (mm): CsCl 130, TEA-Cl 20, EGTA 5, MgATP 5, TrisGTP 0.1 and Hepes (pH 7.2). For perforated-patch experiments, electrodes were filled with a solution containing (mm): CsCl 130, TEA-Cl 20, EGTA 10 and Hepes 5 (pH 7.2). Amphotericin B was first dissolved in DMSO (60 mg ml−1) with the aid of sonication. An aliquot of this stock solution was then added to the pipette solution to yield a final concentration of 0.2 mg ml−1, and then sonicated again. The control extracellular solution contained (mm): NaCl 140, CsCl 5.4, CaCl2 2.5, MgCl2 0.5, glucose 11 and Hepes 5.5 (pH 7.4). Currents due to K+ channel activity were eliminated by using potassium-free solution containing TEA+ and/or Cs+. Setting the Cl− equilibrium potential at 0 mV eliminated the time-independent, cAMP-regulated Cl− current from measurements made at that membrane potential. Myocytes were placed in a 0.5 ml chamber, into which control extracellular solution was introduced at a rate of ∼1 ml min−1. However, cells were exposed to different experimental solutions using a fast flow system, as described previously (Zakharov & Harvey, 1995). This method allows rapid (< 1 s) changes in the extracellular solutions bathing the myocyte. A 3 m potassium chloride-agar bridge was used to ground the bath. All experiments were conducted at 32 °C.
Currents were recorded using an Axopatch 200 voltage-clamp amplifier (Axon Instruments), low-pass filtered at 5 kHz, and sampled at 10 kHz using an IBM-compatible computer with a Digidata 1200 interface and pCLAMP software (Axon Instruments). The series resistance and cell membrane capacitance were compensated before and during experiments. In experiments where the perforated-patch technique was used, current recording did not begin until the uncompensated series resistance reached a level of < 10 MΩ. Visual observation of myocyte contractions associated with voltage-clamp depolarizations activating the Ca2+ current was used to verify that the perforated patch remained intact. The voltage-clamp protocol employed a holding potential of −80 mV. A 50 ms pre-pulse to −30 mV was used to inactivate Na+ channels. The time course of changes in the magnitude of the Ca2+ current was monitored by applying the pre-pulse followed by a 100 ms test pulse to 0 mV once every 5 s. The amplitude of the Ca2+ current was determined by measuring the absolute magnitude of the peak inward current during the step depolarization to 0 mV. All results are expressed as the mean ±s.e.m. of the results obtained from n number of cells. Statistical significance between two groups was defined by Student's t test, P values of < 0.05.
Immunoblotting
Guinea-pig hearts were excised and homogenized with a Polytron in ice-cold phosphate-buffered saline (PBS) containing 2.5 mm EDTA, 1 mm DTT, 0.1 mm phenylmethylsulphonyl fluoride, and 10 μg ml−1 each of aprotinin and leupeptin. The homogenates were filtered through cheesecloth and crude membranes were prepared by isolating the pellet and sedimenting it between 300 and 100 000 g. Proteins were separated by SDS-PAGE using 7.5 % polyacrylamide gels under reducing conditions. Proteins were then transferred to polyvinylidene difluoride membranes and probed with rat-specific antibody generated to the 20 carboxy-terminal amino acids of AC type IV (AC4, Santa Cruz Biotechnology) in the presence and absence of blocking peptide. Goat-antirabbit IgG coupled to horseradish peroxidase was utilized as the secondary antibody. The presence of immunoreactive protein was assessed by enhanced chemiluminescence.
Drugs and chemicals
Acetylcholine hydrochloride (ACh), R(-)-isoprenaline (+)-bitartrate (Iso), histamine hydrochloride (Sigma), NG-monomethyl-l-arginine (l-NMMA), cholera toxin (Calbiochem) and pertussis toxin (PTX, List Biochemical Laboratories) were prepared as aqueous stock solutions. Milrinone, bisindolylmaleimide I (BIM) and wortmannin (Calbiochem) were prepared as stock solutions in DMSO. Forskolin (Calbiochem) was prepared as a stock solution in polyethylene glycol. Atropine (Sigma) was prepared as stock solution in ethanol. All non-aqueous stock solutions were prepared so that final drug concentrations were achieved by at least a 1:1000 dilution. QEHA, a peptide representing the βγ-binding motif (residues 956–982 in the C2a region) of AC2, and SKEE, a peptide representing the cognate region of AC3, which does not bind Gβγ, were kindly provided by Dr Ravi Iyengar at the Mount Sinai School of Medicine in New York City. All other chemicals were obtained from Sigma. Ascorbic acid (50 μm) was added to all external solutions to prevent the oxidative degradation of Iso.
RESULTS
ACh facilitation of β-adrenergic responses
In cardiac myocytes, the L-type Ca2+ current is augmented by cAMP via activation of protein kinase A (PKA) and subsequent protein phosphorylation. This mechanism is typically associated with β-adrenergic receptor activation of AC via the stimulatory G protein Gs. Furthermore, this stimulatory effect of β-adrenergic receptor stimulation can be antagonized by the concurrent activation of muscarinic receptors. However, in atrial myocytes, exposure to and subsequent washout of the muscarinic receptor agonist ACh also produces a cAMP-mediated transient rebound stimulation of the L-type Ca2+ current, even in the absence of β-adrenergic stimulation (Wang & Lipsius, 1995). ACh has also been reported to cause rebound stimulation of the cAMP-regulated Cl− current in ventricular myocytes. Yet, to see this effect it was necessary to first prime the cells with a β-adrenergic receptor agonist such as Iso (Ono & Noma, 1994; Zakharov & Harvey, 1997). In fact, this ACh-induced stimulatory response could be observed in the presence of subthreshold concentrations of Iso, and increasing the concentration of Iso increased the magnitude of the response to ACh, but only up to the point where the magnitude of the stimulatory effect reached that of a maximally stimulating concentration of Iso alone (Zakharov & Harvey, 1997). This suggests that the ACh-induced stimulatory effect in ventricular myocytes is facilitated by β-adrenergic and/or cAMP-dependent responses. Consistent with this idea, ACh produces the same type of stimulatory effect on the L-type Ca2+ current in ventricular myocytes (Fig. 1).
Figure 1. Exposure to and subsequent washout of ACh produced transient rebound stimulation of the L-type Ca2+ current, but only in the presence of submaximally stimulating concentrations of isoprenaline (Iso).

A, time course of changes in amplitude of L-type Ca2+ current under control conditions (a), and during exposure to 0.001 μm Iso alone (b), 0.001 μm Iso plus 1 μm ACh, 0.001 μm Iso following washout of ACh (c) and 1 μm Iso alone (d). The magnitude of the ACh-induced rebound stimulatory effect (R) was calculated by normalizing the magnitude of the rebound response (c − b) to the magnitude of the response to a maximal stimulation concentration of Iso (d − a), according to the equation: R = 100(c − b)/(d − a). Inset, examples of L-type Ca2+ currents recorded under the conditions indicated in A. The dotted line above current traces represents the zero current level. Note: changes in holding current represent parallel regulation of the cAMP-dependent Cl− current. B, changes in the magnitude of the L-type Ca2+ current measured prior to (□) and during the peak of the response following 60–90 s exposure to 1 μm ACh, in the absence and in the presence of 0.0003, 0.001, 0.003 and 1 μm Iso (▪). Measurements were normalized to the magnitude of the response produced by 1 μm Iso alone. Differences between bars at each concentration of Iso represent the magnitude of net ACh-induced stimulatory effect (*P < 0.05; ***P < 0.001; ns, not significant P > 0.4).
Exposure to 1 nm Iso produced submaximal stimulation of the ventricular L-type Ca2+ current, and subsequent addition of 1 μm ACh antagonized this response. Furthermore, washout of ACh resulted in rapid reversal of the inhibitory effect, which then revealed the stimulatory response. This stimulatory response appeared as a transient increase in the amplitude of the Ca2+ current to a level well beyond that observed in the presence of Iso before exposure to ACh (Fig. 1A). The ACh-induced stimulatory effect was measured as the difference between the magnitude of the current at the peak of the rebound response and the magnitude of the current in the presence of Iso alone before exposure to ACh. The size of the stimulatory effect was then normalized to the magnitude of the response to a maximally simulating concentration of Iso (1 μm) observed in the same cell (Zakharov & Harvey, 1997; Belevych & Harvey, 2000). For this and subsequent experiments, cells were exposed to ACh for 60–90 s. We have demonstrated previously that this is sufficient time for the stimulatory response to ACh to reach a steady state (Zakharov & Harvey, 1997). Consistent with this observation, the rebound effect measured in the presence of 1 nm Iso following exposure to ACh for 3 min was 28 ± 7.8 % (n = 3) of that produced by 1 μm Iso alone. This is not significantly different from the size of the rebound effect measured in the presence of 1 nm Iso following exposure to ACh for 60–90 s (P > 0.5; see below).
