

Abstract
In anaesthetised rats, the increase in femoral vascular conductance (FVC) evoked by moderate systemic hypoxia is mediated by adenosine acting on A1 receptors. It is also nitric oxide (NO) dependent: it is attenuated by NO synthase (NOS) inhibition, but restored when baseline FVC is restored by sodium nitroprusside (SNP), a NO donor. However, under these conditions there was in increase in the critical O2 delivery (DO2,crit) at which hindlimb O2 consumption (V̇O2) becomes directly dependent upon O2 delivery (DO2), indicating that V̇O2 is regulated by newly synthesised NO.
In the present study, after NOS inhibition, when baseline FVC was restored with SNP infusion, the increases in FVC evoked by breathing 12 and 8 % O2 were reduced by the A1 receptor antagonist DPCPX, by 60 and 40 %, respectively (n = 8). The A2A receptor antagonist ZM241385 reduced the FVC increase evoked by 12 % O2 (by 45 %, n = 8), but did not alter that evoked by 8 % O2.
DPCPX also reduced the increases in FVC evoked by graded systemic hypoxia, breathing 14–6 % O2 and increased DO2,crit, from 0.64 ± 0.06 to 0.95 ± 0.07 ml O2 min−1 kg−1 (control vs. DPCPX). However, ZM241385 (n = 8) had no effect on the FVC increases or on DO2,crit (0.70 ± 0.02 ml O2 min−1 kg−1, n = 8).
Thus, the increases in FVC evoked by mild to severe systemic hypoxia are mediated by A1 receptors. These responses, which are attributable to proximal arteriolar dilatation, help maintain DO2. Even after NOS inhibition, adenosine still increases FVC via A2A (moderate hypoxia only) and A1 receptors, providing baseline levels of NO are present. Furthermore, adenosine, acting via A1 receptors, is important in determining DO2,crit and therefore in maintaining V̇O2. We propose that this is achieved by A1-evoked dilatation of terminal arterioles and is mediated by increased synthesis of NO.
It is well established that the skeletal muscle of larger mammals such as the dog can maintain O2 consumption (V̇O2) at a steady rate over a wide range of O2 delivery (DO2) values, but that below a critical DO2 level (DO2,crit), V̇O2 declines linearly with DO2 (Duran & Renkin, 1974; Granger et al. 1976; Cain & Chapler, 1979; Samsel & Schumacker, 1988; Curtis et al. 1995). Therefore, the relationship between V̇O2 and DO2 is described by two distinct phases; a delivery-independent phase and a delivery-dependent phase. The delivery-independent phase is thought to rely on the co-ordinated vasodilatation of the terminal arterioles within the microvascular bed of muscle, leading to a more homogeneous distribution of the available O2 and allowing V̇O2 to be maintained (Granger et al. 1976; Harrison et al. 1990). Accordingly, V̇O2 becomes delivery dependent when the terminal arterioles are maximally dilated and can no longer contribute to the regulation of DO2.
We recently demonstrated that this biphasic relationship between V̇O2 and DO2 also exists in the hindlimb muscles of the rat when DO2 is reduced by graded systemic hypoxia (Edmunds & Marshall, 2001). Since the muscle vasodilatation evoked by systemic hypoxia, as reflected by the increase in femoral vascular conductance (FVC), was attenuated by nitro-l-arginine methyl ester (l-NAME)-mediated inhibition of nitric oxide synthase (NOS; Skinner & Marshall, 1996; Bryan & Marshall, 1999b), we investigated the effect of l-NAME on the DO2-V̇O2 relationship (Edmunds & Marshall, 2001). In fact, l-NAME caused such a large decrease in FVC and femoral blood flow (FBF) that DO2 was reduced too much for DO2,crit to be calculated. However, when FBF, and therefore DO2, were subsequently restored by the infusion of a NO donor, then DO2,crit was markedly increased. This suggests that the dilatation of the terminal arterioles, which determines DO2,crit, is normally mediated by NO (Edmunds & Marshall, 2001). However, when a basal level of NO was restored after l-NAME by the infusion of a NO donor, the changes in FVC evoked by graded levels of hypoxia were also fully restored. We therefore proposed that the dilatation of the proximal arterioles, which makes the major contribution to changes in FVC (Froneck & Zweifach, 1975; Hebert & Marshall, 1988), required a basal presence of NO (i.e. it was dependent upon NO) rather than being mediated by NO. The mechanism underlying this NO-dependent dilatation of proximal arterioles remains unclear.
Previous work from our laboratory involving intravital microscopy of skeletal muscle showed that both exogenous adenosine and adenosine released during systemic hypoxia dilate the proximal and terminal arterioles but have preferential effects on terminal arterioles (Mian & Marshall, 1991). We have shown subsequently that infused adenosine can evoke an increase in FVC by stimulating A1 or A2A receptors, but that the increase in FVC evoked by systemic hypoxia was attributable only to A1 receptor stimulation: it was attenuated by a selective A1 receptor antagonist but not an A2A antagonist (Bryan & Marshall, 1999a). Indeed, stimulation of either A1 or A2A receptors can stimulate NOS in human umbilical vein endothelial cells (HUVEC) and in the endothelium of freshly excised rat aorta (Sobrevia et al. 1997; Ray & Marshall, 2000; Rolleston & Marshall, 2000). However, it is also known that adenosine can cause dilatation by stimulating A1 receptors on vascular smooth muscle and opening ATP-sensitive K+ (KATP) channels (Dart & Standen, 1993), as well as by the long-accepted mechanism of stimulating A2A receptors and increasing cAMP (Olsson & Pearson, 1990).
