

Abstract
Using the technique of end-tidal CO2 forcing, we measured the effect of the carbonic anhydrase inhibitor acetazolamide (4 mg kg−1, i.v.) on the CO2 sensitivities of the peripheral and central chemoreflex loops both during hyperoxia and hypoxia in 10 cats anaesthetised with α-chloralose–urethane.
In the control situation, going from hyperoxia (arterial
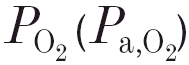 47.40 ± 3.62 kPa, mean ± s.d.) into moderate hypoxia (
47.40 ± 3.62 kPa, mean ± s.d.) into moderate hypoxia (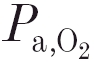 8.02 ± 0.30 kPa) led to an almost doubling of the peripheral CO2 sensitivity (SP): a rise from 0.09 ± 0.07 to 0.16 ± 0.06 l min−1 kPa−1. After acetazolamide, however, lowering the from 46.95 ± 5.19 to 8.02 ± 0.66 kPa did not result in a rise in SP, indicating the absence of a CO2–O2 stimulus interaction.
8.02 ± 0.30 kPa) led to an almost doubling of the peripheral CO2 sensitivity (SP): a rise from 0.09 ± 0.07 to 0.16 ± 0.06 l min−1 kPa−1. After acetazolamide, however, lowering the from 46.95 ± 5.19 to 8.02 ± 0.66 kPa did not result in a rise in SP, indicating the absence of a CO2–O2 stimulus interaction.In hypoxia, acetazolamide reduced SP from 0.16 ± 0.06 to 0.07 ± 0.05 l min−1 kPa−1. In hyperoxia, however, the effect on SP was much smaller (an insignificant reduction from 0.09 ± 0.07 to 0.06 ± 0.05 l min−1 kPa−1).
Acetazolamide reduced both the hyperoxic and hypoxic sensitivities (SC) of the central chemoreflex loop: from 0.45 ± 0.16 to 0.27 ± 0.13 l min−1 kPa−1 and from 0.40 ± 0.16 to 0.26 ± 0.13 l min−1 kPa−1, respectively. In hyperoxia, the apnoeic threshold B (X-intercept of the ventilatory CO2 response curve) decreased from 2.91 ± 0.57 to 0.78 ± 1.9 kPa (P = 0.005). In hypoxia, B decreased from 1.59 ± 1.22 to −0.70 ± 2.99 kPa (P = 0.03).
Because acetazolamide abolished the CO2–O2 interaction, i.e. the expected increase in SP when going from hyperoxia into hypoxia, we conclude that the agent has a direct inhibitory effect on the carotid bodies. The exact mechanism by which the agent exerts this effect will remain unclear until more detailed information becomes available on the identity of the carbonic anhydrase iso-enzymes within the carotid bodies and their precise subcellular distribution.
The carbonic anhydrases are a family of zinc metallo-enzymes catalysing the reversible hydration of CO2. In higher vertebrates including humans, at least 10 different carbonic anhydrase iso-enzymes have been discovered. These isoforms are characterised by different distributions within tissues, kinetic properties, subcellular locations and susceptibility to inhibitors (Sly & Hu, 1995). Hydration and dehydration of CO2 are key events in physiological processes such as respiration, bicarbonate reabsorption in kidney (urinary acidification), the formation of aqueous humour and cerebrospinal fluid and the secretion of fluids in the gastro-intestinal system (Maren, 1967).
Several of our previous studies in the anaesthetised cat were focused on the role of carbonic anhydrase in the control of breathing. One significant finding was that large intravenous doses of the sulfonamides benzolamide and acetazolamide caused a large reduction in the slope of the ventilatory CO2 response curve (Teppema et al. 1995; Wagenaar et al. 1996), and completely abolished the hypoxic response (Teppema et al. 1988, 1992). Using the technique of dynamic end-tidal forcing (DEF) we also found that both during normoxia and moderate hypoxia a low dose (i.e. 4 mg kg−1) of acetazolamide reduced the CO2 sensitivity of the central chemoreflex loop by about 45 %; the sensitivity of the peripheral chemoreflex loop was reduced by 50 and 33 % during moderate hypoxia and normoxia, respectively (Wagenaar et al. 1996, 1998). The effects with low-dose acetazolamide occurred at inhibitor concentrations insufficient to cause effective inhibition of red cell carbonic anhydrase, as indicated by the absence of significant arterial-end-tidal 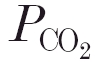 gradients. We attributed the effects of low-dose acetazolamide on the peripheral chemoreflex loop to a direct action of the agent on the carotid bodies. This is not unreasonable because glomus cells are known to contain carbonic anhydrase (Lee & Mattenheimer, 1964; Ridderstråle & Hanson, 1984; Rigual et al. 1985; Nurse, 1990), while enzyme inhibitors have been shown to alter their in vivo and in vitro CO2 responses (Hanson et al. 1981b; Iturriaga et al. 1991; Lahiri et al. 1996; Torrance, 1996). Although it is conceivable that within the carotid bodies the carbonic anhydrases play a role in the (de)hydration of CO2, their precise function is unclear because both the identity and the subcellular location(s) of the isoforms are unknown.