The ability of ACh to produce a stimulatory response was clearly affected by the level of concurrent β-adrenergic stimulation (Fig. 1B). In the absence of Iso, there was no evidence for an ACh-induced stimulatory effect. The magnitude of the Ca2+ current observed following washout of 1 μm ACh was not significantly different from the magnitude of the current measured before exposure to ACh (P > 0.6). The actual difference was −0.75 ± 0.8 % (n = 10) of that produced by exposure to 1 μm Iso in the same cells. However, exposure to even subthreshold concentrations of Iso was sufficient to enable ACh-induced stimulatory responses. In the presence of 0.3 nm Iso, exposure to and subsequent washout of ACh produced a significant stimulatory response (P < 0.05). This is despite the fact that 0.3 nm Iso did not produce a stimulatory effect by itself. The magnitude of the ACh-induced stimulatory response observed in the presence of 0.3 nm Iso was 13 ± 4.8 % (n = 13) of that produced by 1 μm Iso in the same cells. Furthermore, increasing the level of β-adrenergic stimulation augmented the magnitude of the rebound response produced by ACh. In the presence of 1 nm Iso, the magnitude of the stimulatory response to 1 μm ACh was 31 ± 3.7 % (n = 23) of that produced by 1 μm Iso alone. In the presence of 3 nm Iso, the magnitude of the stimulatory response increased to 41 ± 6.2 % (n = 9) of that produced by 1 μm Iso. However, the magnitude of the total current observed following washout of 1 μm ACh in the presence of 3 nm Iso (101 ± 5.4 %) was not significantly different from that observed in the presence of 1 μm Iso alone (P > 0.8). Therefore, this measurement is likely to be an underestimate of the full stimulatory response, since ACh was not able to stimulate the current beyond the level produced by a maximally stimulating concentration of Iso alone. This is supported by the fact that ACh did not elicit a stimulatory response in the presence of 1 μm Iso. The magnitude of the current observed following washout of 1 μm ACh in the presence of 1 μm Iso was 102 ± 4.1 % (n = 6) of that measured in the presence of 1 μm Iso before exposure to ACh. The difference between the effects of 1 μm Iso measured before and following ACh exposure is not statistically significant (P > 0.4). These results confirm the idea that in cardiac ventricular myocytes the stimulatory effect of ACh is due to facilitation of β-adrenergic and/or cAMP-dependent responses. They also demonstrate that in guinea-pig ventricular myocytes, ACh-induced stimulatory mechanisms affect cAMP-regulated Cl− and L-type Ca2+ channels similarly.
These initial experiments were conducted using the conventional configuration of the whole-cell patch clamp technique. This approach has the potential of introducing confounding effects by allowing important components of the cytosol to dialyse out of the cell, leading to distortion of ion channel responses to regulation by G protein-coupled receptors (Kurachi et al. 1989; Zakharov & Harvey, 1995). To determine whether or not cell dialysis might have affected our ability to accurately measure ACh-induced stimulatory responses, we conducted experiments utilizing the perforated-patch variation of the whole-cell voltage-clamp technique, which is known to significantly reduce the experimental impact on signalling mechanisms. Since changes in the relative level of β-adrenergic stimulation can affect the magnitude of the ACh-induced stimulatory response (see Fig. 1), we first determined whether cell dialysis significantly affected β-adrenergic sensitivity of the L-type Ca2+ current. We found that over the time course of our experiments, the stimulatory effect of 1 nm Iso on the L-type Ca2+ current was not significantly altered (P > 0.1). When using the conventional patch-clamp technique, the magnitude of the response to 1 nm Iso was 23 ± 3.0 % (n = 23) of that produced by 1 μm Iso in the same cells. When using the perforated-patch technique, the magnitude of the response to 1 nm Iso was 34 ± 7.1 % (n = 9) of that produced by the maximal stimulatory effect produced by 1 μm Iso. Furthermore, the magnitude of ACh-induced rebound stimulation of the L-type Ca2+ current produced by 1 μm ACh in the presence 1 nm Iso was not significantly affected (P > 0.5). When using the conventional patch-clamp technique, the magnitude of the rebound response was 31 ± 3.7 % of that produced by 1 μm Iso. When using the perforated-patch technique, the magnitude of the rebound response was 31 ± 5.5 % of that produced by 1 μm Iso. Since cell dialysis did not appear to affect these responses, all subsequent experiments were performed using the conventional whole-cell method.
Muscarinic stimulation via a PTX-sensitive G protein
It has been reported that ACh-induced rebound stimulation of the L-type Ca2+ current in atrial myocytes and the cAMP-regulated Cl− current in ventricular myocytes can be elicited not only upon washout of ACh, a muscarinic receptor agonist, but also upon the addition of atropine, a muscarinic receptor antagonist (Wang & Lipsius, 1995; Zakharov & Harvey, 1997). Consistent with these observations, the addition of 10 μm atropine in the continued presence of 1 nm Iso plus 1 μm ACh resulted in a transient stimulation of the L-type Ca2+ current in guinea-pig ventricular myocytes (Fig. 2). Using this approach, the magnitude of the stimulatory response was 26 ± 4.9 % (n = 8) of that produced by 1 μm Iso in the same cells. This is not significantly different from the magnitude of the stimulatory response observed upon washout of ACh in the presence of 1 nm Iso (P > 0.4). This supports the idea that ACh-induced rebound stimulation of the L-type Ca2+ current is mediated by the same muscarinic receptor-dependent mechanism described previously.
Figure 2. Atropine, a muscarinic receptor antagonist, mimics the effect of washing out ACh.

A, time course of changes in amplitude of the L-type Ca2+ current under control conditions (a) and during cumulative exposure to 0.001 μm Iso (b), 1 μm ACh and 10 μm atropine (c) followed by exposure to 1 μm Iso alone (d). B, examples of L-type Ca2+ currents recorded the under conditions indicated in A. The dotted line above current traces represents the zero current level. Note: changes in holding current represent parallel regulation of the cAMP-dependent Cl− current. C, the stimulatory effects of 0.001 μm Iso measured prior to exposure to 1 μm ACh (□) and during the peak of the response caused by atropine washin (▪) were normalized to the magnitude of the response to 1 μm Iso alone ( ). The stimulatory effects of 0.001 μm Iso measured during atropine washin were significantly greater than those measured prior to ACh exposure (**P < 0.01).
). The stimulatory effects of 0.001 μm Iso measured during atropine washin were significantly greater than those measured prior to ACh exposure (**P < 0.01).
It is well established that the ability of ACh to inhibit cardiac β-adrenergic responses is mediated through the PTX-sensitive inhibitory G protein, Gi (Hartzell, 1988). It has also been demonstrated that ACh-induced rebound stimulation of the L-type Ca2+ current in atrial myocytes and the cAMP-activated Cl− current in ventricular myocytes can be prevented by PTX treatment (Wang & Lipsius, 1995; Zakharov & Harvey, 1997). In our experiments, treatment of myocytes with 2 μg ml−1 PTX for at least 2 h at 37 °C also blocked ACh rebound stimulation of the Ca2+ current (Fig. 3). In guinea-pig ventricular myocytes, Iso regulates ion channel function solely through the activation of β1-adrenergic receptors, and it is known that PTX treatment increases the sensitivity of cardiac ion channels to β1-adrenergic receptor stimulation (Hool & Harvey, 1997). Therefore, to avoid saturation of the ACh-induced stimulatory responses we carried out these experiments in the presence of 0.3 nm Iso. Under these conditions, instead of being subthreshold, this concentration of Iso alone activated the L-type type Ca2+ current to a level that was 42 ± 6.5 % (n = 7) of that produced by 1 μm Iso. However, application of 1 μm ACh in the presence of 0.3 nm Iso failed to produce either an inhibitory or a stimulatory effect. The magnitude of the Ca2+ current observed following washout of 1 μm ACh was not significantly different from the magnitude of the current measured before exposure to ACh (P > 0.3). The actual difference was 2 ± 1.8 % (n = 7) of that produced by exposure to 1 μm Iso in the same cells. The results from these experiments are consistent with the idea that ACh-induced rebound stimulation of the L-type Ca2+ current in guinea-pig ventricular myocytes is mediated by the same PTX-sensitive G protein-dependent mechanism involved in ACh-induced rebound stimulation of the same current in atrial cells as well as the cAMP-regulated Cl− current in these same cells.