Thus, the aims of the present study were (1) to test whether the action of adenosine on A1 receptors is responsible for the increase in FVC that persists in response to systemic hypoxia when NOS is inhibited but basal NO is restored with an NO donor, and (2) to test whether adenosine, acting via A1 or A2A receptors, is responsible for the NO-mediated dilatation of terminal arterioles that we deduced maintains V̇O2 in the face of reduced DO2. The hypothesis that stimulation of A2A receptors might contribute to the maintenance of V̇O2 even though they play no role in the increase in FVC evoked by systemic hypoxia (Bryan & Marshall, 1999a) is reasonable because changes in terminal arteriolar tone can occur without significant effects on gross muscle vascular conductance (Hebert & Marshall, 1988).
Some of the results presented in this paper have already been reported in brief (Edmunds & Marshall, 2000).
METHODS
Experiments were performed on male Wistar rats (200–250 g) in which anaesthesia was induced with an oxygen-halothane mixture (3.5 % halothane) and maintained with Saffan (Schering-Plough Animal Health, Welwyn Garden City, UK) delivered at 7–12 mg kg−1 h−1i.v.. during surgery and at 4–8 mg kg−1 h−1i.v.. during the experimental period (Bryan & Marshall, 1999a). The surgery required to record physiological variables was similar to that described previously (Marshall & Davis, 1999; Edmunds & Marshall, 2001). Briefly, arterial blood pressure (ABP) was recorded from the left brachial artery and FBF was recorded from the right femoral artery via a transonic flow probe (0.7 V) connected to a flow meter T106 (Transonic Systems, Ithaca, NY, USA). FVC was computed online as FBF divided by ABP. The trachea was cannulated so that inspired O2 could be altered by changing the mixture of N2 and O2 delivered across the sidearm of the cannula. Samples of arterial blood were taken from a cannula in the right femoral artery, while samples of venous blood from the right hindlimb were taken from a cannula placed in the left femoral vein, and advanced so that the tip lay at the bifurcation of the inferior vena cava. Arterial and venous blood samples (65 μl) were analysed for O2 content using a co-oximeter (IL-682 CO-Oximeter, Instrumentation Laboratory, Lexington, MA, USA). These measurements, together with the measurement of FBF, allowed the calculation of hindlimb DO2 and V̇O2; these parameters are expressed per unit body weight (Edmunds & Marshall, 2001). At the end of each experiment animals were killed by anaesthetic overdose followed by cervical dislocation.
All variables were recorded on an Apple Power Mac computer (4400/160) using MacLab 8/s (AD Instruments, Hastings, West Sussex, UK).
Protocols
Series 1: interactions between adenosine and NO
In group 1 (n = 8 animals), after a 25 min stabilisation period following surgery when animals breathed room air, the inspirate was changed to 12 % or 8 % O2 for 5 min periods. The order was varied between animals and 15 min was allowed for recovery between the hypoxic challenges. The NOS inhibitor l-NAME (10 mg kg−1, i.v..) was then given and 20 min later the hypoxic challenges were repeated. Following this, baseline FVC was restored by continuous infusion of sodium nitroprusside (SNP; 10 μg kg−1 min−1i.a.), and the hypoxic challenges were repeated. The selective adenosine A1 receptor agonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) was then administered (0.1 mg kg−1i.v..) whilst SNP infusion was continued, and 20 min later the hypoxic stimuli were repeated. In group 2 (n = 8), the same protocol was repeated except that the selective adenosine A2A receptor antagonist 4-(2-[7-amino-2-(2-furyl) (triazolo{2,3-a}-[1,3,5]triazin- 5-ylamino]ethyl)phenol (ZM241385) was administered (0.05 mg kg−1i.v..; Bryan & Marshall, 1999a) instead of DPCPX.
Series 2: determination of DO2,crit
In control rats (n = 8), cardiovascular variables were measured continuously while the animals breathed 21 % O2 spontaneously. After a 25 min stabilisation period, the inspirate was changed, for 5 min periods, to a range of different hypoxic mixtures: 14, 12, 10, 9, 8, 7, 6 and sometimes 5 % O2 in N2 (Edmunds & Marshall, 2001). The order of the hypoxic mixtures was randomised and at least 15 min breathing 21 % O2 was allowed between successive periods of hypoxia. During the 5th minute of each hypoxic challenge, blood samples were taken from the femoral vein and artery cannulae, so that hindlimb DO2 and V̇O2 could be calculated at each level of hypoxia. The DO2,crit was calculated as described previously (Samsel & Schumacker, 1988; Edmunds & Marshall, 2001).
In a second group of animals (n = 8), after an initial stabilisation period, DPCPX was administered (0.1 mg kg−1i.v..; Bryan & Marshall, 1999a), and 20 min later the protocol described above was performed. Since preliminary experiments suggested that the value of DO2,crit was increased (data not shown), a slightly different series of hypoxic challenges was used to allow DO2,crit to be calculated accurately: 18, 16, 14, 12, 10, 9, 8, 7 and 6 % inspired O2 in N2. The DPCPX was dissolved in 10 % DMSO/0.1 m NaOH (50/50 v/v) then diluted in saline. Therefore, a further series of experiments (n = 8) was conducted to control for this vehicle.
In a fourth group of animals (n = 8), after an initial stabilisation period ZM241385 was administered (0.05 mg kg−1, i.v..), and after 20 min the initial protocol described was performed. In vivo, the effective half-life for ZM241385 is relatively short (Keddie et al. 1996). Therefore, supplementary doses of ZM241385 (0.05 mg kg−1i.v..) were given every hour after the initial dose.
DPCPX was purchased from Research Biochemicals; ZM241385 was a kind gift from Zeneca Pharmaceuticals, and was dissolved in 3 % polyethylene glycol 400/0.1 m NaOH (50/50 v/v), and then diluted in saline (see Bryan & Marshall, 1999a).
