

Abstract
Whole cell recordings from CA1 pyramidal cells were performed to investigate the interaction between excitatory postsynaptic potentials (EPSPs) or currents (EPSCs), and the slow Ca2+-dependent K+ current, IsAHP. Blockers of the slow afterhyperpolarization (sAHP) such as isoprenaline (ISO) or noradrenaline (NA) reduced the hyperpolarization that followed a short train of EPSPs, and slowed the decay of summated EPSPs or EPSCs.
ISO/NA action on synaptic responses was observed in the absence of action potentials, but was curtailed by Ca2+ chelation (10 mm EGTA in the electrode) and was not observed with a caesium-based recording solution. This suggests the involvement of an ISO/NA-sensitive Ca2+-dependent K+ current without a requirement for regenerative spiking.
An ISO/NA-sensitive sAHP was observed following both NMDA and non-NMDA receptor-mediated EPSP trains in nominally zero Mg2+ medium. Isoprenaline sensitivity was blocked by hyperpolarization during EPSPs or by isradipine, suggesting a requirement for voltage-dependent Ca2+ influx during EPSPs. The data indicate that bursts of EPSPs can activate voltage-gated Ca2+ channels, which trigger IsAHP during synaptic responses.
A decrease in EPSP temporal summation occurred during both spike-evoked sAHPs and persistent activation of sAHP conductance following internal dialysis with diazo-2 (2 mm). At constant membrane potential, diazo-2 caused a decrease in membrane time constant and input resistance and accelerated the rate of EPSP decay. Photolysis of diazo-2 or application of NA reduced the resting sAHP conductance, causing an increased membrane time constant and input resistance in association with an increase in EPSP half-width.
These results indicate that short bursts of EPSPs can activate a Ca2+-dependent K+ current resembling IsAHP, and that activation of this current reduces the postsynaptic response to high-frequency synaptic input. The findings imply that modulation of IsAHP can regulate synaptic efficacy and may influence the threshold for tetanus-induced synaptic plasticity.
In many neurones propagation of synaptic signals from dendrites to the site of output at the soma/axon hillock is accompanied by modulation due to the activity of intrinsic postsynaptic ion channels. Thus, excitatory postsynaptic potentials (EPSPs) can be boosted by voltage-gated Na+ and Ca2+ channels (Magee & Johnston, 1995; Lipowsky et al. 1996) and, conversely, voltage-gated K+ currents attenuate EPSP amplitude (Hoffman et al. 1997). Additionally, summation of EPSPs can be regulated by the hyperpolarization-activated cation current, IH (Magee, 1998). However, little is known about the influence of postsynaptic Ca2+-dependent K+ channels on synaptic responses, although these channels are distributed widely in mammalian neurones.
The slow AHP current (IsAHP) is a voltage-independent, Ca2+-dependent K+ current which follows short bursts of action potentials and restrains repetitive firing in hippocampal pyramidal neurones. In most studies, this current has been activated by the Ca2+ influx associated with action potentials or large depolarizations. There is, however, evidence to suggest that IsAHP should be apparent during repetitive synaptic activity; the β-adrenergic sensitivity of long-term potentiation (LTP) induction thresholds has been suggested to result from involvement of IsAHP in the postsynaptic response to high-frequency synaptic input (Sah & Bekkers, 1996). This possibility has not been examined carefully, although the presence of such a potential has been mentioned briefly (Doze et al. 1991).
During excitatory synaptic input in CA1 pyramidal cells, Ca2+ influx may occur through NMDA receptor channels (Alford et al. 1993; Kovalchuk et al. 2000) or locally activated voltage-gated Ca2+ channels without calcium spiking (Miyakawa et al. 1992; Magee & Johnston, 1995). Therefore, as the first objective of this study, we examined the ability of subthreshold (non-spiking) repetitive excitatory synaptic input to activate Ca2+-dependent K+ current. The results indicate that even without action potentials, synaptic input can generate sufficient Ca2+ influx to elicit Ca2+-dependent K+ current.
Thus during normal cellular activity, if IsAHP is active during synaptic input what are the functional consequences of this K+ conductance for synaptic responses detected at the soma? In CA1 pyramidal cells, a substantial IsAHP-like current can be measured in recordings from the soma and/or in proximal apical dendrites (Andreasen & Lambert, 1995; Lancaster & Batchelor, 2000). This location suggests that the sAHP conductance could shape synaptic input propagating to the soma, although this apical dendritic location is under question (Bekkers, 2000). It is clear that intrinsic conductances can modify synaptic responses (see above), but comparable quantification for the sAHP conductance is more difficult because it requires Ca2+ entry which itself might modify synaptic responses (Kyrozis et al. 1996). To overcome this problem, we have used a novel method to elicit steady-state activation and deactivation of IsAHP without measurable elevation of internal Ca2+ (Lancaster & Batchelor, 2000). This has allowed us to quantify the electrical interaction between EPSPs and IsAHP in CA1 neurones. The results indicate that the characteristics of IsAHP are well suited for modulation of temporal summation.
Some of this work has been published in abstract form (Lancaster et al. 1999, 2000).
METHODS
Hippocampi were obtained from 2- to 3-week-old rats after halothane anaesthesia or cervical dislocation followed by decapitation. The care and use of animals was in accordance with institutional guidelines. Transverse slices (400 μm) were prepared using methods that have been described previously (Pedarzani & Storm 1993; Lancaster & Batchelor, 2000). The bath solution comprised (mm): 119 NaCl, 2.5 KCl, 1.3 MgCl2, 2.0 CaCl2, 1.0 NaH2PO4, 26 NaHCO3, and 11 glucose, equilibrated with 95 % O2-5 % CO2 at a temperature of 28–30 °C or room temperature (20–23 °C). To maximize Ca2+ influx through NMDA channels, which might contribute to activation of Ca2+-dependent K+ channels, Mg2+ was omitted from the external solution (nominally zero Mg2+) in some experiments (Figs 1C–4).
Figure 1. Bursts of excitatory synaptic input generate noradrenaline-sensitive current.

A, averages of 8 consecutive traces before and after noradrenaline (10 μm) application which reduced the hyperpolarization following 5 EPSPs (50 Hz). EPSPs were preceded by brief current pulses to monitor input resistance. Inset, the inhibitory action of noradrenaline (10 μm, applied for 3 min) on the depolarization-evoked IsAHP following an 80 ms voltage command to −10 mV from Vh −60 mV. B, summary data of the action of noradrenaline on the hyperpolarization following a short train of EPSPs as in A (n = 6). C, in voltage clamp, sequential application of 10 μm bicuculline methiodide (bath-applied for 6 min) and noradrenaline (bath-applied for 3 min) caused a slower decay of the inward current following the final EPSC (Vh−55 mV). Da, NMDA receptor-mediated synaptic responses (in 4 μm NBQX, 10 μm bicuculline methiodide) to paired stimuli. The decay of the response is prolonged by 4 μm isoprenaline (Vh−54 mV). Db, a digital subtraction of the two traces in Da, represents the isoprenaline-sensitive current.
Figure 4. An isoprenaline-sensitive slow afterhyperpolarization (sAHP) can be evoked by NMDA receptor-mediated EPSPs.