Figure 3. ACh-induced rebound stimulation of the L-type Ca2+ current is blocked by pertussis toxin (PTX) treatment.

A, time course of changes in amplitude of the L-type Ca2+ current in PTX-treated cell under control conditions (a), and during exposure to 0.0003 μm Iso alone (b), 0.0003 μm Iso plus 1 μm ACh, 0.0003 μm Iso following washout of ACh (c) and 1 μm Iso alone (d). B, examples of L-type Ca2+ currents recorded under the conditions indicated in A. The dotted line above current traces represents the zero current level. Note: changes in holding current represent parallel regulation of the cAMP-dependent Cl− current. C, the stimulatory effects of 0.0003 μm Iso were measured in PTX-treated cells prior to (□) and following (▪) exposure to 1 μm ACh and then normalized to the magnitude of the response to 1 μm Iso alone ( ). The stimulatory effects of 0.3 nm Iso measured following exposure to ACh were not significantly different from those measured prior to ACh exposure (ns, P > 0.3).
). The stimulatory effects of 0.3 nm Iso measured following exposure to ACh were not significantly different from those measured prior to ACh exposure (ns, P > 0.3).
NOS and PDE-III-independent responses
Despite the similarities between the ACh-induced stimulatory responses observed in cat atrial and guinea-pig ventricular myocytes, previous work has suggested that the actual signalling pathway responsible for the ACh-induced rebound stimulation of L-type Ca2+ channel function in atrial myocytes involves the NO-dependent production of cGMP and cGMP-dependent inhibition of PDE III. The resulting decrease in cAMP metabolism could then explain the resulting stimulatory effects. However, in guinea-pig ventricular myocytes, we demonstrated that this pathway is unlikely to explain ACh-induced rebound stimulation of the cAMP-regulated Cl− current. Furthermore, we found that rebound stimulation of the L-type Ca2+ current is intact in ventricular myocytes obtained from mice in which there has been targeted disruption of the gene responsible for the expression of NOS type III, the predominant isoform of NOS expressed in the heart (Belevych & Harvey, 2000). Consistent with these latter observations, we have found that treatment of guinea-pig ventricular myocytes with the NOS inhibitor l-NMMA did not inhibit ACh-induced rebound stimulation of the L-type Ca2+ current (Fig. 4). Wang & Lipsius (1995) have demonstrated that acute exposure to 100 μml-NMMA was sufficient to completely block ACh-induced rebound stimulation of the L-type Ca2+ current in cat atrial myocytes. We treated our cells with 1 mml-NMMA for a period beginning at least 1 h before and continuing through the duration of the patch-clamp experiments. Under these conditions the magnitude of the stimulatory response produced by exposure to 1 μm ACh in the presence of 1 nm Iso was 27 ± 5.2 % (n = 13) of that produced by a maximally stimulating concentration of Iso alone. This is not significantly different from the magnitude of the rebound response observed in the absence of l-NMMA (P > 0.5). These data support the idea that the lack of NO involvement in ACh-induced stimulatory responses in guinea-pig ventricular myocytes is not unique to the cAMP-regulated Cl− current.
Figure 4. NG-monomethyl-l-arginine (l-NMMA), a nitric oxide synthase inhibitor, does not affect ACh-induced rebound stimulation of the L-type Ca2+ current.

A, time course of changes in amplitude of the L-type Ca2+ current under control conditions (a), and during exposure to 0.001 μm Iso alone (b), 0.001 μm Iso plus 1 μm ACh, 0.001 μm Iso following washout of ACh (c) and 1 μm Iso alone (d). The cell was pretreated with 1 mml-NMMA for more than 1 h prior to and continuing through the patch-clamp experiment. B, examples of L-type Ca2+ currents recorded under the conditions indicated in A. The dotted line above current traces represents the zero current level. Note: changes in holding current represent parallel regulation of the cAMP-dependent Cl− current. C, the stimulatory effects of 0.001 μm Iso were measured in l-NMMA-treated cells prior to (□) and following (▪) exposure to 1 μm ACh and then normalized to the magnitude of the response to 1 μm Iso alone ( ). The stimulatory effects of 0.001 μm Iso measured following exposure to ACh were significantly greater than those measured prior to ACh exposure (***P < 0.001).
). The stimulatory effects of 0.001 μm Iso measured following exposure to ACh were significantly greater than those measured prior to ACh exposure (***P < 0.001).
In addition to being unable to block ACh-induced rebound stimulation of the Ca2+ current with l-NMMA, we were also unable to block these effects with milrinone (Fig. 5), a specific inhibitor of PDE III activity (Harrison et al. 1986). Cells were exposed to 10 μm milrinone, a concentration that is at least 1000-fold greater than the IC50 for inhibiting this enzyme (Fischmeister & Hartzell, 1991), and 2-fold greater than the concentration used to completely block facilitation of the L-type Ca2+ current by maximally effective concentrations of exogenous cGMP (Ono & Trautwein, 1991). Although this concentration of milrinone by itself did not stimulate the Ca2+ current, it did increase the sensitivity of the Ca2+ current to stimulation by Iso. However, it did not block the rebound stimulatory response produced by ACh. In the presence of 1 nm Iso, 1 μm ACh produced a stimulatory response that was 29 ± 5.3 % (n = 4) of that produced by 1 μm Iso alone. This is not significantly different from the magnitude of the stimulatory response observed in the absence of milrinone (P > 0.8). However, the total magnitude of the current observed in milrinone-treated cells following washout of ACh in the presence of 1 nm Iso (103 ± 3.3 %) was not significantly different from that produced by 1 μm Iso alone in the same cells (P > 0.3), suggesting that the rebound response had saturated. When these experiments were repeated using 0.3 nm Iso, the rebound response did not saturate. Under these conditions, the magnitude of the stimulatory response elicited by 1 μm ACh was 31 ± 8.7 % (n = 11) of that produced by 1 μm Iso. This is about 2.5 times larger than the magnitude of the stimulatory response produced by 1 μm ACh in the presence of 0.3 nm Iso in cells that were not pretreated with milrinone (see Fig. 1). Although the difference is not statistically significant (P = 0.07), an increase in the magnitude of the response is not unexpected since total PDE activity was partially inhibited. The fact that milrinone did not block the rebound response supports the idea that ACh-induced stimulatory effects in guinea-pig ventricular myocytes do not involve the inhibition of PDE III activity.
Figure 5. Milrinone, a specific type III phosphodiesterase (PDE III) inhibitor, does not block ACh-induced rebound stimulation of L-type Ca2+ current.

A, time course of changes in amplitude of L-type Ca2+ current under control conditions (a), and during exposure to 0.0003 μm Iso alone (b), 0.0003 μm Iso plus 1 μm ACh, 0.0003 μm Iso following washout of ACh (c) and 1 μm Iso alone (d). The cell was exposed to milrinone (10 μm) starting at the beginning of the patch-clamp experiment. Inset, examples of L-type Ca2+ currents recorded under conditions indicated in A. The dotted line above the current traces represents the zero current level. Note: changes in holding current represent parallel regulation of cAMP-dependent Cl− current. B, the stimulatory effects of 0.0003 and 0.001 μm Iso were measured in milrinone-treated cells prior to (□) and following (▪) exposure to 1 μm ACh and then normalized to the magnitude of the response to 1 μm Iso alone ( ). The stimulatory effects of 0.001 and 0.0003 μm Iso measured following exposure to ACh were significantly greater than those measured prior to ACh exposure (*P < 0.01 and **P < 0.05, respectively).
). The stimulatory effects of 0.001 and 0.0003 μm Iso measured following exposure to ACh were significantly greater than those measured prior to ACh exposure (*P < 0.01 and **P < 0.05, respectively).