Statistical analysis of data
All data are expressed as means ±s.e.m. Changes in FVC were computed as the integrated FVC in conductance units, for the 5 min period during the hypoxic stimulus minus the integrated baseline FVC measured for 5 min before hypoxia (see Edmunds & Marshall, 2001). In series 1 experiments, differences in the FVC integral were determined with repeated-measures ANOVA. In all cases, the level of statistical significance was set at P < 0.05. In series 2 experiments, the effects of hypoxia on baseline levels of mean arterial pressure (MAP) and FBF within groups were analysed using Student's paired t test. Changes in MAP and FVC were compared at each level of hypoxia between different groups of animals by using one-way ANOVA for multiple comparisons, and Tukey's post hoc test for differences at particular levels of hypoxia. The DO2,crit values were compared using Student's unpaired t test. Data from control animals in series 2 have been published before (Edmunds & Marshall, 2001).
RESULTS
Series 1: interactions between adenosine and NO
In group 1, in the absence of any drug, each level of systemic hypoxia (12 and 8 % inspired O2) evoked a pronounced increase in FVC, indicating hindlimb vasodilatation (Fig. 1), accompanied by a marked decrease in MAP (data not shown). After l-NAME, baseline MAP increased and FVC decreased (Table 1), which is indicative of peripheral and hindlimb vasoconstriction, respectively. Infusion of SNP after l-NAME restored baseline FVC to values similar to those seen prior to the infusion of l-NAME, although MAP fell disproportionately (Table 1). Nevertheless, as we described recently (Edmunds & Marshall, 2001), infusion of SNP completely restored the hypoxia-evoked increases in FVC, despite continued NOS blockade (Fig. 1). Under these conditions, DPCPX attenuated the hypoxia-evoked increases in FVC by about 60 and 40 % during 12 % O2 and 8 % O2, respectively (Fig. 3). The baseline cardiovascular variables for group 2 are also shown in Table 1; they followed a similar pattern to those in group 1. Similarly, FVC increased in response to each level of hypoxia (Fig. 2). This effect was attenuated by l-NAME, and restored by infusion of SNP (Fig. 2). Interestingly, subsequent infusion of ZM241385 partially attenuated the increase in FVC evoked by 12 % inspired O2, but not that evoked by 8 % inspired O2.
Figure 1. Efects of l-NAME, l-NAME with SNP and l-NAME with SNP and DPCPX on the changes in FVC evoked by hypoxia.
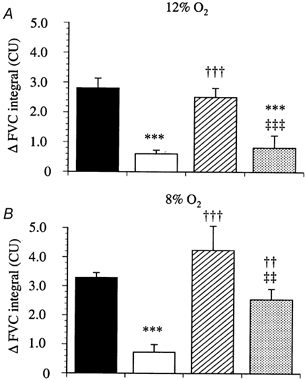
Each column represents the mean ±s.e.m. of the change in integrated FVC (in conductance units, CU) after 12 % inspired O2 (A) and 8 % inspired O2 (B), for control responses (▪), after l-NAME (10 mg kg−1, □), after l-NAME and SNP (10 μg kg−1 min−1,  ) and after l-NAME, SNP and DPCPX (0.1 mg kg−1,
) and after l-NAME, SNP and DPCPX (0.1 mg kg−1,  ). ***P < 0.001 vs. control; ††P < 0.01, †††P < 0.001 vs.l-NAME; ‡‡P < 0.01, ‡‡‡P < 0.001 vs.l-NAME + SNP.
). ***P < 0.001 vs. control; ††P < 0.01, †††P < 0.001 vs.l-NAME; ‡‡P < 0.01, ‡‡‡P < 0.001 vs.l-NAME + SNP.
Table 1.
Baseline cardiovascular variables in series 1 experiments after l-NAME (10 mg kg−1), l-NAME with SNP (10 μg kg−1 min−1) and l-NAME ith SNP and either DPCPX (0.1 mg kg−1, group 1) or ZM241385 (0.05 mg kg−1, group 2)
| MAP (mmHg) | FBF (ml min−1) | FVC (ml min−1 mmHg−1) | |
|---|---|---|---|
| Group 1 | |||
| Control | 120 ± 3 | 2.0 ± 0.5 | 0.0162 ± 0.0014 |
| l-NAME | 143 ± 2 *** | 1.1 ± 0.1 ** | 0.0077 ± 0.0007 *** |
| l-NAME + SNP | 104 ± 4 ***††† | 1.5 ± 0.1 | 0.0139 ± 0.0010 †† |
| l-NAME + SNP + DPCPX | 120 ± 2 †††‡‡‡ | 1.7 ± 0.2 † | 0.0137 ± 0.0018 †† |
| Group 2 | |||
| Control | 119 ± 5 | 2.1 ± 0.2 | 0.0175 ± 0.0021 |
| l-NAME | 151 ± 3 *** | 1.2 ± 0.1 * | 0.0078 ± 0.0007 *** |
| l-NAME + SNP | 111 ± 3 *††† | 1.5 ± 0.1 | 0.0152 ± 0.0012 † |
| l-NAME + SNP + ZM241385 | 121 ± 6 ††† | 1.8 ± 0.2 | 0.0149 ± 0.0020 † |
Values are means ±s.e.m. MAP, mean arterial pressure; FBF, femoral blood flow; FVC, femoral vascular conductance.
Significantly different vs. control
significantly different vs.l-NAME
significantly different vs.l-NAME with SNP, where 1, 2 and 3 symbols indicate p < 0.05, p < 0.01 and p < 0.001, respectively.
Figure 3. Changes in MAP and FVC evoked by graded systemic hypoxia.