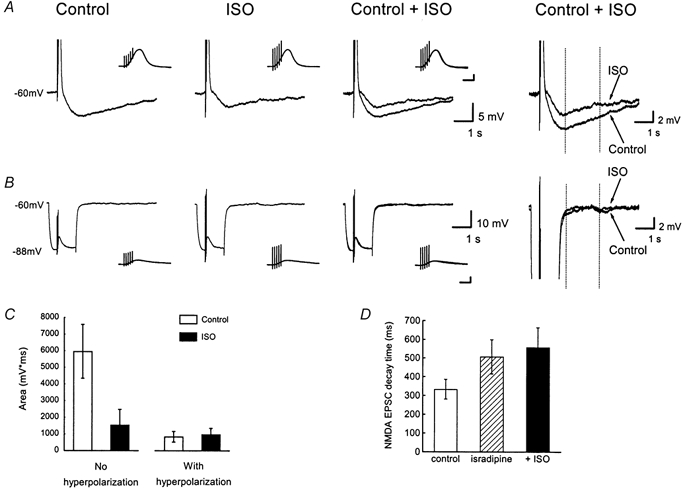
The slices were maintained at room temperature in a nominally Mg2+-free medium designed to isolate NMDA receptor-mediated responses (see text). A, a train of five evoked EPSPs (at 50 Hz) was followed by a sAHP (Control). Bath application of 10 μm isoprenaline (ISO) inhibited the sAHP. To the right, the traces before and after application of ISO are shown on an expanded scale. The vertical dotted lines indicate the integration period over which the sAHP area was measured (columns in C). In each case, the cell was held at −60 mV prior to stimulation. The insets in A and B show the EPSP trains at an expanded time scale. Scale bars: 10 mV, 100 ms. B, when the identical train of five EPSPs was superimposed on a hyperpolarizing current pulse, there was no sAHP. Same cell as in A. The insets show the EPSP trains at an expanded time scale. Scale bars: 10 mV, 100 ms. C, summary data from 6 experiments of the kind shown in A and B. The sAHP area was measured by integration between the vertical dotted lines in right-hand panels of A and B. □, control medium. ▪, after isoprenaline application. D, summary data (n = 5) of EPSC train decay during voltage clamp experiments similar to Fig. 2B. Columns show the 90–10 % decay time of EPSCs under control conditions (□), following application of isradipine (10 μm,  ) and sequential application of isoprenaline (ISO, 10 μm, ▪).
) and sequential application of isoprenaline (ISO, 10 μm, ▪).
For isolation of NMDA receptor-mediated synaptic responses, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulphonamide (NBQX; 3–5 μm) or 6,7-dinitroquinoxaline-2,3-dione (DNQX; 10 μm; both from Tocris, Bristol, UK) and the free base (or methiodide salt, Fig. 1) of bicuculline (10 μm, Tocris, Bristol, UK) were added to the bath. For isolation of non-NMDA (AMPA/kainate) receptor-mediated synaptic responses, d- or dl-aminophosphonovalerate (AP5, 50 or 100 μm, respectively, Tocris) and the free base (or methiodide salt, Fig. 1) of bicuculline (10 μm) were added to the bath. When inhibition was blocked, the Schaffer collateral- commissural pathway to CA1 was cut at CA2 for recordings at 28–30 °C, to prevent bursting activity. In some experiments GABAA receptors were blocked with picrotoxin (100 μm), GABAB receptors were blocked with 2-hydroxysaclofen (200 μm, Tocris), metabotropic glutamate receptors were blocked by (S)-α-methyl-4-carboxyphenylglycine (MCPG; 0.5 mm, Tocris) and the β-adrenergic agonist-sensitive current, IH, was blocked using ZD 7288 (10 μm, Tocris). Noradrenaline or isoprenaline bitartrate was added from freshly made stock solution. Chemicals were obtained from Sigma unless stated otherwise.
The standard solution inside whole cell recording pipettes was (mm): 150 KMeSO4 (Phase Separations, Wales, and ICN Biomedicals, Aurora, OH, USA), 10 KCl, 10 Hepes, 0–4 NaCl, 4 Mg2ATP, 0.4 Na3GTP. This solution was adjusted to 280–290 mosmol l−1 (pH 7.35–7.4). In some experiments 140 KMeSO4 was used, or the following compounds were added to the pipette solution: diazo-2 (2 mm, Molecular Probes, Eugene, OR, USA), KEGTA (10 mm, pH 7.4), QX-314 (0.5–2 mm, Alomone Labs, Jerusalem). The data in Fig. 2 were obtained with pipette solutions in which caesium gluconate replaced KMeSO4. Field responses were recorded using pipettes filled with the external solution. The Schaffer collateral- commissural pathway to CA1 was stimulated with an electrode placed in the stratum radiatum. An Axopatch-1D (Axon Instruments, Foster City CA, USA) was used for voltage clamp recordings. In most experiments 80 % compensation of the series resistance (6–20 MΩ) was used. Data were filtered at 5 or 2 kHz and collected using a split clock with a frequency of 10 kHz for the synaptic waveform. Current clamp data were collected using an Axoclamp-2A or Axopatch-1D (Axon Instruments).
Figure 2. Internal Cs+ blocks the noradrenergic modulation of EPSCs.
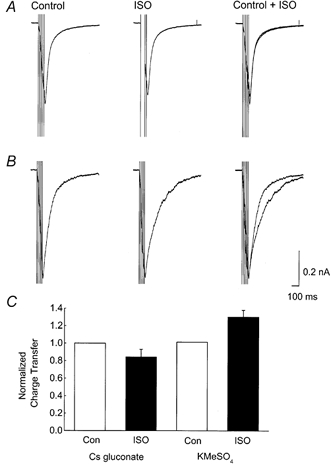
A, examples of NMDA receptor-mediated EPSCs (5 at 50 Hz) recorded with a caesium gluconate-based intracellular solution to suppress K+ currents, before (Control) and during (ISO) application of 10 μm isoprenaline. There was no measurable difference in the synaptic current between the two conditions (superimposed records: Control and ISO). B, NMDA receptor-mediated EPSCs recorded with a KMeSO4-based intracellular solution before (Control) and during (ISO) application of 10 μm isoprenaline. Under these conditions, an isoprenaline-sensitive outward current was observed. C, summary data showing that the Cs+-based intracellular solution (n = 5) blocked the isoprenaline-sensitive increase in charge transfer observed with KMeSO4 electrodes (n = 9).
Membrane time constants were obtained from single exponential fits to the charging curve generated by 30 ms 0.1 nA hyperpolarizing current pulses (from resting membrane potentials ∼-65 mV). Input resistances were also estimated from these data; these isochronal (30 ms), values are underestimates since membrane charging is incomplete. Diazo-2 photolysis was performed using the 1 ms discharge from a xenon arc bulb (filtered with a UG11) of a Rapp flashlamp (Hi-Tech Scientific, Salisbury, UK) which illuminated the CA1 region. Data analysis and acquisition used pCLAMP 6/7 (Axon Instruments) and Origin (MicroCal Software Inc., Northampton, MA, USA). Summary data are plotted as means ±s.e.m. Statistical significance was assessed using a Student's paired or unpaired t test as appropriate.