Gs dependence of the muscarinic stimulatory response
If ACh-induced stimulatory responses do not involve NO-dependent regulation of PDE III activity, what other mechanism can explain such effects? One possibility is that muscarinic receptor activation of the PTX-sensitive inhibitory G protein Gi may directly stimulate AC4 activity via a Gβγ-dependent mechanism. This hypothesis is based on the fact that Gβγ can directly activate AC2 and AC4 (Gao & Gilman, 1991; Tang & Gilman, 1991; Federman et al. 1992). In cardiac myocytes, AC5 and AC6 mRNA levels are most abundant. However, AC4 mRNA has also been detected (Gao & Gilman, 1991; Espinasse et al. 1999). Furthermore, only AC5 and AC6 are inhibited by Gαi (Chen & Iyengar, 1993; Taussig et al. 1994). Therefore, muscarinic receptor activation of Gi could antagonize β-adrenergic responses by inhibiting AC5 and/or AC6 while at the same time facilitating β-adrenergic responses by stimulating AC4. If this is true, then the ability of ACh to produce stimulatory effects in guinea-pig ventricular myocytes should depend not only on there being sufficient basal levels of cAMP, but perhaps more importantly it should depend on there being sufficient basal Gs activation, since Gβγ-dependent stimulation of AC4 should only occur in the presence of activated Gαs (Gao & Gilman, 1991; Federman et al. 1992). To test this hypothesis, we determined whether or not ACh-induced stimulatory responses could be observed when using Gs-dependent as well as Gs-independent means of priming the cells.
We first looked for the ability of ACh to produce a stimulatory response in the presence of histamine. In the heart, activation of both H2-histaminergic and β-adrenergic receptors regulates L-type Ca2+ channel activity via Gs-dependent stimulation of AC (Hescheler et al. 1987). Consistent with this idea, exposure to and subsequent washout of ACh in the continuous presence of a submaximally stimulating concentration of histamine resulted in significant rebound stimulation of L-type Ca2+ current (Fig. 6). In the presence of 30 nm histamine, the stimulatory effect produced by 1 μm ACh was 42 ± 11 % (n = 7) of that produced by a maximally effective concentration of histamine (10 μm) in the same cells.
Figure 6. ACh can produce transient rebound stimulation of L-type Ca2+ currents in the presence of submaximally stimulating concentrations of histamine.
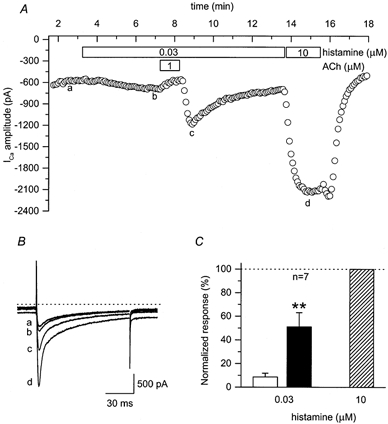
A, time course of changes in amplitude of L-type Ca2+ current under control conditions (a), and during exposure to 0.03 μm histamine alone (b), 0.03 μm histamine plus 1 μm ACh, 0.03 μm histamine following washout of ACh (c) and 10 μm histamine alone (d). B, examples of L-type Ca2+ current recorded under conditions indicated in A. The dotted line above current traces represents the zero current level. Note: changes in holding current represent parallel regulation of the cAMP-dependent Cl− current. C, stimulatory effects of 0.03 μm histamine were measured prior to (□) and following (▪) exposure to 1 μm ACh and then normalized to the magnitude of the response to 10 μm histamine alone ( ). The stimulatory effects of 0.03 μm histamine measured following exposure to ACh were significantly greater than those measured prior to ACh exposure (**P < 0.01).
). The stimulatory effects of 0.03 μm histamine measured following exposure to ACh were significantly greater than those measured prior to ACh exposure (**P < 0.01).
A similar response was obtained when Gs was activated via a receptor-independent mechanism using cholera toxin. ACh produced a rebound stimulatory response in cells that were first treated with 1 μg ml−1 cholera toxin for at least 2 h at 37 °C. Basal L-type Ca2+ current density increased from 8.6 ± 0.6 (n = 7) to 41 ± 3.1 pA pF−1 (n = 12) in cholera-toxin-treated cells. The increase in basal current is due at least in part to an increase in cAMP production caused by direct activation of Gs by the toxin (Gilman, 1987). Consistent with this idea, exposure of cholera toxin-treated cells to 10 μm ACh resulted in significant inhibition of the basal Ca2+ current, an effect that was not observed in control cells (Fig. 7). However, the Ca2+ current did not appear to be maximally activated since exposure of cholera toxin-treated cells to 1 μm Iso still produced a stimulatory response. Figure 7B demonstrates that washout of ACh produced a transient rebound increase in the amplitude of Ca2+ current to a level significantly higher than that observed before ACh application. The amplitude of the Ca2+ current observed following washout of ACh increased by 19 ± 5.0 % (n = 12) over baseline. This was not significantly different from the magnitude of the Ca2+ current response to 1 μm Iso in cholera toxin-treated cells ( 17 ± 13.6 %, n = 3, P > 0.8). In control cells, exposure to and washout of 10 μm ACh did not produce a stimulatory effect (Fig. 7A). The amplitude of the Ca2+ current measured in control cells following ACh washout was 97 ± 2.5 % (n = 7) of that observed before ACh application. These data are consistent with the hypothesis that ACh-induced rebound effects require the presence of activated Gαs.
Figure 7. Treatment with cholera toxin reveals ACh-induced rebound stimulation of basal L-type Ca2+ currents.

A and B, time courses of changes in amplitude of the L-type Ca2+ current under control conditions (a), and during (b) and following (c) washout of 10 μm ACh in control myocytes (A) and myocytes treated with cholera toxin (B). Insets, examples of L-type Ca2+ currents recorded under the conditions indicated in A and B. The dotted lines above current traces represent the zero current level.
Next we looked for ACh-induced rebound responses in cells that were primed using forskolin (Fig. 8). This compound activates AC directly via a Gs-independent mechanism (Hurley, 1999). If elevated cAMP production alone is sufficient to enable ACh to produce its stimulatory effect, then one should see rebound responses in the presence of forskolin. Exposure of guinea-pig ventricular myocytes to 0.1 and 0.3 μm forskolin produced a submaximal, concentration-dependent stimulation of the L-type Ca2+ current. Furthermore, addition of 1 μm ACh reversed the forskolin-induced response. However, upon washout of ACh, there was no rebound stimulation of the current. In the presence of 0.1 μm forskolin, the magnitude of the Ca2+ current following washout of ACh was 101 ± 2.0 % (n = 7) of that observed just prior to the application of ACh. In the presence of 0.3 μm forskolin, the magnitude of the Ca2+ current following washout of ACh was 98 ± 2.5 % (n = 7) of that observed just prior to the application of ACh. These data support the conclusion that ACh-induced stimulatory responses occur only in the presence of Gs activation, which is consistent with the idea that Gβγ activation of AC4 may be involved.
Figure 8. ACh does not produce a stimulatory effect on L-type Ca2+ currents in the presence of submaximal stimulating concentrations of forskolin.
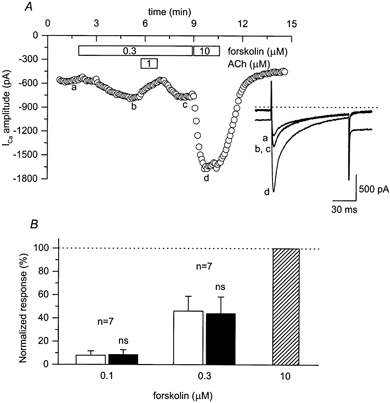
A, time course of changes in amplitude of L-type Ca2+ current under control conditions (a), and during exposure to 0.3 μm forskolin alone (b), 0.3 μm forskolin plus 1 μm ACh, 0.3 μm forskolin following washout of ACh (c) and 10 μm forskolin alone (d). Inset, examples of L-type Ca2+ currents recorded under the conditions indicated in A. The dotted line above current traces represents the zero current level. Note: changes in holding current represent parallel regulation of cAMP-dependent Cl− current. B, stimulatory effects of 0.1 and 0.3 μm forskolin measured prior to (□) and following (▪) exposure to 1 μm ACh and normalized to the magnitude of the response to 10 μm forskolin alone ( ). The stimulatory effects of 0.1 and 0.3 μm forskolin measured following exposure to ACh were not significantly different from those measured prior to ACh exposure (ns, P > 0.6 and P > 0.4, respectively).
). The stimulatory effects of 0.1 and 0.3 μm forskolin measured following exposure to ACh were not significantly different from those measured prior to ACh exposure (ns, P > 0.6 and P > 0.4, respectively).