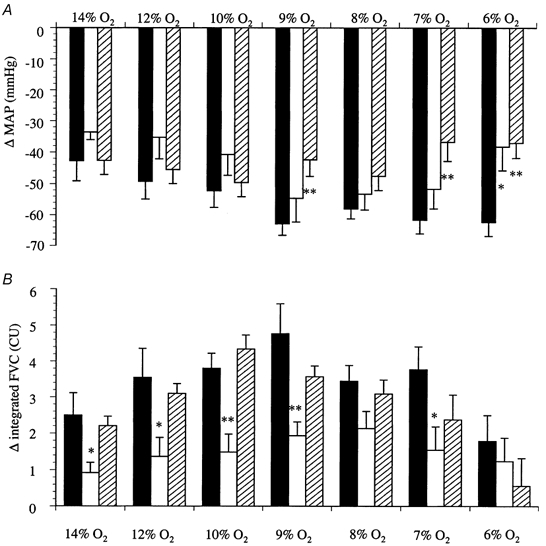
Each column represents the mean ±s.e.m. of the change in MAP (A) or integrated FVC (B) recorded over 5 min periods of hypoxia; the percentage O2 in the inspirate is indicated for each column. Control animals, ▪; after DPCPX (0.1 mg kg−1, i.v..), □; and after ZM243186 (0.05 mg kg−1, i.v..),  . *P < 0.05 and **P < 0.01, when compared with control.
. *P < 0.05 and **P < 0.01, when compared with control.
Figure 2. Effects of l-NAME, l-NAME with SNP and l-NAME with SNP and ZM243185 on the changes in FVC evoked by hypoxia.
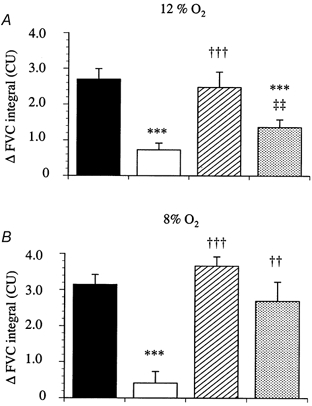
Each column represents the mean ±s.e.m. of the change in integrated FVC after 12 % inspired O2 (A) and 8 % inspired O2 (B), for control responses (▪), after l-NAME (10 mg kg−1, □), after l-NAME and SNP (10 μg kg−1 min−1,  ) and after l-NAME, SNP and ZM243185 (0.05 mg kg−1,
) and after l-NAME, SNP and ZM243185 (0.05 mg kg−1,  ). ***P < 0.001 vs. control; ††P < 0.01, †††P < 0.001 vs.l-NAME; ‡‡P < 0.01 vs.l-NAME + SNP.
). ***P < 0.001 vs. control; ††P < 0.01, †††P < 0.001 vs.l-NAME; ‡‡P < 0.01 vs.l-NAME + SNP.
Series 2: determination of DO2,crit
Control animals
The changes in cardiovascular parameters induced by graded hypoxia have been described in detail previously (Marshall & Metcalfe, 1988; Edmunds & Marshall, 2001), and are shown in Fig. 3. As expected, each level of hypoxia caused a decrease in MAP (P < 0.05; Fig. 3) and an increase in FVC (P < 0.01; Fig. 3). In spite of a decrease in MAP during hypoxia, FBF was generally well maintained due to the increase in FVC (Table 2). However, when breathing 6 % O2 the increase in FVC was not sufficient to sustain FBF (21 % O2vs. 6 % O2: 1.8 ± 0.2 vs. 1.0 ± 0.2 ml min−1, P < 0.05; Table 2). As described previously (Edmunds & Marshall, 2001), DO2 decreased progressively with the severity of the hypoxic challenge. V̇O2 was maintained during moderate levels of hypoxia, and was thus delivery independent (Fig. 4A). However, during more severe hypoxic challenges, when DO2 was reduced to lower values, V̇O2 declined and thus became delivery dependent. Calculation of DO2,crit from each individual animal gave a value of 0.64 ± 0.06 ml O2 min−1 kg−1 (Fig. 4A; Edmunds & Marshall, 2001).
Table 2.
MAP and FBF during air breathing and at the 5th minute of each hypoxic challenge for control, DPCPX (0.1 mg kg−1) and ZM241385 (0.05 mg kg−1) groups
| Control | DPCPX | ZM241385 | ||
|---|---|---|---|---|
| MAP (mmHg) | Before drug | — | 118 ± 4 | 114 ± 3 |
| 21 % O2 | 119 ± 2 | 129 ± 3 † | 118 ± 3 | |
| 14 % O2 | 77 ± 4 | 89 ± 10 | 78 ± 3 | |
| 12 % O2 | 71 ± 4 | 83 ± 6 | 73 ± 3 | |
| 10 % O2 | 67 ± 3 | 71 ± 7 | 72 ± 3 | |
| 9% O2 | 59 ± 4 | 68 ± 6 | 72 ± 2 | |
| 8% O2 | 62 ± 2 | 76 ± 5 | 70 ± 3 | |
| 7% O2 | 50 ± 4 | 72 ± 7 | 72 ± 4 | |
| 6% O2 | 60 ± 4 | 78 ± 4 * | 80 ± 4 * | |
| FBF (ml min−1) | Before drug | — | 2.1 ± 0.2 | 1.9 ± 0.1 |
| 21 % O2 | 1.8 ± 0.2 | 2.1 ± 0.1 | 2.0 ± 0.2 | |
| 14 % O2 | 1.9 ± 0.1 | 1.8 ± 0.1 | 1.9 ± 0.1 | |
| 12 % O2 | 1.7 ± 0.2 | 1.8 ± 0.2 | 2.0 ± 0.1 | |
| 10 % O2 | 2.0 ± 0.2 | 1.7 ± 0.2 | 2.3 ± 0.1 | |
| 9% O2 | 2.0 ± 0.2 | 1.8 ± 0.2 | 2.2 ± 0.1 | |
| 8% O2 | 1.7 ± 0.2 | 2.1 ± 0.2 | 1.9 ± 0.2 | |
| 7% O2 | 1.8 ± 0.2 | 1.5 ± 0.1 | 1.8 ± 0.2 | |
| 6% O2 | 1.0 ± 0.2 | 1.0 ± 0.1 | 1.5 ± 0.2 |
Values are means ±s.e.m.