RESULTS
Activation of IsAHP during synaptic responses
Whole cell patch clamp recordings were obtained from the somata of CA1 pyramidal cells (n = 71) in transverse hippocampal slices from young rats. The recording conditions were designed to be optimal for preserving IsAHP in a form similar to that observed following action potentials with sharp microelectrode recording (see Methods). This current was completely and reversibly inhibited by 10 μm noradrenaline (Fig. 1A inset) or isoprenaline (ISO; 2–10 μm). This is a well-defined action via β-adrenergic receptors, cAMP and protein kinase A (PKA; Madison & Nicoll, 1986; Pedarzani & Storm, 1993). Together with appropriate controls, this characteristic sensitivity to β-adrenergic agonists can be used to probe for activation of IsAHP during synaptic responses.
In order to test the action of noradrenaline on excitatory synaptic input, short bursts (5 stimuli at 50 Hz) of synaptic stimulation were used to evoke EPSPs under conditions of minimal pharmacological intervention. It was, however, necessary to block concurrent activation of GABAA receptors (using picrotoxin, 100 μm), which are PKA sensitive. Under these conditions the EPSPs were followed by a hyperpolarization that was partially sensitive to noradrenaline (10 μm, Fig. 1A). The hyperpolarization area was measured over a 200 ms time window starting when membrane potential reached the resting value on the EPSP decay. The summary data in Fig. 1B (n = 6) show that noradrenaline reduced the hyperpolarization by 47 % which suggests that some IsAHP might be activated following a train of EPSPs. The noradrenaline-resistant component may be due to synaptically activated GABAB receptors or voltage-dependent channels modulated by the depolarization.
Maintaining a reasonable driving force for a K+ conductance without allowing the summated EPSPs to evoke action potentials was often intractable, so further experiments (Figs 1C–5) were performed with a low concentration of QX-314 (0.5–2 mm) in the recording pipette to suppress spiking. In whole cell voltage clamp (holding potential ∼-60 mV), trains of five stimuli at 50 Hz produced excitatory postsynaptic currents (EPSCs). Bath application of noradrenaline (10 μm) produced a slowing of the decay of the synaptic response following the peak of the last EPSC (Fig. 1C, n = 7). The possible involvement of GABAA receptors in the noradrenaline action (Andreasen & Lambert, 1991) can be excluded since the effect of noradrenaline was observed in the presence of 10 μm bicuculline, which clearly blocked the early inhibitory phase of the synaptic response (Fig. 1C). Noradrenaline or 8Br-cAMP has no effect on GABAB receptor-mediated synaptic responses in these cells (Newberry & Nicoll, 1984; Doze et al. 1991), and in our experiments the action of isoprenaline was not inhibited by the GABAB receptor blocker 2-hydroxysaclofen (200 μm, n = 9, data not shown). The time course of the current that was suppressed by β-adrenergic stimulation is illustrated in Fig. 1D, which shows NMDA receptor-mediated synaptic responses before and after bath-application of 4 μm isoprenaline (ISO, Fig. 1Da). Digital subtraction of the two traces in Fig. 1Da shows that the isoprenaline-sensitive component is a slowly decaying outward current lasting for ∼500 ms (Fig. 1Db). A similar β-adrenergic/noradrenaline-sensitive current was observed in five cells.
Figure 5. An isoprenaline-sensitive slow afterhyperpolarization (sAHP) can be evoked by non-NMDA receptor-mediated EPSPs.

The slices were bathed in medium designed to isolate non-NMDA receptor-mediated synaptic responses (see text). A, a train of five EPSPs (50 Hz) was followed by a sAHP (Control). The sAHP was blocked by bath application of 10 μm isoprenaline (ISO). Superimposed and amplified traces are shown to the right. In each case, the cell was held at −60 mV prior to stimulation. The insets in A and B show the EPSP trains at an expanded time scale. Scale bars: 10 mV, 100 ms. B, when the identical train of five EPSPs was superimposed on a hyperpolarizing current pulse injected into the cell, there was no sAHP, although the amplitude of the EPSPs were enhanced by the hyperpolarization. Same cell as in A (EPSP trains with and without hyperpolarizing pulse were alternated before and after application of ISO). C, summary data from 3 experiments of the kind shown in A and B. The sAHP area was measured by integration between the vertical dotted lines in right-hand panels of A and B). □, control medium. ▪, after isoprenaline application.
To test if the outward current was dependent on internal K+, a series of experiments similar to the one illustrated in Fig. 2A were performed. NMDA receptor-mediated EPSCs were recorded in a nominally Mg2+-free medium containing 10 μm DNQX, and caesium gluconate replaced KMeSO4 as the basis for the internal solution in order to suppress K+ currents. When Cs+-containing electrodes were used, isoprenaline application (Fig. 2A, ISO, 10 μm) caused no significant alteration in the charge carried during and following a train of NMDA receptor-mediated EPSCs (5 shocks at 50 Hz). Figure 2B shows records from a similar control experiment with the normal, KMeSO4-based, internal solution illustrating a clear ISO-sensitive current. Summary data are plotted in Fig. 2C. The charge was measured by integrating over a 1 s time window starting from the last stimulus artefact in the train. Similar results were obtained at both room temperature and 30 °C and the results have been normalized to control values and pooled. With Cs+ electrodes the total charge transfer fell to 84 ± 8 % (n = 5) of control following isoprenaline application whereas in similar experiments with KMeSO4-based internal medium, isoprenaline increased the synaptic charge transfer to 131 ± 8 % of control (n = 9).
IsAHP can be blocked by using Ca2+ chelators in the recording electrode. Accordingly, if the action of noradrenaline on synaptic responses is mediated by IsAHP, it should be sensitive to inclusion of the Ca2+ chelator EGTA (10 mm) in the recording electrode. Under these conditions IsAHP, evoked by depolarizing current injection, was abolished (Fig. 3A) and the noradrenergic action on pharmacologically intact synaptic responses was also abolished or significantly inhibited (Fig. 3B and C). To quantify this action, synaptic responses were divided into two measurement windows (dashed lines in Fig. 3B). A 100 ms measurement window starting from the beginning of the inward synaptic current was used to calculate synaptic charge. The difference current between averaged (6–9 sweeps) control and drug traces during the subsequent 300 ms time window was used to calculate the noradrenaline-sensitive charge. This method underestimates the noradrenaline-sensitive charge transfer, which begins within the previous time window, but allowed an objective quantification of the data. Within the limits of this analysis, 10 mm EGTA caused a 57 % inhibition of noradrenaline-sensitive charge transfer (Fig. 3C, control 6.3 ± 1.4 pC compared to 2.7 ± 0.5 pC with EGTA; n = 7, P = 0.03, unpaired t test). This was not accounted for by reduced synaptic charge in the presence of EGTA, since this was not significantly different from control values (Fig. 3D, control −23.2 ± 4.0 pC, compared to −30.6 ± 1.2 pC in EGTA; n = 7, P = 0.1, unpaired t test).
Figure 3. Internal EGTA inhibits the noradrenergic modulation of EPSCs.

A, each sample trace contains (left to right) a 5 mV hyperpolarizing command (to monitor access and input resistance), the response to 5 stimuli (50 Hz) and IsAHP evoked by an 80 ms depolarization to −10 mV. The IsAHP was absent when electrodes contained 10 mm EGTA (right trace). B, expansions of the synaptic responses of the cells in A. The action of noradrenaline (10 μm) of prolonging the inward current (left trace) was absent in the presence of 10 mm EGTA internally (right trace). Dashed lines indicate the measurement windows used for integration (see text). C, noradrenaline-sensitive charge transfer (100–400 ms integral of subtracted traces) was significantly reduced by the presence of 10 mm EGTA (n = 7, P = 0.03, unpaired t test). D, the synaptic charge (0–100 ms integral) is not significantly altered by the EGTA (n = 7, P = 0.1, unpaired t test).