Gβγ dependence of the muscarinic stimulatory response
If ACh-induced stimulatory responses are due to Gβγ activation of AC4, then it should be possible to antagonize such effects by introducing an excess of the peptide QEHA, which represents the Gβγ binding sequence of AC2. This peptide has been shown to bind Gβγ and block Gβγ-mediated activation of a variety of responses, including stimulation of AC2 (Chen et al. 1995). For these experiments, 100 μm QEHA was added to the pipette solution and allowed to diffuse into the cell for 8–12 min before the effects of ACh were tested. Figure 9A demonstrates an example of the response to 1 μm ACh in a cell dialysed with 100 μm QEHA. Exposure to 1 μm ACh inhibited the stimulatory response to 1 nm Iso. However, washout of ACh did not produce rebound stimulation of the Ca2+ current. On average, the magnitude of the ACh-induced rebound response measured in cells dialysed with QEHA was 13 ± 4.2 % (n = 9) of that produced by 1 μm Iso (Fig. 9B). In time-matched control experiments, ACh-induced stimulation of the Ca2+ current was 27 ± 4.6 % (n = 11) of that produced by 1 μm Iso. These results indicate that QEHA significantly attenuated the magnitude of the ACh-induced stimulatory response (P < 0.05). This effect of QEHA cannot be explained by a change in β-adrenergic responsiveness, since dialysing cells with QEHA did not significantly affect the response to 1 nm Iso (P > 0.5). The magnitude of the Ca2+ current response to 1 nm Iso was 32 ± 5.8 % of that produced by 1 μm Iso in cells dialysed with QEHA and 36 ± 4.7 % of that produced by 1 μm Iso in control cells.
Figure 9. Cell dialysis with the Gβγ-binding peptide QEHA significantly reduced ACh-induced rebound stimulation of L-type Ca2+ currents.

A, time course of changes in amplitude of L-type Ca2+ current under control conditions (a), and during exposure to 0.001 μm Iso alone (b), 0.001 μm Iso plus 1 μm ACh, 0.001 μm Iso following washout of ACh (c) and 1 μm Iso alone (d). 100 μm QEHA was included in the pipette solution. B, examples of L-type Ca2+ current recorded under the conditions indicated in A. The dotted line above current traces represents the zero current level. Note: changes in the holding current represent parallel regulation of the cAMP-dependent Cl− current. C, net ACh-induced rebound effect measured following 8–12 min of cell dialysis with control pipette solution (□) and pipette solution containing 100 μm QEHA (▪). Rebound stimulation was elicited by washing out 1 μm ACh in the presence of 1 nm Iso and normalized to the magnitude of the response to 1 μm Iso alone. The net ACh-induced stimulatory effect was calculated as described in Fig. 1. Cell dialysis with QEHA significantly reduced the net rebound effect of ACh (* P < 0.05).
Another set of control experiments was performed by dialysing cells with a pipette solution containing the peptide SKEE. This peptide represents the analogous region of AC3, an isoform of AC that does not bind Gβγ (Chen et al. 1995). For these experiments, 100 μm SKEE was added to the pipette solution and allowed to dialyse into the cell for 8–12 min before the effects of ACh were tested. The magnitude of ACh-induced rebound stimulation of the Ca2+ current was 38 ± 2.7 % (n = 3; data not shown) of that produced by 1 μm Iso. This is significantly greater than the magnitude of the stimulatory response observed in cells dialysed with QEHA (P < 0.01), but it is not significantly different from the magnitude of the stimulatory response observed in the absence of QEHA (P > 0.2). These results support our hypothesis that ACh-induced stimulatory effects can be mediated through the direct activation of AC by Gβγ in the presence of activated Gαs.
Although the stimulatory effect of ACh is obvious upon washout of the agonist, it may still affect the response observed in the presence of the agonist. As we have suggested previously (Zakharov & Harvey, 1997), the net response to ACh most likely represents a balance between its stimulatory and inhibitory effects. In this case, one might expect that the attenuation of the ACh stimulatory response would result in a more pronounced inhibitory effect. Analysis of the inhibitory effects of ACh in the presence of QEHA revealed that in cells dialysed with QEHA, 1 μm ACh inhibited the response to 1 nm Iso by 68 ± 6.7 % (n = 9). However, in the time-matched control cells, 1 μm ACh antagonized the stimulatory effect of 1 nm Iso only by 43 ± 4.2 % (n = 11). This indicates that in cells dialysed with QEHA, there is a statistically significant increase in the ability of ACh to inhibit β-adrenergic responses (P < 0.05).
It has been demonstrated that mRNA for AC4 is present in cardiac tissue (Gao & Gilman, 1991; Espinasse et al. 1999). However, if AC4 is involved in the stimulatory effect of ACh, it would be important to demonstrate the presence of AC4 protein. Immunoblot analysis of guinea-pig ventricle membrane preparations (n = 3) consistently established the presence of a protein that reacts with a specific anti-AC4 antibody (Murthy & Makhlouf, 1997). A single immunogenic band with a molecular mass greater than 200 kDa was identified (Fig. 10), which corresponds to the reported molecular mass of AC4 found in other guinea-pig tissues (Liu et al. 1999). Furthermore, pre-incubation of the anti-AC4 antibody with its specific blocking peptide abolished the immunoreaction. These data indicate that AC4 is present at the protein level in guinea-pig ventricular myocytes.
Figure 10. Adenylyl cyclase type 4 (AC4) is present in guinea-pig ventricular myocytes.

AC4 immunoreactive protein is identified in guinea-pig heart crude membrane preparations following SDS-PAGE and electrophoretic transfer to polyvinylidene difluoride membranes. An immunogenic band greater than 200 kDa is identified that is absent in preparations treated with AC4-specific blocking peptide. Immunoreactive proteins were visualized by enhanced chemiluminescence. Each lane contains 70 μg of crude membrane protein.
The data presented thus far support the idea that the ACh-induced stimulatory effect involves a Gβγ-dependent mechanism. The requirement for activation of Gs and the evidence that AC4 is present in guinea-pig ventricular myocytes further supports the idea that the ACh-induced stimulatory response may actually involve the Gβγ-dependent activation of AC4. However, it is conceivable that other Gβγ-dependent mechanisms could be involved, including Gβγ-dependent stimulation of phospholipase C activity and subsequent activation of protein kinase C (PKC; Clapham & Neer, 1997). The ability of PKC to stimulate AC5 and/or AC6 in a Gs-dependent manner might then explain the stimulatory effect of ACh. To test this possibility, we studied the effects of ACh in the presence of the PKC inhibitor bisindolylmaleimide I (BIM) (Fig. 11). These experiments were carried out using myocytes that were exposed to 300 nm BIM, starting 1 h before and continuing through the completion of the patch-clamp experiment. This concentration of BIM is 30-fold greater than the IC50 for inhibition of PKC activity (Toullec et al. 1991). Furthermore, we have used this concentration of BIM previously to inhibit PKC-dependent responses in guinea-pig ventricular myocytes (Middleton & Harvey, 1998). However, BIM did not block the ability of 1 μm ACh to produce a stimulatory response in the presence of 1 nm Iso. The magnitude of the ACh-induced stimulatory response was 31 ± 6.3 % of that produced by 1 μm Iso in the same cells. This is not significantly different from the magnitude of the ACh-induced stimulatory response observed in the absence of BIM (P > 0.8).
Figure 11. ACh-induced rebound stimulation of L-type Ca2+ currents is not affected by bisindolylmaleimide I (BIM), a protein kinase C inhibitor.

A, time course of changes in amplitude of L-type Ca2+ current under control conditions (a), during exposure to 0.001 μm Iso alone (b), 0.001 μm Iso plus 1 μm ACh, 0.001 μm Iso following washout of ACh (c) and 1 μm Iso alone (d). The cell was pretreated with 0.3 μm BIM for at least 8 min prior to the beginning of the patch-clamp experiment. B, examples of L-type Ca2+ currents recorded under the conditions indicated in A. The dotted line above current traces represents the zero current level. C, the stimulatory effects of 0.001 μm Iso were measured in BIM-treated cells prior to (□) and following (▪) exposure to 1 μm ACh and then normalized to the magnitude of the response to 1 μm Iso alone ( ). Stimulatory effects of 0.001 μm Iso measured following exposure to ACh were significantly greater than those measured prior to ACh exposure (***P < 0.001).
). Stimulatory effects of 0.001 μm Iso measured following exposure to ACh were significantly greater than those measured prior to ACh exposure (***P < 0.001).