P < 0.05 vs. control
p < 0.05 for before DPCPX vs. after DPCPX.
Figure 4. Effects of graded systemic hypoxia on DO2 and V̇O2 of the rat hindlimb in control animals and after administration of adenosine antagonists.
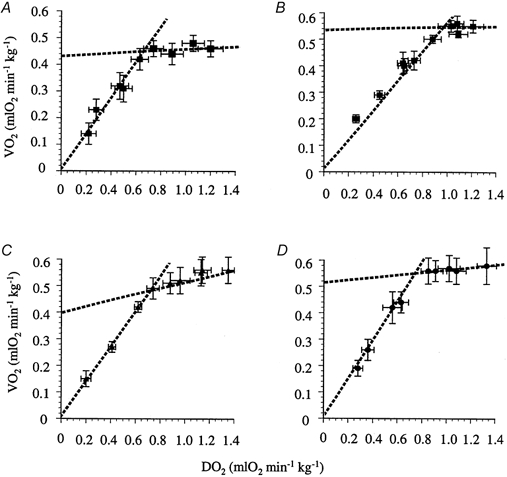
Each value represents the mean ±s.e.m. for V̇O2 and DO2 during air breathing or after 5 min periods of hypoxia. A, control animals. B, animals treated with DPCPX (0.1 mg kg−1). C, animals treated with the vehicle for DPCPX. D, animals treated with ZM243186 (0.05 mg kg−1).
Effects of DPCPX
Administration of the selective adenosine A1 receptor antagonist DPCPX resulted in a small, but significant increase in baseline MAP (Table 2), indicating that under resting conditions MAP is under the tonic influence of adenosine. Baseline FVC was not altered by DPCPX (0.0171 ± 0.0012 vs. 0.0159 ± 0.0011 ml min−1 mmHg−1, before vs. after DPCPX, P > 0.05), indicating that the increase in MAP seen with DPCPX was not the result of skeletal muscle vasoconstriction. In consequence, FBF was slightly elevated when compared with that of control animals, although this difference did not achieve statistical significance (Table 2). DPCPX blunted the hypoxia-evoked increase in FVC by approximately 50 % (P < 0.001 control group vs. DPCPX group; Fig. 3), in agreement with Bryan & Marshall (1999a). The vehicle for DPCPX did not alter the FVC response to hypoxia (data not shown; see Bryan & Marshall, 1999a). The changes in MAP evoked by hypoxia were slightly smaller after DPCPX than in control animals (P < 0.01, control group vs. DPCPX group). This effect was most apparent during the most severe hypoxic challenge (Fig. 3).
During air breathing after DPCPX, hindlimb V̇O2 appeared to be slightly higher than in control animals, although this difference did not achieve statistical significance (0.46 ± 0.03 vs. 0.55 ± 0.02 ml O2 min−1 kg−1, P = 0.41; Fig. 4A vs. B). As indicated in Methods, two milder levels of hypoxia were included in the DPCPX group; this allowed the accurate calculation of the delivery-independent portion of the DO2–V̇O2 relationship. Thus, after administration of DPCPX each level of systemic hypoxia resulted in decreased hindlimb DO2, and delivery-dependent and delivery-independent phases in the DO2–V̇O2 relationship were observed. DO2,crit was calculated to be 0.95 ± 0.07 ml O2 min−1 kg−1 (Fig. 4B). This was significantly greater than the DO2,crit calculated for control animals (0.64 ± 0.06 ml O2 min−1 kg−1, P < 0.01 vs. DPCPX). In animals given the vehicle for DPCPX, again two distinct phases were observed in the relationship between DO2 and V̇O2 (Fig. 4C); DO2,crit in these animals was calculated to be 0.70 ± 0.05 ml O2 min−1 kg−1. This was not significantly different from that calculated in control animals, but was different from the DO2,crit calculated after the infusion of DPCPX (P < 0.01).
Effects of ZM241385
The effects of ZM241385 on baseline values are shown in Table 1; ZM241385 did not alter MAP, FBF or FVC (0.0164 ± 0.0013 vs. 0.0165 ± 0.0011 ml min−1 mmHg−1, before vs. after ZM241385). As the severity of the hypoxic challenges increased, ZM241385 progressively attenuated the evoked decrease in MAP (Fig. 3). However, ZM241385 did not alter the increase in FVC evoked by hypoxia (Fig. 3; see Bryan & Marshall, 1999a). Again, the biphasic relationship between DO2 and V̇O2 was observed (Fig. 4D); DO2,crit was calculated to be 0.73 ± 0.02 ml O2 min−1 kg−1, a value that was not different from the DO2,crit measured in control animals.
DISCUSSION
Changes in femoral vascular conductance
Adenosine has been proposed to be a mediator of the hypoxia-evoked vasodilatation in skeletal muscle (Mian & Marshall, 1991; Skinner & Marshall, 1996; Bryan & Marshall, 1999b), in the mesenteric circulation (Mian & Marshall, 1995), within the coronary circulation (Deussen et al. 1986; Nakhostine & Lamontagne, 1993, 1994) as well as in the cerebral circulation (Armstead, 1997; Coney & Marshall, 1998). So, a general role for adenosine in hypoxia-evoked vasodilatation is widely accepted. Within hindlimb muscle we have shown previously that the adenosine component of the change in FVC evoked by one level of moderate systemic hypoxia (8 % inspired O2) is mediated by adenosine A1 receptors (see Results, and Bryan & Marshall, 1999a). The present study confirms and extends these observations, demonstrating a role for adenosine A1 receptors in the hypoxia-evoked increase in FVC over a wide range of hypoxic challenges (14–6 % O2). We have also extended our evidence that the increase in FVC is not altered by the selective adenosine A2A receptor antagonist ZM241385 (Bryan & Marshall, 1999a) over the same wide range of hypoxic challenges. Nevertheless, ZM241385 did attenuate partially the decrease in MAP evoked by some of the more severe levels of hypoxia, suggesting that the adenosine released by systemic hypoxia acts on A2A receptors in tissues other than muscle.