The hyperpolarization-activated current, IH, is enhanced by noradrenaline in a PKA-independent manner (Pedarzani & Storm, 1995). Any contribution from this effect to our results was minimal for the following reasons. Firstly, somatic holding potentials were maintained at values (∼-60 mV) where IH is small. Secondly, the recording electrode contained 1–2 mm QX-314, which would reduce IH (Perkins & Wong, 1995) in addition to the Na+ current, INa (higher concentrations could not be used without causing block of IsAHP; see Lancaster & Batchelor, 2000). Thirdly, the action of isoprenaline was blocked when the recording electrode contained caesium, which does not block IH. Finally, the action of isoprenaline on the synaptic responses persisted after selective block of IH with ZD 7288 (10 μm, n = 9; see Fig. 4 and Fig. 5).
CA1 pyramidal cells also have a Ca2+-activated K+ current that is generated by apamin/bicuculline-sensitive SK potassium channels. This current has a faster time course than IsAHP and is not inhibited by noradrenaline or isoprenaline (Stocker et al. 1999). In agreement with these distinctions we have observed that the noradrenaline-sensitive outward current, associated with synaptic input, persisted in the presence of bicuculline methiodide (Fig. 1) and was not mimicked by an application of apamin, which blocked the apamin-sensitive current following voltage steps (n = 3, data not shown).
Thus, four observations suggest that the noradrenaline- and isoprenaline-sensitive outward current following a train of EPSPs is a Ca2+-activated K+ current that is identical to IsAHP: (1) the sensitivity to internal Cs+, (2) the sensitivity to the Ca2+ chelator EGTA, (3) inhibition by isoprenaline and (4) insensitivity to ZD 7288. In order to maximize the possible Ca2+ influx through NMDA receptor channels, most of the experiments described above used a nominally zero Mg2+ medium. Thus a question arises about the source of the Ca2+ that triggers IsAHP. Does it come from Ca2+ influx through the activated NMDA receptor channels, and/or via voltage-gated Ca2+ channels activated by the EPSPs? This latter possibility is still open since the voltage clamp applied at the soma, will not be fully effective at the dendrites where the synapses are located, and so does not exclude Ca2+ entry via dendritic voltage-gated channels activated by synaptic potentials.
To help define the source of Ca2+ for IsAHP we performed experiments testing the voltage dependence of the Ca2+ influx during NMDA receptor-mediated EPSPs or EPSCs (Fig. 4). The slices were bathed in medium without added Mg2+ with 10 μm DNQX to block AMPA-type glutamate receptors. The remaining EPSPs were fully blocked by AP-5 (50–100 μm; data not shown). In addition, 10 μm ZD 7288, 500 μm MCPG, 200 μm 2-hydroxysaclofen and 10 μm bicuculline were included in the medium to block IH, mGluR, GABAB and GABAA receptors, respectively. Under these conditions, a train of five evoked EPSPs (at 50 Hz) was followed by a sAHP that was reduced by bath application of 10 μm isoprenaline (ISO, Fig. 4A). The sAHP component associated with synaptic responses in Fig. 4 and Fig. 5 had a slower time course than in the other experiments because the data were obtained at room temperature, which greatly prolonged the sAHP (but was associated with a reduced inhibition by isoprenaline). The slower time course allowed comparison of EPSP trains with and without a superimposed hyperpolarizing current pulse; the sAHP was then measured following the return of the membrane potential only at a latency where an isoprenaline-sensitive sAHP was clearly present in control conditions (between the vertical dotted lines in Fig. 4A, right panel). EPSP trains with and without hyperpolarizing pulses were alternated before and after application of isoprenaline. When the train of five evoked EPSPs was superimposed on hyperpolarization, the sAHP and isoprenaline sensitivity were abolished (Fig. 4B). Figure 4C shows summary data from six experiments of this kind. The columns represent the area of the sAHP which was integrated over the 2 s window illustrated by the dotted lines in the right-hand panels of Fig. 4A and B. When the EPSPs were evoked at −60 mV, isoprenaline reduced the sAHP area from 5964 ± 1623 to 1549 ± 931 mV ms (P < 0.01, paired t test). Isoprenaline had no significant effect when EPSPs were evoked at strongly hyperpolarized membrane potentials (control 841 ± 323 mV ms, compared to 989 ± 351 mV ms in isoprenaline, P = 0.54).
This result suggests that some Ca2+ influx during the NMDA receptor-mediated EPSPs is voltage gated. However, hyperpolarization would be predicted to increase the driving force for the excitatory synaptic current, but a reduction in the NMDA receptor-mediated EPSP amplitude during the hyperpolarizing pulses was observed (Fig. 4). This might result from Mg2+ contamination in the salts we used to make nominally Mg2+-free medium, or NMDA receptor-mediated EPSP amplitude may involve intrinsic, voltage-dependent currents (see Discussion), although AMPA receptor-mediated EPSPs were not attenuated by hyperpolarization (Fig. 5). Since the hyperpolarization experiment was not fully conclusive, we performed further experiments in which NMDA receptor-mediated responses were tested for isoprenaline sensitivity (similar to Fig. 2B) after application of the L-type Ca2+ channel blocker isradipine (a gift from Novartis, Norge AS, Norway). In a series of voltage clamp experiments at room temperature, isradipine (10 μm) was applied for 10–20 min followed by addition of 10 μm isoprenaline. The decay of summated EPSCs was significantly slower after isradipine application (Fig. 4D). The 90–10 % decay times were 333 ± 53 ms for controls and 507 ± 92 ms after isradipine exposure (P = 0.04, paired t test, n = 5). Subsequent addition of isoprenaline was without significant effect (decay time 558 ± 106 ms). We observed, however, that isradipine application was associated with some rundown of synaptic responses, which compromises the interpretation of these data.
To test further for voltage-dependent effects, and perform an independent test of whether the sAHP required NMDA receptor channels, we performed a comparable series of experiments with non-NMDA EPSPs (Fig. 5). In these experiments, the NMDA receptor blocker dl-AP5 (100 μm) was added to the extracellular medium (in addition to 10 μm ZD 7288, 500 μm MCPG, 200 μm 2-hydroxysaclofen and 10 μm bicuculline, as in Fig. 4). Again the train of five EPSPs was followed by a distinct sAHP, which was sensitive to 10 μm isoprenaline (ISO, Fig. 5A). In contrast, when the cell was hyperpolarized during the EPSP train, the sAHP was again abolished (Fig. 5B), in spite of the fact that the amplitudes of the EPSPs were enhanced by the hyperpolarization, as expected from the increased driving force (Fig. 5B, insets). The action of isoprenaline on EPSPs with and without hyperpolarization was again compared using the 2 s measurement window, as in Fig. 4. At −60 mV the sAHP area was reduced by isoprenaline from 4047 ± 676 mV ms to 699 ± 350 mV ms (P = 0.01, unpaired t test, control value n = 4, isoprenaline value n = 3). There was no detectable effect of isoprenaline following the much larger EPSPs evoked from hyperpolarized potentials (control 1676 ± 545 mV ms, ISO 1529 ± 272 mV ms, P = 0.8). This result strongly suggests that the triggering of the sAHP was dependent upon a certain absolute level of depolarization, as expected if the sAHP is triggered by Ca2+ influx through voltage-gated Ca2+ channels opened by the EPSPs.