gradients. We attributed the effects of low-dose acetazolamide on the peripheral chemoreflex loop to a direct action of the agent on the carotid bodies. This is not unreasonable because glomus cells are known to contain carbonic anhydrase (Lee & Mattenheimer, 1964; Ridderstråle & Hanson, 1984; Rigual et al. 1985; Nurse, 1990), while enzyme inhibitors have been shown to alter their in vivo and in vitro CO2 responses (Hanson et al. 1981b; Iturriaga et al. 1991; Lahiri et al. 1996; Torrance, 1996). Although it is conceivable that within the carotid bodies the carbonic anhydrases play a role in the (de)hydration of CO2, their precise function is unclear because both the identity and the subcellular location(s) of the isoforms are unknown.
One way to study possible direct effects of the agents on the peripheral chemoreceptors in a whole animal preparation is to focus on the interaction between CO2 and O2, which is known to originate in the carotid bodies (Lahiri & Delaney, 1975; Fitzgerald & Deghani, 1982). At the level of minute ventilation, this interaction is translated into steeper CO2 ventilatory response curves at lower 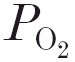 levels. An effect of carbonic anhydrase inhibitors on the CO2–O2 interaction within the carotid bodies has not been systematically investigated. Our previous experiments with low-dose acetazolamide were performed in separate animals, and thus did not allow a study of the CO2–O2 interaction within animals. Therefore, the aim of this study in the anaesthetised cat was to determine the effect of 4 mg kg−1 acetazolamide on the CO2 sensitivities of the peripheral and central chemoreflex loops both during hyperoxia and moderate hypoxia within the animals. In this way we should be able to see if the inhibitor affects the expected increase in peripheral chemoreflex gain on going from hyperoxia into moderate hypoxia. We used the DEF technique to separate the CO2 sensitivities of the peripheral and central chemoreflex loops from each other (Degoede et al. 1985).
levels. An effect of carbonic anhydrase inhibitors on the CO2–O2 interaction within the carotid bodies has not been systematically investigated. Our previous experiments with low-dose acetazolamide were performed in separate animals, and thus did not allow a study of the CO2–O2 interaction within animals. Therefore, the aim of this study in the anaesthetised cat was to determine the effect of 4 mg kg−1 acetazolamide on the CO2 sensitivities of the peripheral and central chemoreflex loops both during hyperoxia and moderate hypoxia within the animals. In this way we should be able to see if the inhibitor affects the expected increase in peripheral chemoreflex gain on going from hyperoxia into moderate hypoxia. We used the DEF technique to separate the CO2 sensitivities of the peripheral and central chemoreflex loops from each other (Degoede et al. 1985).
METHODS
Animals and measurements
Experiments were performed in 10 female adult cats (weight 2.8–4.2 kg). The Ethical Committee for Animal Experiments of the University of Leiden approved the use of animals and the experimental protocols. The animals were sedated with 10 mg kg−1 ketamine hydrochloride (i.m.). Anaesthesia was induced with 2 % sevoflurane in 30 % O2 in N2. The right femoral artery and vein were cannulated, 20 mg kg−1α-chloralose and 100 mg kg−1 urethane were slowly administered intravenously, and the volatile anaesthetic was gradually withdrawn. About 1 h later, an infusion of a α-chloralose- urethane solution was started at a rate 1.0–1.5 mg kg−1 h−1α-chloralose and 5.0–7.5 mg kg−1 h−1 urethane. This regimen is sufficient to suppress the pain-withdrawal reflex but light enough to preserve the corneal reflex. In previous studies we found that the maintenance of this condition has little effect on the ventilatory response to changes in end-tidal (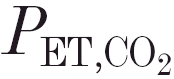 ; Teppema et al. 1997). The animals were monitored throughout the experiment to ensure an adequate level of anaesthesia.
; Teppema et al. 1997). The animals were monitored throughout the experiment to ensure an adequate level of anaesthesia.