In addition, we have confirmed that the hypoxia-evoked increase in FVC is severely attenuated by inhibition of NOS (Skinner & Marshall, 1996; Bryan & Marshall, 1999b) and that the increase in FVC (proximal arteriolar vasodilatation) can be restored by the infusion of a NO donor (Edmunds & Marshall, 2001). A major new finding of the present study is that this restored increase in FVC was attenuated by the A1 receptor antagonist DPCPX. Interestingly, although ZM241385 did not alter the restored increase in FVC evoked by 8 % inspired O2, it did attenuate the restored increase in FVC evoked by 12 % inspired O2. This finding, coupled with the observation that the increase in FVC evoked by adenosine infusion was almost abolished by NOS inhibition (Skinner & Marshall, 1996; Bryan & Marshall, 1999b), suggests that the tonic endothelial release of NO resulting mainly from shear stress (Buga et al. 1991) somehow sensitises the underlying smooth muscle to the vasodilator influence of adenosine.
It should be noted that NO, acting via cGMP, has been shown to sensitise smooth muscle to the actions of the vasodilators that cause an increase in cAMP (De Wit et al. 1994). Both A1 and A2A receptors are present on the vascular smooth muscle of skeletal muscle tissue (Rådegran & Hellsten, 2000). This observation could therefore explain our results with respect to ZM241385, because A2A receptors would be expected to cause vasodilatation by a direct action on vascular smooth muscle via an increase in cAMP. However, this is an unlikely explanation of the results concerning DPCPX, since A1 receptor stimulation would be expected to reduce rather than increase vascular smooth muscle cAMP levels. An alternative possibility is that adenosine acts directly on the vascular smooth muscle A1 receptors that are coupled to vasodilator KATP channels (Dart & Standen, 1993). This is because cGMP, a well-known effector of NO, facilitates the opening of KATP channels (Kubo et al. 1994). Therefore, a synergy between smooth muscle adenosine A1 receptors and shear-activated endothelial NO release is possible. Taken together, these findings lead to the obvious conclusion that the A1-mediated dilatation of proximal arterioles, which is responsible for the increase in FVC evoked by systemic hypoxia, is dependent upon the existence of a basal level of NO, rather than being mediated by an increased synthesis of NO (as we originally proposed; Bryan & Marshall, 1999b).
However, the alternative possibility is that our original proposal was at least partially correct, but that when adenosine A1 receptor-stimulated synthesis of NO is prevented, the action of adenosine on vascular smooth muscle A1 receptors can predominate providing that some NO is present. This alternative proposal is attractive because adenosine A1 receptors are present on skeletal muscle vascular endothelium (Rådegran & Hellsten, 2000) and when stimulated can induce the release of NO from endothelial cells by acting on A1 receptors (Ray & Marshall, 2000; Rolleston & Marshall, 2000). This proposal would, of course, suggest that redundancy exists in the vasodilator signals evoked by hypoxia. This would not be surprising given the importance of maintaining the O2 supply to tissue cells. Interestingly, as mentioned above, adenosine A2A receptors are also able to stimulate the release of NO from endothelial cells (Sobrevia et al. 1997; Ray & Marshall, 2000; Rolleston & Marshall, 2000). During systemic hypoxia, A2A-mediated NO release is not an important component of the adenosine-evoked vasodilatation of either the proximal arterioles or the terminal arterioles (see below).
Effects on DO2 and V̇O2
The second major novel finding of the present study is that DPCPX caused a substantial increase in the DO2,crit, calculated during progressive systemic hypoxia, from 0.64 ± 0.06 ml O2 min−1 kg−1 in control animals to 0.95 ± 0.07 ml O2 min−1 kg−1. This represents a severe disruption in the ability of the hindlimb vasculature to maintain its V̇O2 during decreases in DO2 produced by graded levels of acute systemic hypoxia. The maintenance of V̇O2 during periods of reduced DO2 has been attributed to dilatation of the terminal arterioles (Granger et al. 1976), and the adenosine receptor antagonist 8-phenyltheophylline, which is non-selective between A1 and A2 receptors, reduced the responses of the terminal arterioles that dilated during hypoxia, without altering the responses of terminal arterioles that constricted (Main & Marshall, 1991). Therefore, the present results suggest that adenosine acts on A1 receptors on terminal arterioles as an essential component in the defence of V̇O2 when DO2 is decreased by systemic hypoxia. The heterogeneous mixture of constriction and A1 receptor-mediated dilatation of terminal arterioles allows a redistribution of blood flow and a more homogeneous distribution of O2 within muscle (Harrison et al. 1990; Mian & Marshall, 1991; Marshall, 1995).
In our previous study (Edmunds & Marshall, 2001), when a basal level of NO was maintained by infusion of a NO donor under conditions of NOS blockade, DO2,crit was significantly greater than that measured under control conditions. This indicates that dilatation of the terminal arterioles that maintain V̇O2 requires an increase in the synthesis of NO. The DO2,crit calculated after DPCPX in the present study compares well with that calculated after NOS inhibition (0.95 ± 0.07 ml O2 min−1 kg−1 after DPCPX and 0.96 ± 0.07 ml O2 min−1 kg−1 during NOS inhibition; Edmunds & Marshall, 2001). Thus, it is now reasonable to propose that this terminal arteriolar vasodilatation is, at least in part, mediated by adenosine acting on A1 receptors and stimulating NOS. The evidence that adenosine A1 receptors are present on skeletal muscle vascular endothelium (Rådegran & Hellsten, 2000) and cause HUVECs and rat aorta endothelial cells to release NO when stimulated (Ray & Marshall, 2000; Rolleston & Marshall, 2000) is consistent with this proposal.