Synaptic responses during IsAHP activity
The results above demonstrate that a current with the characteristics of IsAHP can be activated as a component of synaptic responses. An important and closely related issue concerns the reciprocal modulation of EPSPs by the slow AHP. To evaluate the influence of the sAHP conductance on synaptic responses, the K+ conductance ideally should be at a steady state; this would minimize other variables such as membrane potential, postsynaptic Ca2+ dynamics and the transient nature of the current itself. Recent work using the photolabile BAPTA derivative diazo-2 in the recording electrode showed that this compound caused steady state activation of an outward current, indistinguishable from IsAHP, without bulk Ca2+ elevation. The outward current occluded IsAHP and it was inhibited by noradrenaline or photolysis of the diazo-2 (Lancaster & Batchelor, 2000).
These Ca2+-independent effects of flash photolysis on outward current are complete within 1–2 s (Lancaster & Batchelor, 2000) and permit simple quantification of synaptic potentials before and after deactivation of steady state sAHP conductance within single cells. The time course of the action of diazo-2 on synaptic responses and passive electrical properties is presented in Fig. 6. The data were obtained from simultaneous whole cell (current injection was used to maintain a membrane potential of −70 mV) and field potential recordings (traces in Fig. 6A), which controlled for changes in synaptic responses unrelated to the manipulation in the whole cell recording. The input resistance of the cell decreased within the first 10 min of the recording as the outward current developed (Fig. 6B).
Figure 6. Slow AHP conductance modulates excitatory synaptic responses.

A, superimposed whole cell recordings (averages, n = 6) showing input resistance and EPSPs for the minute prior and subsequent to a UV flash, which reduces the steady state AHP conductance. After the UV flash, the input resistance increased and EPSPs showed a slower decay. Inset shows corresponding field recordings. B, experimental time course of input resistance measured at the end of a 30 ms (0.1 nA) hyperpolarization. Left panel in C, normalized input resistance in the minute before and after photolysis (pre-UV and post-UV respectively). Right panel in C, comparison of normalized membrane time constants before and after photolysis. D, whole cell EPSP half-width (normalized to the minute before a flash) decreases during onset of the K+ conductance and is increased following a UV flash. E–G, field EPSP half-width, whole cell EPSP slope and EPSP amplitude are unaffected by flash photolysis.
A single UV flash to photolyse the diazo-2 caused an instantaneous increase in the input resistance (arrow in Fig. 6B) as the resting K+ conductance was deactivated. This was accompanied by a slower decay of the EPSPs (Fig. 6A and D). The traces in Fig. 6A show synaptic responses before and after photolysis for whole cell and field recordings. The increase of intracellular EPSP half-width was not associated with changes in the population response (Fig. 6A, inset). There were, however, significant changes in passive electrotonic properties of cells (Fig. 6C) estimated from 0.1 nA, 30 ms current steps (before any detectable voltage sag due to IH). Immediately after photolysis of diazo-2, input resistance increased from 101 ± 2 to 127 ± 7 %, (n = 5) and the membrane time constant increased from 98 ± 6 to 175 ± 21 % (n = 6). The summary data (Fig. 6D) indicate that EPSP half-width (normalized to the minute before photolysis) increased from 97 ± 4 to 129 ± 2 % (n = 6) immediately after photolysis. This occurred without corresponding changes in the field EPSP half-width (Fig. 6E). By contrast, neither amplitude nor initial slope measurements of the intracellular EPSP - the conventional measures of synaptic strength - were altered by photolysis during these experiments (Fig. 6F and G).
In order to establish the pharmacology of the diazo-2-induced current and to provide a physiological context for regulation of slow AHP current, a similar analysis of the action of isoprenaline was carried out. These results are shown in Fig. 7. The voltage traces (Fig. 7A) show intracellular and (below) field recordings before and after a 2 min application of 2 μm isoprenaline. The summary data (Fig. 7B, n = 6) show the time course of the half-width measurements during the experiment. This decreased from 25 ± 0.6 ms (mean ±s.e.m. for minute 2) to 15 ± 0.3 ms (minute 12). Subsequent isoprenaline application caused a significant increase in EPSP half-width to 21 ± 0.7 ms (15 min data, Fig. 7B, P < 0.001). There was also an associated increase in input resistance from 61 ± 0.4 to 86 ± 0.4 MΩ (Fig. 7E, P < 0.001). Isoprenaline caused no changes in the half-width of field EPSPs or the intracellular EPSP slope (Fig. 7C and D).
Figure 7. EPSPs during noradrenergic block of steady state K+ conductance.

All graphs show summary data (mean ±s.e.m.) from the same 6 cells. A, long traces show superimposed whole cell recordings (average of 6 sweeps) before and after isoprenaline (2 μm) application, which increases input resistance and prolongs EPSP decay. The shorter traces are two corresponding field recordings which are unaltered by isoprenaline. B, summary data (n = 6) showing experimental time course of whole cell EPSP half-width, which increased from 15 to 21 ms following isoprenaline administration. C, experimental time course of field EPSP half-width was unaltered by isoprenaline. D, experimental time course of whole cell EPSP slope was unaltered by isoprenaline. E, input resistance before (Con, 61 MΩ) and after (ISO, 86 MΩ) isoprenaline action.
The alteration of the EPSP decay rate by the K+ conductance is predicted to have prominent consequences for temporal summation. This was tested using diazo-2 as well as physiological activation of the sAHP by action potentials. The summation of five EPSPs at 50 Hz was measured close to the peak of sAHPs evoked by five action potentials and compared to the same synaptic stimulus given during a matching hyperpolarization caused by current injection (Fig. 8A inset). The expanded traces in Fig. 8A show EPSPs under these two conditions (thick trace during current injection). These two manipulations were alternated to control for any slow, systematic changes in synaptic strength. In all six cells studied, EPSPs during the sAHP showed consistently less summation than during matching passive hyperpolarization (Fig. 8B). The mean area of the synaptic response during current injection was 504 ± 56 mV ms compared to 384 ± 49 mV ms during the sAHP (Fig. 8C; n = 6, P < 0.01, paired t test).
Figure 8. AHP conductance suppresses temporal integration.
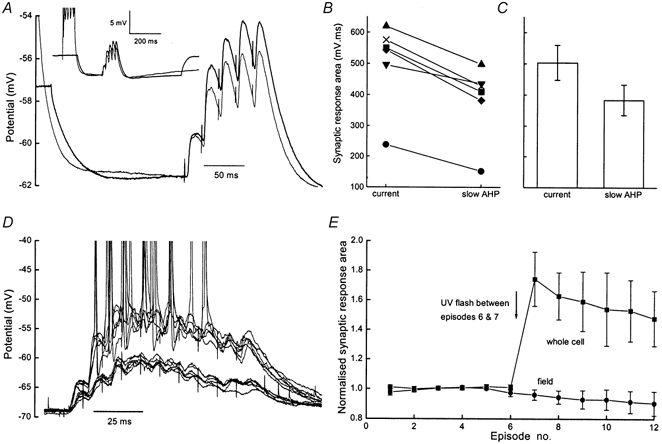
A, the traces are averages (n = 20) from a single recording in which the burst of EPSPs was alternately superimposed on the sAHP evoked by action potentials (thin trace) or a matching, passive hyperpolarization caused by current injection (thick trace). Inset shows a fuller time course with the synaptic response at the peak of the sAHP. B, area under the synaptic response in six cells when EPSP bursts were superimposed on hyperpolarization caused by current injection (current) and action potential evoked sAHP (slow AHP). C, mean values (±s.e.m.) of area under the EPSPs during the slow AHP or matching hyperpolarization. D, 12 superimposed traces showing the whole cell response to 10 stimuli (100 Hz) in the presence of 50 μmd-AP5 and 100 μm picrotoxin. In the presence of diazo-2, a UV flash (after trial 6) caused a substantial enhancement of the subsequent 6 trials. Membrane potential after the flash maintained by current injection. E, summary data (mean ±s.e.m., normalized to the first 6 responses) from 6 slices in which the area under the simultaneously recorded whole cell and field responses was calculated. A UV flash between episodes 6 and 7 (arrow) enhanced the whole cell responses from 101 ± 2 to 174 ± 18 %.