The trachea was cannulated at the midcervical level and connected to a respiratory circuit. Tidal volume was measured electronically by integrating airway gas flow obtained from a pneumotachograph (number 0 flow transducer, Fleisch, Lausanne, Switzerland) connected to a differential pressure transducer (Statham PM 197, Los Angeles, CA, USA). The respiratory fractions of O2 and CO2 were continuously measured with a Datex gas monitor (Multicap, Helsinki, Finland), which was calibrated with gas mixtures of known composition. The inspiratory gas concentrations were made with computer-steered mass flow controllers (type AFC 260, Advanced Semiconductor Materials, DeBilt, Netherlands). The animals were connected to an extra-corporeal circuit (ECC) for continuous blood gas measurement. Using the ECC, blood from the cannulated left femoral artery was pumped back via the right femoral vein with a flow of 6 ml min−1. Arterial pH, and in the blood passing the extra corporeal circuit were measured continuously with a pH electrode (Radiometer E-5037-0, Copenhagen, Denmark) calibrated with phosphate buffers, a CO2 electrode (General Electric A312AB, Milwaukee, WI, USA) and a home-made Clark-type O2 electrode, respectively, which were calibrated with water equilibrated with CO2–O2-N2 gas mixtures delivered by a gas mixing pump (Wösthoff, Bochum, Germany). The transport delay from the lungs to the CO2 electrode was approximately 50 s. The CO2 electrode was recalibrated every 2 h and corrections for drift were made when necessary. Arterial blood pressure was measured using a Statham pressure transducer (Statham P23AC).
Rectal temperature was measured and controlled within 1 °C. All signals were recorded on polygraphs, converted to digital values (sample frequency 100 Hz) and processed by a PC. All signals were stored on a breath-by-breath basis.
Experimental design
Using the technique of end-tidal CO2 forcing, we performed step changes in end-tidal before and after intravenous infusion of 4 mg kg−1 acetazolamide (Diamox, AHP Pharma, Hoofddorp, The Netherlands; 2 mg ml−1 in saline) against two different background conditions, namely hyperoxia (mean 47.40 ± 3.62 and 46.95 ± 5.19 kPa before and after administration of the inhibitor, respectively) and moderate hypoxia. Because the CO2 sensitivity of the peripheral chemoreflex loop increases with decreasing levels of , the hypoxic DEF runs in particular were performed at equal levels of arterial before and after acetazolamide (mean 8.02 ± 0.30 and 8.02 ± 0.66 kPa, respectively). In each condition (control-hyperoxia, control-hypoxia, acetazolamide-hyperoxia, acetazolamide-hypoxia) two to four dynamic end-tidal forcing (DEF) runs were performed and the dynamic ventilatory responses analysed (see below). The pattern during a DEF run was as follows. After a 10–15 min period of steady-state ventilation at constant (about 0.5 kPa above the apnoeic threshold), the was increased by 1–1.5 kPa in a stepwise fashion and kept constant for 7 min. Thereafter the was returned to its previous value and maintained for another 7 min.
After the termination of the experiments, the animals were humanely killed without recovery by way of an intravenous infusion of 10 ml pentobarbital.
Dynamic end-tidal forcing
The steady-state relation of inspiratory ventilation  to at constant can be described by:
to at constant can be described by:
where SP is the carbon dioxide sensitivity of the peripheral chemoreflex loop, SC is the carbon dioxide sensitivity of the central chemoreflex loop, and B is the apnoeic threshold or extrapolated at zero . The sum of SP and SC is the overall carbon dioxide sensitivity.
For the analysis of the dynamic response of ventilation to a step-wise change in we used a two-compartment model (Degoede et al. 1985):
where τP and τC are the time constants of the peripheral and central chemoreflex loops, respectively, P(t) and (t) are the outputs of the peripheral and central chemoreflex loops, respectively, [t - TP] is the stimulus to the peripheral chemoreflex loop delayed by the peripheral transport delay time (TP), and [t - TC] = the stimulus to the central chemoreflex loop delayed by the central transport delay time (TC).
To allow the time constant of the ventilatory on-transient to be different from that of the off-transient τC is written as:
where τon is the time constant of the ventilatory on-transient, τoff is the time constant of the off-transient, and x = 1 when is high, while x = 0 when is low.