The present study has not addressed the issue of whether either A1 or A2A receptors still contribute to setting DO2,crit during NOS inhibition when a background level of NO is replaced by infusion of SNP (Edmunds & Marshall, 2001). The fact that these receptors contribute to the hypoxia-induced increase in FVC under these conditions (see above) raises this possibility. Technical problems, including the use of multiple drugs and increased duration of the experiments, make such studies very difficult to undertake successfully. However, we can say that if there is redundancy in the vasodilator signal at the level of the terminal arterioles, as we have proposed for the proximal arterioles (see above), then it is unable to restore DO2,crit to values observed in control animals.
The action of adenosine on A2A receptors was implicated in the muscle vasodilatation-associated muscle contraction in anaesthetised cats (Poucher, 1996) and in the cerebral vasodilatation effected by acute systemic hypoxia in rats (Coney & Marshall, 1998). Exogenous adenosine can stimulate A2A receptors to evoke an increase in FVC (Bryan & Marshall, 1999a) and A2A receptor stimulation can increase the synthesis of NO by HUVECs or rat aorta endothelium (Sobrevia et al. 1997; Ray & Marshall, 2000; Rolleston & Marshall, 2000). However, the present results not only confirm that there is no apparent role for adenosine A2A receptors in the hypoxia-evoked increase in FVC (see above and Bryan & Marshall, 1999a), but also, given the lack of effect of ZM241385 on DO2,crit, demonstrate that there is no functional role for these receptors in the dilatation of the terminal arterioles during systemic hypoxia.
In summary, we have shown an integrated physiological consequence of adenosine release in the hindlimb during systemic hypoxia. Graded systemic hypoxia results in a reduction in muscle DO2, which is partially compensated for by an adenosine-evoked increase in muscle vascular conductance that is mediated via adenosine A1 receptors. This acts to maintain muscle blood flow, and therefore limits the fall in DO2, in the face of a large decrease in MAP. This adenosine-mediated vasodilatation of proximal arterioles probably requires a basal presence of NO, but not necessarily an increase in the release of NO; it can be mediated by A2A (moderate hypoxia) or A1 receptors when NOS is blocked. Our results also indicate that adenosine, acting via A1 receptors but not A2A receptors, plays an important role in the co-ordinated terminal arteriolar response to hypoxia that is required for V̇O2 to be maintained in the face of a reduced DO2. We suggest that this action of A1 receptors is likely to be mediated by stimulation of NOS and release of NO.
Acknowledgments
This work was funded by the British Heart Foundation.
References
- Armstead WM. Role of nitric oxide, cyclic nucleotides, and the activation of ATP-sensitive K+ channels in the contribution of adenosine to hypoxia-induced pial artery dilation. Journal of Cerebral Blood Flow and Metabolism. 1997;17:100–108. doi: 10.1097/00004647-199701000-00013. [DOI] [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Adenosine receptor subtypes and vasodilatation in rat skeletal muscle during systemic hypoxia: a role for A1 receptors. Journal of Physiology. 1999a;514:151–162. doi: 10.1111/j.1469-7793.1999.151af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Cellular mechanisms by which adenosine induces vasodilatation in rat skeletal muscle: significance for systemic hypoxia. Journal of Physiology. 1999b;514:163–175. doi: 10.1111/j.1469-7793.1999.163af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buga GM, Gold ME, Fukuto JM, Ignarro LJ. Shear stress-induced release of nitric oxide from endothelial cells grown on beads. Hypertension. 1991;17:187–193. doi: 10.1161/01.hyp.17.2.187. [DOI] [PubMed] [Google Scholar]
- Cain SM, Chapler CK. Oxygen extraction by canine hindlimb during hypoxic hypoxia. Journal of Applied Physiology. 1979;46:1023–1028. doi: 10.1152/jappl.1979.46.6.1023. [DOI] [PubMed] [Google Scholar]
- Coney AM, Marshall JM. Role of adenosine and its receptors in the vasodilatation induced in the cerebral cortex of the rat by systemic hypoxia. Journal of Physiology. 1998;509:507–518. doi: 10.1111/j.1469-7793.1998.507bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis SE, Vallet B, Winn MJ, Caufield JB, King CE, Chapler CK, Cain SM. Role of the vascular endothelium in O2 extraction during progressive ischemia in canine skeletal muscle. Journal of Applied Physiology. 1995;79:1351–1360. doi: 10.1152/jappl.1995.79.4.1351. [DOI] [PubMed] [Google Scholar]
- Dart C, Standen NB. Adenosine-activated potassium current in smooth muscle cells isolated from the pig coronary artery. Journal of Physiology. 1993;471:767–786. doi: 10.1113/jphysiol.1993.sp019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deussen A, Moser G, Schrader J. Contribution of coronary endothelial cells to cardiac adenosine production. Pflügers Archiv. 1986;406:608–614. doi: 10.1007/BF00584028. [DOI] [PubMed] [Google Scholar]
- De Wit C, Bismarck P, Pohl U. Synergistic action of vasodilators that increase cGMP and cAMP in the hamster cremaster microcirculation. Cardiovascular Research. 1994;28:1513–1518. doi: 10.1093/cvr/28.10.1513. [DOI] [PubMed] [Google Scholar]
- Duran WN, Renkin EM. Oxygen consumption and blood flow in resting mammalian skeletal muscle. American Journal of Physiology. 1974;226:173–177. doi: 10.1152/ajplegacy.1974.226.1.173. [DOI] [PubMed] [Google Scholar]