In further experiments the action of diazo-2 was allowed to stabilize, after which 12 trains of 10 stimuli at 100 Hz were delivered to the CA1 afferents at 30 s intervals. Unlike the data reported above, these experiments were performed in 50 μmd-AP5 and 100 μm picrotoxin to minimize stimulus-induced plasticity of fast excitatory and inhibitory transmission. A single UV flash was delivered to the slice between episodes 6 and 7 (arrow in Fig. 8E). Figure 8D shows the twelve intracellular responses corresponding to the six pre- and six post-flash tetani. Deactivation of the steady state AHP conductance converted a non-spiking to a spiking input while field responses were unaltered (individual data not shown, summarized in Fig. 8E). Pooled data for the normalized area of the synaptic potential (100 ms integral) show an increase of 74 ± 18 % for the intracellular response (n = 6, Fig. 8E) following photolysis.
DISCUSSION
The main conclusion of the present study is that the Ca2+ dependent K+ current known as IsAHP can be activated by repetitive excitatory input in CA1 pyramidal cells, even in the absence of action potentials in the postsynaptic cell. This current then contributes to the profile of the postsynaptic response to high-frequency synaptic input.
This conclusion is supported by the following observations. (1) At room temperature, trains of EPSPs or EPSCs were accompanied by slow afterhyperpolarizations, or slow outward currents respectively, which were reduced by β-adrenergic receptor agonists known to suppress IsAHP. (2) The β-adrenergic agonists altered the decay of responses following the last EPSP/EPSC in a train as predicted if a slow outward current like IsAHP was suppressed. (3) These effects were reduced or abolished by loading the postsynaptic cells with Cs+, thus blocking K+ currents, or EGTA, thus inhibiting Ca2+-dependent currents. (4) In contrast, the EPSP-evoked sAHP/outward current was resistant to blockers of other postsynaptic outward currents (GABAA, GABAB) and the β-adrenergic agonist-sensitive current IH.
It is highly unlikely that activity of A-current can explain our results. Most of IA would be inactivated at the holding potentials used. Furthermore, IA activates and inactivates too rapidly to cause such long latency effects on the EPSP train or produce the slow AHPs that were observed (Fig. 1). Finally, IA would not be sensitive to internal EGTA (Fig. 2). The high density of IA in the dendrites and its inhibition by NA/ISO (Hoffman & Johnston, 1998) may, if anything, partly mask the effect on IsAHP. Any suppression of IA could lead to larger dendritic EPSPs and larger Ca2+ influx and thereby promote activation of any remaining IsAHP.
It is notable from the data in Fig. 1 that an sAHP evoked by spikes or large depolarizations has a different time course from the current activated during the synaptic response. One important consideration is the dynamics of the Ca2+ transient under these different conditions. Spiking or voltage jumps will generate widespread Ca2+ influx in the soma and dendritic tree (due to back-propagation). Subthreshold synaptic input will generate Ca2+ influx in more restricted dendritic regions and the amplitude of the Ca2+ elevation is likely to be smaller. The surface-to-volume ratio in dendrites causes Ca2+ transients in CA1 pyramidal cell dendrites to rise and decay faster than in the soma (Helmchen et al. 1996), so spiking will generate a larger and slower Ca2+ signal than synaptic input and the difference in the time course of isoprenaline-sensitive current is consistent with this. Of course we do not really know where the sAHP conductance is primarily located so the correlation between Ca2+ signals and time course remains rather speculative.
The IsAHP associated with synaptic responses was detected without the occurrence of somatic action potentials. The possible sources of Ca2+ elevation are NMDA receptor channels, internal stores (Alford et al. 1993; Emptage et al. 1999; Kovalchuk et al. 2000), subthreshold Ca2+ channel activity (Miyakawa et al. 1992; Markram & Sakmann, 1994; Magee & Johnston, 1995) and dendritic spikes which are electrically isolated from the soma (Golding & Spruston, 1998). In our experiments, the β-adrenergic-sensitive sAHP was prevented by hyperpolarizing the cell during the EPSP train. This would reduce the opening of voltage-activated Ca2+ channels but would be predicted, if anything, to enhance Ca2+ influx through NMDA receptor channels since the experiments were performed without added Mg2+.
This result suggests that Ca2+ influx which generates sAHP following NMDA receptor-mediated EPSPs is voltage gated. Unexpectedly, a reduction in the NMDA receptor-mediated EPSP amplitude during the hyperpolarizing pulses was observed, although hyperpolarization would be predicted to increase the driving force for the excitatory synaptic current. A possible explanation is that the EPSPs were attenuated by an outward current (possibly IA in the dendrites; Hoffman et al. 1998) that was enhanced by the hyperpolarization due to removal of inactivation, or that boosting of the EPSPs by voltage-gated Na+ or Ca2+ currents was reduced (Magee & Johnston, 1995; Lipowsky et al. 1996). This seems less likely, since the AMPA receptor-mediated EPSPs were not attenuated by hyperpolarization, although this might be ascribed to the faster time course of the AMPA receptor-mediated EPSPs, which would activate less voltage-gated current than longer lasting NMDA receptor-mediated EPSPs. The observation that NMDA and AMPA receptor-mediated EPSPs evoked sAHPs of comparable amplitude speaks against this possibility, at least for Ca2+ channels. An alternative explanation is that Mg2+ contamination, calculated to be approximately 13 μm, from the salts we use to make our nominally Mg2+-free medium may confer a weak, negative slope conductance to the NMDA receptor current.
Pharmacological means of identifying the voltage-dependent Ca2+ channels responsible for the sAHP have shown involvement of L-type channels (Tanabe et al. 1998), although not all studies have reported complete block of the sAHP with L channel blockers (Hu & Storm, 1999; Shah & Haylett, 2000). We did not observe complete block of the sAHP by the L-type Ca2+ channel blocker isradipine, but there was a concurrent loss of isoprenaline-sensitive current in NMDA receptor-mediated EPSCs. This does not exclude the possibility that Ca2+ influx through NMDA receptors can activate the sAHP, but does suggest that there was no absolute requirement for NMDA receptor-mediated Ca2+ influx in our experiments.