Gβγ can also produce stimulation of the L-type Ca2+ current through a phosphoinositide-3 (PI3) kinase-dependent signalling pathway (Viard et al. 1999). There is also evidence that M2 muscarinic receptors are coupled to PI3 kinase via a βγ-dimer derived from Gi/Go proteins (Wang et al. 1999). To test the possible contribution of PI3 kinase to ACh-induced rebound stimulation of the L-type Ca2+ current, we studied the effect of ACh in the presence of wortmannin, a specific PI3-kinase inhibitor (Ui et al. 1995). Cells were pretreated with 100 nm wortmannin for up to 40 min prior to and continuing through the patch-clamp experiment. This concentration of wortmannin is 20-fold higher than the IC50 for inhibition of PI3 kinase activity (Okada et al. 1994). The net rebound response produced by 1 μm ACh in the presence of 1 nm Iso in wortmannin-treated cells was 43 ± 12.6 % (n = 6; data not shown) of that elicited by 1 μm Iso. This is not significantly different from the magnitude of ACh stimulatory effect measured under control conditions (P > 0.2). To ensure that wortmannin had reached an effective level in the cytosol, cells were dialysed with a pipette solution containing 1 μm wortmannin; 1 μm wortmannin was also added to external solution. Under these conditions, the stimulatory effect of ACh (1 μm) measured in the presence of 1 nm Iso was 38 ± 7.0 % (n = 4; data not shown) of the effect produced by 1 μm Iso. This is also not significantly different from the magnitude of the ACh-induced stimulatory effect measured under control conditions (P > 0.4).
DISCUSSION
Muscarinic receptor activation can produce rebound stimulatory effects on the L-type Ca2+ current in guinea-pig ventricular myocytes (Song et al. 1998). This was confirmed in the present study by demonstrating that rebound responses can be elicited not only upon withdrawal of exposure to ACh, but also by exposure to the muscarinic receptor antagonist atropine in the continued presence of ACh. The present study goes further and demonstrates that this stimulatory effect is due to the facilitation of cAMP-dependent responses. This conclusion is supported by the fact that ACh-induced stimulatory effects were not observed in the absence of β-adrenergic stimulation. In addition, increasing the level of β-adrenergic stimulation increased the magnitude of the stimulatory response, but only up to the point were it reached the magnitude of the response elicited by a maximally stimulating concentration of Iso alone. These findings demonstrate that in guinea-pig ventricular myocytes, the L-type type Ca2+ current and the cAMP-regulated Cl− current both respond to the stimulatory effects of ACh in the same cAMP-dependent manner (Zakharov & Harvey, 1997). This observation is also consistent with the fact that ACh-induced rebound stimulation of the L-type Ca2+ current in cat atrial myocytes involves a cAMP-dependent mechanism (Wang & Lipsius, 1995). One difference, however, is that the ACh-induced rebound responses observed in atrial myocytes can be elicited in the absence of β-adrenergic stimulation. This is consistent with the observation that atrial myocytes exhibit a greater level of basal AC activity.
Despite the fact that ACh-induced stimulatory responses involve a cAMP-dependent mechanism, there appears to be a distinct difference in the signalling pathway responsible for the ACh-induced increase in cAMP in the different preparations. In cat atrial myocytes, ACh-induced stimulation of the L-type Ca2+ current is believed to involve the activation of NOS via a PTX-sensitive G protein and subsequent cGMP-dependent inhibition of PDE III (Wang et al. 1998). We have demonstrated previously that in guinea-pig ventricular myocytes, ACh-induced stimulation of the cAMP-regulated Cl− current involves a PTX-sensitive G protein, but it is not linked to a NOS/PDE-III-dependent mechanism (Zakharov & Harvey, 1997). The present study demonstrates that ACh-induced stimulation of the L-type Ca2+ current in guinea-pig ventricular myocytes also involves a PTX-sensitive G protein that is activating a NOS/PDE-III-independent mechanism. This indicates that the previously described differences in the signalling pathways responsible for ACh-induced stimulatory responses are not due to differences in how Ca2+ channels and Cl− channels are regulated. Our results support the idea that there are differences in the signalling mechanisms responsible for the ACh-induced stimulation of ion channel activity in atrial and ventricular myocytes. Although such differences might be explained on the basis of species-specific signalling mechanisms, this seems unlikely since we have already demonstrated that the NO signalling pathway is not involved in ACh-induced stimulatory responses in mouse ventricular myocytes (Belevych & Harvey, 2000).
The observation that ACh did not produce a stimulatory response in the absence of β-adrenergic stimulation (see Fig. 1) suggests that it is necessary to prime the cells by elevating cAMP to some critical level in order to see such effects. However, an increase in cAMP production alone is clearly not sufficient to enable muscarinic receptor stimulation to elicit a stimulatory response. This is demonstrated by the lack of ACh-induced rebound stimulation in the presence of forskolin. Exposure to 0.1 or 0.3 μm forskolin increased basal cAMP production, as demonstrated by the stimulatory effect it had on the Ca2+ current. Furthermore, subsequent exposure to ACh inhibited this stimulatory response. However, there was no evidence of rebound stimulation upon washout of ACh (see Fig. 8). In addition to demonstrating that increased cAMP production alone is not enough to enable ACh to elicit its stimulatory effect, it also supports the conclusion that the stimulatory response to muscarinic receptor stimulation is not due to the inhibition of PDE activity or a reduction in cAMP breakdown. In fact, such experiments eliminate any mechanism that involves the regulation of the β-adrenergic signalling pathway at any point downstream of AC.
The fact that ACh-induced rebound stimulation could be observed in the presence of 0.3 nm Iso might be used to argue that a significant elevation of cAMP levels is actually unnecessary, since this concentration of the β-adrenergic agonist by itself did not affect the magnitude of the Ca2+ current (see Fig. 1). However, it is possible that cAMP levels were increased by this concentration of Iso, but that they had not yet reached the threshold necessary to affect L-type Ca2+ channel function. In any case, the fact that exposure to Iso, but not forskolin, enabled ACh to produce a stimulatory effect indicates that this muscarinic response requires the activation of some component of the β-adrenergic signalling pathway that is associated specifically with receptor activation. The finding that the ACh-induced stimulatory response could also be observed in the presence of histamine indicates that this component is also activated by H2-histaminergic receptors. The likely candidate is Gs, since both receptors are known to couple to this stimulatory G protein (Hescheler et al. 1987). This conclusion is further supported by the fact that ACh-induced rebound stimulation was observed when Gs was activated directly by cholera toxin (see Fig. 7).
The activation of Gs, like other heterotrimeric G proteins, is associated with the exchange of GDP for the GTP bound to the α subunit. This results in the dissociation of Gαs and Gβγ subunits. Activated Gαs can then bind directly to and stimulate all isoforms of AC. In cardiac myocytes, there is evidence for expression of AC4, AC5, AC6 and AC7 (Defer et al. 2000). However, it is commonly assumed that AC5 and AC6 are the only isoforms of any consequence in cardiac tissue (Ishikawa & Homcy, 1997; Espinasse et al. 1999). This is largely based on the finding that mRNA levels for these two isoforms are the most abundant. Furthermore, AC5 and AC6, but not AC4 and AC7, can be inhibited by direct binding of the activated Gα subunit of the inhibitory G protein Gi, which may explain the ability of M2 muscarinic receptor activation to antagonize β-adrenergic responses via a PTX-sensitive G protein. However, mRNA levels do not necessarily correspond with the level of protein expression. Although it is not known what the relative levels of AC5 and AC6 vs. AC4 and/or AC7 are, in the present study we have found evidence that AC4 can be detected at the protein level (see Fig. 10). We observed an immunoreactive band with an apparent molecular mass of > 200 kDa, which is consistent with the 220 kDa protein identified as AC4 in guinea-pig myenteric neurones (Liu et al. 1999). Based on the amino acid composition, the predicted molecular masses of most cloned AC isoforms is around 120 kDa (Taussig & Gilman, 1995). The significant difference between the predicted and experimentally observed molecular masses of AC4 has been attributed to the fact that the protein undergoes glycosylation and that it is believed to associate tightly with some G proteins. As demonstrated previously in guinea-pig neurones, treating membrane preparations to remove N-linked glycosylation and prevent G protein association causes a shift of labelled protein to the expected molecular mass (Liu et al. 1999). The idea that guinea-pig cardiac myocytes express significant levels of AC4 protein is consistent with the previous immunohistochemical evidence that the AC4 antibody specifically labels cardiac tissue (Schulze & Buchwalow, 1998).