- Edmunds NJ, Marshall JM. A role for adenosine A1 receptors in the regulation of hindlimb oxygen consumption during graded systemic hypoxia in the rat. Journal of Physiology. 2000;525.P:15–16P. [Google Scholar]
- Edmunds NJ, Marshall JM. Vasodilatation, oxygen delivery and oxygen consumption in rat hindlimb during systemic hypoxia: roles of nitric oxide. Journal of Physiology. 2001;532:251–259. doi: 10.1111/j.1469-7793.2001.0251g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froneck K, Zweifach BW. Microvascular pressure distribution in skeletal muscle and the effect of vasodilatation. American Journal Physiology. 1975;228:791–796. doi: 10.1152/ajplegacy.1975.228.3.791. [DOI] [PubMed] [Google Scholar]
- Granger HJ, Goodman AH, Granger ND. Role of resistance and exchange vessels in local microvascular control of skeletal muscle oxygenation in the dog. Circulation Research. 1976;38:379–385. doi: 10.1161/01.res.38.5.379. [DOI] [PubMed] [Google Scholar]
- Harrison DK, Kessler M, Knauff SK. Regulation of capillary blood flow and oxygen supply in skeletal muscle in dogs during hypoxemia. Journal of Physiology. 1990;420:431–446. doi: 10.1113/jphysiol.1990.sp017921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert MT, Marshall JM. Direct observations of the effects of baroreceptor stimulation on skeletal muscle circulation of the rat. Journal of Physiology. 1988;400:45–59. doi: 10.1113/jphysiol.1988.sp017109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keddie JR, Poucher SM, Shaw GR, Brooks R, Collis MG. In vivo characterisation of ZM 241385, a selective adenosine A2A receptor antagonist. European Journal of Pharmacology. 1996;301:107–113. doi: 10.1016/0014-2999(96)00020-9. [DOI] [PubMed] [Google Scholar]
- Kubo M, Nakaya Y, Matsuoka S, Saito K, Kuroda Y. Atrial natriuretic factor and isosorbide dinitrate modulate the gating of ATP-sensitive K+ channels in cultured vascular smooth muscle cells. Circulation Research. 1994;74:471–476. doi: 10.1161/01.res.74.3.471. [DOI] [PubMed] [Google Scholar]
- Marshall JM. Skeletal muscle vasculature and systemic hypoxia. News in Physiological Sciences. 1995;1:163–167. [Google Scholar]
- Marshall JM, Davis WR. The effects of acute and chronic systemic hypoxia on muscle oxygen supply and oxygen consumption in the rat. Experimental Physiology. 1999;84:57–68. doi: 10.1111/j.1469-445x.1999.tb00072.x. [DOI] [PubMed] [Google Scholar]
- Marshall JM, Metcalfe JD. Analysis of the cardiovascular changes induced in the rat by graded levels of systemic hypoxia. Journal of Physiology. 1988;407:385–403. doi: 10.1113/jphysiol.1988.sp017422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mian R, Marshall JM. The role of adenosine in dilator responses induced in arterioles and venules of rat skeletal-muscle by systemic hypoxia. Journal of Physiology. 1991;443:499–511. doi: 10.1113/jphysiol.1991.sp018847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mian R, Marshall JM. The role of adenosine in mediating vasodilatation in mesenteric circulation of the rat in acute and chronic hypoxia. Journal of Physiology. 1995;489:225–234. doi: 10.1113/jphysiol.1995.sp021044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakhostine N, Lamontagne D. Adenosine contributes to hypoxia-induced vasodilatation through ATP-sensitive K+ channel activation. American Journal of Physiology. 1993;265:H1289–1293. doi: 10.1152/ajpheart.1993.265.4.H1289. [DOI] [PubMed] [Google Scholar]
- Nakhostine N, Lamontange D. Contribution of prostaglandins in hypoxia-induced vasodilatation in isolated rabbit hearts. Relation to adenosine and KATP channels. Pflügers Archiv. 1994;428:526–532. doi: 10.1007/BF00374574. [DOI] [PubMed] [Google Scholar]
- Olsson RA, Pearson JD. Cardiovascular purinoceptors. Physiological Reviews. 1990;70:761–845. doi: 10.1152/physrev.1990.70.3.761. [DOI] [PubMed] [Google Scholar]
- Poucher SM. The role of the A2A adenosine receptor subtype in functional hyperaemia in the hindlimb of anaesthetized cats. Journal of Physiology. 1996;492:495–503. doi: 10.1113/jphysiol.1996.sp021324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rådegran G, Hellsten Y. Adenosine and nitric oxide in exercise-induced human skeletal muscle vasodilatation. Acta Physiologica Scandinavica. 2000;168:575–591. doi: 10.1046/j.1365-201x.2000.00705.x. [DOI] [PubMed] [Google Scholar]
- Ray C, Marshall JM. Interactions of adenosine, nitric oxide and the cyclo-oxygenase pathway in freshly excised rat aorta. Journal of Physiology. 2000;528.P:111P. [Google Scholar]
- Rolleston S, Marshall JM. The ability of adenosine to generate nitric oxide in human umbilical vein endothelial cells. Journal of Physiology. 2000;523.P:151P. [Google Scholar]
- Samsel RW, Schumacker PT. Determination of the critical O2 delivery from experimental data: sensitivity to error. Journal of Applied Physiology. 1988;64:2074–2082. doi: 10.1152/jappl.1988.64.5.2074. [DOI] [PubMed] [Google Scholar]
- Skinner MR, Marshall JM. Studies on the roles of ATP, adenosine and nitric oxide in mediating muscle vasodilatation induced in the rat by acute systemic hypoxia. Journal of Physiology. 1996;495:553–560. doi: 10.1113/jphysiol.1996.sp021615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobrevia L, Yudilevich DL, Mann GE. Activation of A2A-purinoceptors by adenosine stimulates L-arginine transport (system y+) and nitric oxide synthesis in human fetal endothelial cells. Journal of Physiology. 1997;499:135–140. doi: 10.1113/jphysiol.1997.sp021916. [DOI] [PMC free article] [PubMed] [Google Scholar]