Clearly, however, the β-adrenergic-sensitive sAHP was resistant to blockers of either AMPA receptors or NMDA receptors alone. This also suggests that the Ca2+ influx through voltage-activated Ca2+ channels, possibly L-type channels, was a sufficient source of Ca2+ to trigger IsAHP in response to a train of EPSPs/EPSCs. Our experiments do not distinguish between subthreshold activation of Ca2+ channels and electrically remote calcium spiking. Our data also do not exclude the possibility that the Ca2+ signal may be amplified by Ca2+-induced Ca2+ release (Emptage et al. 1999; Hu & Storm, 1999). The observation that the synaptically generated IsAHP was resistant to blockade of postsynaptic metabotropic glutamate receptors (mGluR) seems to rule out the possibility of mGluR stimulation of IP3-dependent Ca2+ release during tetanic stimuli (Nakamura et al. 1999; Partridge & Valenzuela, 1999) as a trigger for the IsAHP.
Voltage-dependent channels in pyramidal cell dendrites are controlling factors in the propagation of both action potentials (Schwindt & Crill, 1997; Hoffmann et al. 1997; Magee & Carruth, 1999) and EPSPs or EPSP waveforms (Magee, 1998; Urban & Barrionuevo, 1998; Cash & Yuste, 1999). Comparable data for Ca2+-dependent K+ currents are rather sparse, but the TEA- and voltage-sensitive Ca2+-dependent K+ current, IC (mediated by BK channels) can affect signal propagation in some cell types (Schwindt & Crill, 1997; Wessel et al. 1999). There are no systematic studies on voltage-independent Ca2+-dependent K+ current, despite the likelihood that slow AHP current is to some extent located in the proximal apical dendrites of CA1 pyramidal cells (Andreasen & Lambert, 1995; but see Bekkers, 2000), where it may modulate synaptic input (Sah & Bekkers, 1996).
The uncertainty about the location of IsAHP raises the possibility that the diazo-2-induced current may not have the same spatial distribution as sAHP evoked by action potentials. However, both spike-evoked sAHPs and the diazo-2-induced outward current (presumptive IsAHP) are similarly capable of modifying EPSPs generated at apical dendritic sites; this functional similarity is consistent with similar spatial distribution. This result seems to be in agreement with Sah & Bekkers (1996) who concluded that AHP current had greater influence on EPSPs in stratum radiatum than in stratum oriens. Previously the diazo-2-induced current was determined to be < 100 μm from the soma (Lancaster & Batchelor, 2000), and comparable regions of the apical dendrites show clear Ca2+ elevation following action potentials which normally activate the sAHP (Helmchen et al. 1996). The use of diazo-2 allowed us to eliminate two important variables from the study of IsAHP. First, the transient nature of the K+ conductance change itself and second the requirement for repetitive elevation of [Ca2+]i which indirectly might modify synaptic responses (Kyrozis et al. 1996).
The presence of a large sAHP conductance close to the soma will cause a shunt, which might contribute to electrical separation of dendritic and somatic regions (Golding & Spruston, 1998) following action potentials. This location further implies that most excitatory synaptic input will also be subject to the influence of post-spike sAHPs. Our observations indicate that this influence is exerted primarily as a consequence of changes in passive properties of the cell membrane, which affect the decay rate of EPSPs without clear effects on EPSP amplitude. Such an effect is predicted for potentials within passive dendrites of variable input resistance (Koch, 1999). Additionally the EPSP amplitude at the somatic recording site might also be expected to increase during block of the sAHP conductance. Although some of our data showed EPSP amplitude changes (Fig. 8) this was not a consistent observation and, therefore, was not reflected in the pooled data (Fig. 6G). This may be due to a predominance of capacitative attenuation rather than ohmic shunting during the early part of the EPSPs, a large synaptic conductance compared to sAHP conductance, and/or a possible involvement of active conductances, such as persistent Na+ current, which boost EPSP amplitude (Lipowsky et al. 1996; Andreasen & Lambert, 1999). These factors would render amplitude measurements less sensitive to slow membrane conductance changes. This apparent insensitivity of EPSP slope and amplitude to the sAHP conductance will minimize the implications for neuronal output in response to low-frequency synaptic input.
The action of sAHP conductance primarily on the decay rate of EPSPs allows for a preferential modification of high-frequency synaptic input. Any transmitter-induced inhibition of IsAHP, by monoamines, acetylcholine or peptide transmitters (Storm et al. 2000), will lead to greater membrane depolarization and an increased probability of cell firing during high-frequency excitatory input than during comparable low-frequency input. Our data are consistent with the proposal (Sah & Bekkers, 1996) that the β-noradrenergic sensitivity of LTP induction threshold derives from the AHP conductance, although a later study has cast doubt on the conclusions of the earlier work (Bekkers, 2000). We have provided convergent evidence that the sAHP current can indeed be activated by high-frequency synaptic input. The much larger depolarization that is achieved after block of the sAHP (Fig. 8) will allow greater Ca2+ influx through NMDA receptors, which could contribute to a shift in LTP and long-term depression (LTD) induction thresholds.
Acknowledgments
This work was supported by the Wellcome Trust, the Sir Jules Thorn Charitable Trust, an EU Research Training Grant (BIO4CT975106), the Norwegian Research Council (NFR/MH), and the Nansen and Jahre Foundations.
References
- Alford S, Frenguelli BG, Schofield JG, Collingridge GL. Characterization of Ca2+ signals induced in hippocampal CA1 neurones by the synaptic activation of NMDA receptors. Journal of Physiology. 1993;469:693–716. doi: 10.1113/jphysiol.1993.sp019838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen M, Lambert JDC. Noradrenaline receptors participate in the regulation of GABAergic inhibition in area CA1 of the rat hippocampus. Journal of Physiology. 1991;439:649–669. doi: 10.1113/jphysiol.1991.sp018686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen M, Lambert JDC. The excitability of CA1 pyramidal cell dendrites is modulated by a local Ca2+-dependent K+-conductance. Brain Research. 1995;698:193–203. doi: 10.1016/0006-8993(95)00910-i. [DOI] [PubMed] [Google Scholar]
- Andreasen M, Lambert JDC. Somatic amplification of distally generated subthreshold EPSPs in rat hippocampal neurones. Journal of Physiology. 1999;519:85–100. doi: 10.1111/j.1469-7793.1999.0085o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM. Distribution of slow AHP channels on hippocampal CA1 pyramidal neurons. Journal of Neurophysiology. 2000;83:1756–1759. doi: 10.1152/jn.2000.83.3.1756. [DOI] [PubMed] [Google Scholar]
- Cash S, Yuste R. Linear summation of excitatory inputs by CA1 pyramidal neurons. Neuron. 1999;22:383–394. doi: 10.1016/s0896-6273(00)81098-3. [DOI] [PubMed] [Google Scholar]
- Doze VA, Cohen GA, Madison DV. Synaptic localization of adrenergic disinhibition in the rat hippocampus. Neuron. 1991;6:889–900. doi: 10.1016/0896-6273(91)90229-s. [DOI] [PubMed] [Google Scholar]
- Emptage N, Bliss TVP, Fine A. Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron. 1999;22:115–124. doi: 10.1016/s0896-6273(00)80683-2. [DOI] [PubMed] [Google Scholar]