The idea that AC4 is present at functionally relevant levels is important because being highly homologous to AC2, it does not appear to be inhibited by Gαi (Gao & Gilman, 1991; Taussig et al. 1994). Furthermore, it can actually be stimulated by Gβγ, albeit only in the presence of Gαs (Gao & Gilman, 1991; Federman et al. 1992). Therefore, the derivation of Gβγ from a PTX-sensitive G protein (see Fig. 3) could explain the ACh-induced Gαs-dependent facilitation of cAMP-dependent responses. Indeed, scavenging free Gβγ with QEHA peptide significantly attenuated the stimulatory effect that ACh has on the L-type Ca2+ current (see Fig. 9). We have demonstrated previously that in guinea-pig ventricular myocytes the muscarinic stimulatory response, like the muscarinic inhibitory response, involves activation of M2 muscarinic receptors (Zakharov & Harvey, 1997). Therefore, the response to muscarinic receptor stimulation is likely to represent the balance between two signalling mechanisms activated by the same receptor: (1) Gαi inhibition of Gαs-stimulated AC5 and/or AC6 and (2) Gβγ facilitation of Gαs-stimulated AC4 and possibly AC7. Although it has not been demonstrated directly that AC7 can be stimulated by Gβγ, this isoform of AC is structurally similar to AC2 and AC4.
The fact that the muscarinic stimulatory responses can be observed as rebound stimulation upon removal of the agonist indicates that the inhibitory effect turns on and off rapidly, while the stimulatory effect turns on and off much more slowly. We have demonstrated previously that the stimulatory effect of 1 μm ACh observed in the presence of 1 nm Iso turns on with a time constant of 34 s (Zakharov & Harvey, 1997). The exact reason for the difference in the kinetics of the two processes is not clear. However, it might be explained, at least in part, by the ability of the Gα-binding regions of AC to act as regulators of G protein signalling. They possess the properties of both guanine nucleotide exchange factors and GTPase activating proteins (Scholich et al. 1999; Wittpoth et al. 2000). This would be expected to speed both the onset and the offset of Gαi-mediated inhibitory effects, but not Gβγ-mediated stimulatory effects. Speeding the inactivation of Gαi by facilitating the hydrolysis of bound GTP might then be expected to terminate Gβγ-mediated responses with a similar time course due to reassociation of the heterotrimeric G protein. The fact that Gβγ-mediated stimulatory responses do not deactivate as quickly as the inhibitory effects would suggest that individual G proteins do not elicit both inhibitory and stimulatory actions. In fact, it is possible that different isoforms of AC are found in unique microdomains, although this hypothesis has yet to be investigated.
Another important question is whether or not the stimulatory effect of ACh contributes to the net response observed in the presence of muscarinic receptor activation. In general, the inhibitory effect is dominant. However, the ability of muscarinic receptor stimulation to antagonize the β-adrenergic regulation of cardiac ion-channel activity is a competitive interaction (Hescheler et al. 1986). Therefore, anything that facilitates β-adrenergic responses should act to overcome the inhibitory effect of ACh. In support of this idea, we have reported previously that continued exposure to ACh results in a time-dependent relaxation or escape from the inhibitory effect that ACh has on β-adrenergically regulated ion-channel activity (Zakharov & Harvey, 1997). This would be consistent with the slowly developing stimulatory response helping to overcome the inhibitory response. It suggests that the stimulatory response contributes to the net steady-state effect observed during exposure to muscarinic agonists. More direct evidence favouring this idea was also found in the present study, in experiments where cells were dialysed with QEHA. Not only did this Gβγ-binding peptide antagonize the stimulatory response to muscarinic receptor stimulation, it also resulted in an increase in the magnitude of the inhibitory effect that ACh had on the β-adrenergically stimulated L-type Ca2+ current.
Acknowledgments
The authors thank Dr Ravi Iyengar for providing QEHA and SKEE, and Montelle Sanders for excellent technical assistance. This work was supported by grants from the American Heart Association and National Institutes of Health (AG16658 and HL68170).
References
- Belevych AE, Harvey RD. Muscarinic inhibitory and stimulatory regulation of the L-type Ca2+ current is not altered in cardiac ventricular myocytes from mice lacking endothelial nitric oxide synthase. Journal of Physiology. 2000;528:279–289. doi: 10.1111/j.1469-7793.2000.00279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Sims C, Harvey RD. Acetylcholine-induced rebound stimulation of the cardiac L-type Ca2+ current mediated by direct activation of adenylate cyclase. Biophysical Journal. 2001;80:635a–636a. [Google Scholar]
- Burke GH, Calaresu FR. An experimental analysis of the tachycardia that follows vagal stimulation. Journal of Physiology. 1972;226:491–510. doi: 10.1113/jphysiol.1972.sp009995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Devivo M, Dingus J, Harry A, Li J, Sui J, Carty DJ, Blank JL, Exton JH, Stoffel RH, Inglese J, Lefkowitz RJ, Logothetis DE, Hildebrandt JD, Iyengar R. A region of adenylyl cyclase 2 critical for regulation by G protein βγ subunits. Science. 1995;268:1166–1169. doi: 10.1126/science.7761832. [DOI] [PubMed] [Google Scholar]
- Chen J, Iyengar R. Inhibition of cloned adenylyl cyclases by mutant-activated Gi-alpha and specific suppression of type 2 adenylyl cyclase inhibition by phorbol ester treatment. Journal of Biological Chemistry. 1993;268:12253–12256. [PubMed] [Google Scholar]
- Clapham DE, Neer EJ. G protein βγ subunits. Annual Review of Pharmacology and Toxicology. 1997;37:167–203. doi: 10.1146/annurev.pharmtox.37.1.167. [DOI] [PubMed] [Google Scholar]
- Defer N, Best-Belpomme M, Hanoune J. Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. American Journal of Physiology — Renal Physiology. 2000;279:F400–416. doi: 10.1152/ajprenal.2000.279.3.F400. [DOI] [PubMed] [Google Scholar]
- Espinasse I, Iourgenko V, Richer C, Heimburger M, Defer N, Bourin MC, Samson F, Pussard E, Giudicelli JF, Michel JB, Hanoune J, Mercadier JJ. Decreased type VI adenylyl cyclase mRNA concentration and Mg2+- dependent adenylyl cyclase activities and unchanged type V adenylyl cyclase mRNA concentration and Mn2+-dependent adenylyl cyclase activities in the left ventricle of rats with myocardial infarction and longstanding heart failure. Cardiovascular Research. 1999;42:87–98. doi: 10.1016/s0008-6363(98)00283-1. [DOI] [PubMed] [Google Scholar]
- Federman AD, Conklin BR, Schrader KA, Reed RR, Bourne HR. Hormonal stimulation of adenylyl cyclase through Gi-protein βγ subunits. Nature. 1992;356:159–161. doi: 10.1038/356159a0. [DOI] [PubMed] [Google Scholar]
- Fischmeister R, Hartzell HC. Cyclic AMP phosphodiesterases and Ca2+ current regulation in cardiac cells. Life Sciences. 1991;48:2365–2376. doi: 10.1016/0024-3205(91)90369-m. [DOI] [PubMed] [Google Scholar]
- Gao BN, Gilman AG. Cloning and expression of a widely distributed (type IV) adenylyl cyclase. Proceedings of the National Academy of Sciences of the USA. 1991;88:10178–10182. doi: 10.1073/pnas.88.22.10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman AG. G proteins: transducers of receptor-generated signals. Annual Review of Biochemistry. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- Gilmour RF, Zipes DP. Positive inotropic effect of acetylcholine in canine cardiac Purkinje fibers. American Journal of Physiology. 1985;249:H735–H740. doi: 10.1152/ajpheart.1985.249.4.H735. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Harrison SA, Chang ML, Beavo JA. Differential inhibition of cardiac cyclic nucleotide phosphodiesterase isozymes by cardiotonic drugs. Circulation. 1986;73:III109–III116. [PubMed] [Google Scholar]
- Hartzell HC. Regulation of cardiac ion channels by catecholamines, acetylcholine and second messenger systems. Progress in Biophysics and Molecular Biology. 1988;52:165–247. doi: 10.1016/0079-6107(88)90014-4. [DOI] [PubMed] [Google Scholar]
- Hescheler J, Kameyama M, Trautwein W. On the mechanism of muscarinic inhibition of the cardiac Ca current. Pflügers Archiv. 1986;407:182–189. doi: 10.1007/BF00580674. [DOI] [PubMed] [Google Scholar]
- Hescheler J, Tang M, Jastorff B, Trautwein W. On the mechanism of histamine induced enhancement of the cardiac Ca2+ current. Pflügers Archiv. 1987;410:23–29. doi: 10.1007/BF00581891. [DOI] [PubMed] [Google Scholar]
- Hollenberg M, Carriere S, Barger AB. Biphasic action of acetylcholine on ventricular myocardium. Circulation Research. 1965;16:527–536. doi: 10.1161/01.res.16.6.527. [DOI] [PubMed] [Google Scholar]
- Hool LC, Harvey RD. Role of β1- and β2-adrenergic receptors in regulation of Cl− and Ca2+ channels in guinea-pig ventricular myocytes. American Journal of Physiology. 1997;273:H1669–1676. doi: 10.1152/ajpheart.1997.273.4.H1669. [DOI] [PubMed] [Google Scholar]
- Hurley JH. Structure, mechanism, and regulation of mammalian adenylyl cyclase. Journal of Biological Chemistry. 1999;274:7599–7602. doi: 10.1074/jbc.274.12.7599. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Homcy CJ. The adenylyl cyclases as integrators of transmembrane signal transduction. Circulation Research. 1997;80:297–304. doi: 10.1161/01.res.80.3.297. [DOI] [PubMed] [Google Scholar]
- Kurachi Y, Asano Y, Takikawa R, Sugimoto T. Cardiac Ca current does not run down and is very sensitive to isoprenaline in the nystatin-method of whole cell recording. Naunyn Schmiedebergs Archives of Pharmacology. 1989;340:219–222. doi: 10.1007/BF00168972. [DOI] [PubMed] [Google Scholar]
- Levy MN. Sympathetic-parasympathetic interactions in the heart. Circulation Research. 1971:437–445. doi: 10.1161/01.res.29.5.437. [DOI] [PubMed] [Google Scholar]
- Linden J. Enhanced cAMP accumulation after termination of cholinergic action in the heart. FASEB Journal. 1987;1:119–124. doi: 10.1096/fasebj.1.2.2440752. [DOI] [PubMed] [Google Scholar]
- Liu CY, Jamaleddin AJ, Zhang H, Christofi FL. FlCRhR/cyclic AMP signaling in myenteric ganglia and calbindin-D28 intrinsic primary afferent neurons involves adenylyl cyclases I, III and IV. Brain Research. 1999;826:253–269. doi: 10.1016/s0006-8993(99)01269-x. [DOI] [PubMed] [Google Scholar]
- Middleton LM, Harvey RD. PKC regulation of cardiac CFTR Cl− channel function in guinea pig ventricular myocytes. American Journal of Physiology. 1998;275:C293–C302. doi: 10.1152/ajpcell.1998.275.1.C293. [DOI] [PubMed] [Google Scholar]
- Murthy KS, Makhlouf GM. Differential coupling of muscarinic m2 and m3 receptors to adenylyl cyclases V/VI in smooth muscle. Concurrent m2-mediated inhibition via Gαi3 and m3-mediated stimulation via Gβγq. Journal of Biological Chemistry. 1997;272:21317–21324. doi: 10.1074/jbc.272.34.21317. [DOI] [PubMed] [Google Scholar]
- Okada T, Sakuma L, Fukui Y, Hazeki O, Ui M. Blockage of chemotactic peptide-induced stimulation of neutrophils by wortmannin as a result of selective inhibition of phosphatidylinositol 3-kinase. Journal of Biological Chemistry. 1994;269:3563–3567. [PubMed] [Google Scholar]
- Ono K, Noma A. Autonomic regulation of cardiac chloride current. Japanese Journal of Physiology. 1994;44:S193–S198. [PubMed] [Google Scholar]
- Ono K, Trautwein W. Potentiation by cyclic GMP of β-adrenergic effect on Ca2+ current in guinea-pig ventricular cells. Journal of Physiology. 1991;443:387–404. doi: 10.1113/jphysiol.1991.sp018839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. Journal of Neuroscience Methods. 1991;37:14–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Scholich K, Mullenix JB, Wittpoth C, Poppleton HM, Pierre SC, Lindorfer MA, Garrison JC, Patel TB. Facilitation of signal onset and termination by adenylyl cyclase. Science. 1999;283:1328–1331. doi: 10.1126/science.283.5406.1328. [DOI] [PubMed] [Google Scholar]
- Schulze W, Buchwalow IB. Adenylyl cyclase in the heart: an enzymocytochemical and immunocytochemical approach. Microscopy Research and Technique. 1998;40:473–478. doi: 10.1002/(SICI)1097-0029(19980301)40:6<473::AID-JEMT7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Song Y, Shryock JC, Belardinelli L. Potentiating effect of acetylcholine on stimulation by isoproterenol of L-type Ca2+ current and arrhythmogenic triggered activity in guinea pig ventricular myocytes. Journal of Cardiovascular Electrophysiology. 1998;9:718–726. doi: 10.1111/j.1540-8167.1998.tb00959.x. [DOI] [PubMed] [Google Scholar]
- Tang WJ, Gilman AG. Type-specific regulation of adenylyl cyclase by G protein βγ subunits. Science. 1991;254:1500–1503. doi: 10.1126/science.1962211. [DOI] [PubMed] [Google Scholar]
- Taussig R, Gilman AG. Mammalian membrane-bound adenylyl cyclases. Journal of Biological Chemistry. 1995;270:1–4. doi: 10.1074/jbc.270.1.1. [DOI] [PubMed] [Google Scholar]
- Taussig R, Tang WJ, Hepler JR, Gilman AG. Distinct patterns of bidirectional regulation of mammalian adenylyl cyclases. Journal of Biological Chemistry. 1994;269:6093–6100. [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, Duhamel L, Charon D, Kirilovsky J. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. Journal of Biological Chemistry. 1991;266:15771–15781. [PubMed] [Google Scholar]
- Ui M, Okada T, Hazeki K, Hazeki O. Wortmannin as a unique probe for an intracellular signalling protein, phosphoinositide 3-kinase. Trends in Biochemical Sciences. 1995;20:303–307. doi: 10.1016/s0968-0004(00)89056-8. [DOI] [PubMed] [Google Scholar]
- Viard P, Exner T, Maier U, Mironneau J, Nurnberg B, Macrez N. Gβγ dimers stimulate vascular L-type Ca2+ channels via phosphoinositide 3-kinase. FASEB Journal. 1999;13:685–694. doi: 10.1096/fasebj.13.6.685. [DOI] [PubMed] [Google Scholar]
- Wang YG, Huser J, Blatter LA, Lipsius SL. Withdrawal of acetylcholine elicits Ca2+-induced delayed afterdepolarizations in cat atrial myocytes. Circulation. 1997;96:1275–1281. doi: 10.1161/01.cir.96.4.1275. [DOI] [PubMed] [Google Scholar]
- Wang YG, Lipsius SL. Acetylcholine elicits a rebound stimulation of Ca2+ current mediated by pertussis toxin-sensitive G protein and cAMP- dependent protein kinase A in atrial myocytes. Circulation Research. 1995;76:634–644. doi: 10.1161/01.res.76.4.634. [DOI] [PubMed] [Google Scholar]
- Wang YG, Lipsius SL. A cellular mechanism contributing to postvagal tachycardia studied in isolated pacemaker cells from cat right atrium. Circulation Research. 1996;79:109–114. doi: 10.1161/01.res.79.1.109. [DOI] [PubMed] [Google Scholar]
- Wang YG, Rechenmacher CE, Lipsius SL. Nitric oxide signaling mediates stimulation of L-type Ca2+ current elicited by withdrawal of acetylcholine in cat atrial myocytes. Journal of General Physiology. 1998;111:113–125. doi: 10.1085/jgp.111.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Dhulipala PD, Li L, Benovic JL, Kotlikoff MI. Coupling of M2 muscarinic receptors to membrane ion channels via phosphoinositide 3-kinase gamma and atypical protein kinase C. Journal of Biological Chemistry. 1999;274:13859–13864. doi: 10.1074/jbc.274.20.13859. [DOI] [PubMed] [Google Scholar]
- Wittpoth C, Scholich K, Bilyeu JD, Patel TB. Adenylyl cyclase regulates signal onset via the inhibitory GTP-binding protein, Gi. Journal of Biological Chemistry. 2000;275:25915–25919. doi: 10.1074/jbc.M001687200. [DOI] [PubMed] [Google Scholar]
- Zakharov SI, Harvey RD. Altered β-adrenergic and muscarinic response of CFTR Cl− current in dialyzed cardiac myocytes. American Journal of Physiology. 1995;268:H1795–1802. doi: 10.1152/ajpheart.1995.268.5.H1795. [DOI] [PubMed] [Google Scholar]
- Zakharov SI, Harvey RD. Rebound stimulation of the cAMP-regulated Cl− current by acetylcholine in guinea-pig ventricular myocytes. Journal of Physiology. 1997;499:105–120. doi: 10.1113/jphysiol.1997.sp021914. [DOI] [PMC free article] [PubMed] [Google Scholar]