- Golding NL, Spruston N. Dendritic sodium spikes are variable triggers of axonal action potentials in hippocampal CA1 pyramidal neurons. Neuron. 1998;21:1189–1200. doi: 10.1016/s0896-6273(00)80635-2. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Imoto K, Sakmann B. Ca2+ buffering and action potential-evoked Ca2+ signalling in dendrites of pyramidal neurons. Biophysical Journal. 1996;70:1069–1081. doi: 10.1016/S0006-3495(96)79653-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Johnston D. Down-regulation of transient K+ channels in dendrites of hippocampal CA1 pyramidal neurons by activation of PKA and PKC. Journal of Neuroscience. 1998;18:3521–3528. doi: 10.1523/JNEUROSCI.18-10-03521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- Hu H, Storm JF. Coupling of Ca2+ channels to BK- and SK-type channel activation in rat CA1 hippocampal pyramidal cells. Society for Neuroscience Abstracts. 1999;25:453. [Google Scholar]
- Koch C. Biophysics of Computation. Oxford, UK: Oxford University Press; 1999. [Google Scholar]
- Kovalchuk Y, Eilers J, Lisman JE, Konnerth A. NMDA receptor-mediated subthreshold Ca2+ signals in spines of hippocampal neurons. Journal of Neuroscience. 2000;20:1791–1799. doi: 10.1523/JNEUROSCI.20-05-01791.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrozis A, Albuquerque C, Gu J, MacDermott AB. Ca2+-dependent inactivation of NMDA receptors: fast kinetics and high Ca2+ sensitivity in rat dorsal horn neurons. Journal of Physiology. 1996;495:449–463. doi: 10.1113/jphysiol.1996.sp021606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Batchelor AM. Novel action of BAPTA series chelators on intrinsic K+ currents in rat hippocampal neurones. Journal of Physiology. 2000;522:231–246. doi: 10.1111/j.1469-7793.2000.t01-1-00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Hu H, Ramakers GMJ, Storm JF. Repetitive synaptic activity recruits Ca2+-dependent outward current in CA1 pyramidal cells. Journal of Physiology. 1999;518.P:148P. [Google Scholar]
- Lancaster B, Hu H, Ramakers GMJ, Storm JF. Electrical interaction between synaptic input and steady-state K+ current in CA1 pyramidal cells. Journal of Physiology. 2000;527.P:117P. doi: 10.1111/j.1469-7793.2001.00809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipowsky R, Gillesen T, Alzheimer C. Dendritic Na+ channels amplify EPSPs in hippocampal CA1 pyramidal cells. Journal of Neurophysiology. 1996;76:2181–2191. doi: 10.1152/jn.1996.76.4.2181. [DOI] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Cyclic adenosine 3′,5′-monophosphate mediates β-receptor actions of noradrenaline in rat hippocampal pyramidal cells. Journal of Physiology. 1986;372:245–259. doi: 10.1113/jphysiol.1986.sp016007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. Journal of Neuroscience. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Carruth M. Dendritic voltage-gated ion channels regulate the action potential firing mode of hippocampal CA1 pyramidal neurons. Journal of Neurophysiology. 1999;82:1895–1901. doi: 10.1152/jn.1999.82.4.1895. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. Synaptic activation of voltage-gated channels in the dendrites of hippocampal pyramidal neurons. Science. 1995;268:301–304. doi: 10.1126/science.7716525. [DOI] [PubMed] [Google Scholar]
- Markram H, Sakmann B. Calcium transients in dendrites of neocortical neurons evoked by single subthreshold excitatory postsynaptic potentials via low-voltage-activated calcium channels. Proceedings of the National Academy of Sciences of the USA. 1994;91:5207–5211. doi: 10.1073/pnas.91.11.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa H, Ross WN, Jaffe D, Callaway JC, Lasser-Ross N, Lisman JE, Johnston D. Synaptically activated increases in Ca2+ concentration in hippocampal CA1 pyramidal cells are primarily due to voltage-gated Ca2+ channels. Neuron. 1992;9:1163–1173. doi: 10.1016/0896-6273(92)90074-n. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Barbara J-G, Nakamura K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727–737. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- Newberry NR, Nicoll RA. A bicuculline-resistant inhibitory postsynaptic potential in rat hippocampal pyramidal cells in vitro. Journal of Physiology. 1984;348:239–254. doi: 10.1113/jphysiol.1984.sp015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge LD, Valenzuela CF. Ca2+ store-dependent potentiation of Ca2+-activated non-selective cation channels in rat hippocampal neurones in vitro. Journal of Physiology. 1999;521:617–627. doi: 10.1111/j.1469-7793.1999.00617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF. PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 1993;11:1023–1035. doi: 10.1016/0896-6273(93)90216-e. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF. Protein kinase A-independent modulation of ion channels in the brain by cyclic AMP. Proceedings of the National Academy of Sciences of the USA. 1995;92:11716–11720. doi: 10.1073/pnas.92.25.11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins KL, Wong RKS. Intracellular QX-314 blocks the hyperpolarization-activated inward current Iq in hippocampal CA1 pyramidal cells. Journal of Neurophysiology. 1995;73:911–915. doi: 10.1152/jn.1995.73.2.911. [DOI] [PubMed] [Google Scholar]
- Sah P, Bekkers JM. Apical dendritic location of slow afterhyperpolarization current in hippocampal pyramidal neurons: implications for the integration of long-term potentiation. Journal of Neuroscience. 1996;16:4537–4542. doi: 10.1523/JNEUROSCI.16-15-04537.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwindt PC, Crill WE. Modification of current transmitted from apical dendrite to soma by blockade of voltage- and Ca2+-dependent conductances in rat neocortical pyramidal neurons. Journal of Neurophysiology. 1997;78:187–198. doi: 10.1152/jn.1997.78.1.187. [DOI] [PubMed] [Google Scholar]
- Shah M, Haylett DG. Ca2+ channels involved in the generation of the slow afterhyperpolarization in cultured rat hippocampal pyramidal neurons. Journal of Neurophysiology. 2000;83:2554–2561. doi: 10.1152/jn.2000.83.5.2554. [DOI] [PubMed] [Google Scholar]
- Stocker M, Krause M, Pedarzani P. An apamin-sensitive Ca2+-activated K+ current in hippocampal pyramidal neurons. Proceedings of the National Academy of Sciences of the USA. 1999;96:4662–4667. doi: 10.1073/pnas.96.8.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF, Pedarzani P, Haug TM, Winther T. Modulation of K+ channels in hippocampal neurons: transmitters acting via cyclic AMP enhance the excitability through kinase-dependent and -independent modulation of AHP- and h-channels. In: Kuba K, Higashida H, Brown DA, Yoshioka T, editors. Slow Synaptic Responses and Modulation. Tokyo: Springer-Verlag; 2000. pp. 78–92. [Google Scholar]
- Tanabe M, Gähwiler BH, Gerber U. L-type Ca2+ channels mediate the slow Ca2+-dependent afterhyperpolarization current in rat CA3 pyramidal cells in vitro. Journal of Neurophysiology. 1998;80:2268–2273. doi: 10.1152/jn.1998.80.5.2268. [DOI] [PubMed] [Google Scholar]
- Urban NN, Barrionuevo G. Active summation of excitatory postsynaptic potentials in hippocampal CA3 pyramidal neurons. Proceedings of the National Academy of Sciences of the USA. 1998;95:11450–11455. doi: 10.1073/pnas.95.19.11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel R, Kristan WB, Kleinfeld D. Dendritic Ca2+-activated K+ conductances regulate electrical signal propagation in an invertebrate neuron. Journal of Neuroscience. 1999;19:8319–8326. doi: 10.1523/JNEUROSCI.19-19-08319.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]