In most experiments a small drift in ventilation was present. We therefore included a drift term (Ct) in our model. The total ventilatory response (t) is made up of the contributions of the central and peripheral chemoreflex loops and Ct:
The parameters of the model were estimated by fitting the model to the breath-by-breath data using a least-squares method. To obtain optimal time delays a ‘grid search’ was applied, and all combinations of TP and TC, with increments of 1 s and with TP≤TC, were tried until a minimum in the residual sum of squares was obtained. The minimum time delay was chosen, arbitrarily, to be 1 s; the τP was constrained to be at least 0.3 s.
Statistical analysis
To compare the means of the values obtained from the analysis of the DEF runs in the control situation with those obtained after acetazolamide infusion, analysis of variance was performed on individual data. The level of significance was set at P = 0.05 or lowered in the case of multiple comparisons. Results are given as means of the mean per cat ± s.d.
RESULTS
The mean parameter values obtained from the optimal fits during hyperoxia, before (32 DEF runs) and after (27 DEF runs) administration of 4 mg kg−1 acetazolamide are summarised in Table 1.
Table 1.
Effects of low-dose acetazolamide on respiratory parameters during hyperoxia
| Control | Acetazolamide | P | |
|---|---|---|---|
| B (kPa) | 2.91 ± 0.57 | 0.78 ± 1.9 | 0.005 |
| SC (l min-1 kPa-1) | 0.45 ± 0.16 | 0.27 ± 0.13 | 0.001 |
| SP (l min-1 kPa-1) | 0.09 ± 0.07 | 0.06 ± 0.05 | 0.123 |
| τP (s) | 5.91 ± 4.28 | 7.27 ± 5.76 | 0.741 |
| τon (s) | 83.60 ± 47.60 | 88.56 ± 25.28 | 0.756 |
| τoff (s) | 108.65 ± 45.04 | 128.98 ± 46.86 | 0.362 |
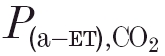 (kPa) (kPa) |
0.05 ± 0.57 | 0.43 ± 0.81 | 0.029 |
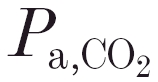 (kPa) (kPa) |
47.40 ± 3.62 | 46.95 ± 5.19 | 0.633 |
Parameter values obtained from the optimal fits in the analysis of DEF runs performed during hyperoxia (32 control runs and 27 runs after acetazolamide). B, the apnoeic threshold or X-intercept of the ventilatory CO2 response curve. SC and SP, the CO2 sensitivities of the central and peripheral chemoreflex loops, respectively. τp, time constant of the peripheral chemoreflex loops. τon and τoff, central time constants of responses to CO2 on-and off transients, respectively. , arterial–end-tidal difference.
Before infusion of the inhibitor, the mean arterial tension was maintained at 47.40 ± 3.62 kPa. After acetazolamide, the DEF runs were performed at a mean of 46.95 ± 5.19 kPa.
Acetazolamide caused a small increase in the mean arterial-to-end-tidal difference from 0.05 to 0.43 kPa. The mean value of the apnoeic threshold B decreased from 2.91 to 0.78 kPa. The CO2 sensitivity SC of the central chemoreflex loop decreased significantly from a mean value of 0.45 to 0.27 l min−1 kPa−1. The sensitivity SP of the peripheral chemoreflex loop, however, was less clearly affected by the inhibitor: a decrease from 0.09 to 0.06 l min−1 kPa−1 which did not reach significance. Figure 1 displays the effects of acetazolamide on SC and SP in all individual animals in scatter diagrams. No significant effects of acetazolamide on the peripheral and central time constants were observed (see Table 1). Also, no acetazolamide-induced differences in peripheral and central transport delay times were found.
Figure 1. Effect of 4 mg kg−1 acetazolamide (i.v.) on the hyperoxic CO2 sensitivities of the peripheral and central chemoreflex loops.
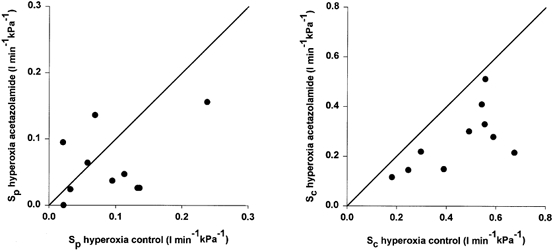
Effect of acetazolamide (4 mg kg−1, i.v.) on the CO2 sensitivities of the peripheral (SP, left panel) and central (SC, right panel) chemoreflex loops during hyperoxia. Each animal is represented by one symbol. Diagonal line represents line of identity.
The effects of acetazolamide on the parameter values obtained during moderate hypoxia are summarised in Table 2 (32 DEF runs before and 29 runs after infusion). To prevent an effect of changes in on the sensitivity of the peripheral chemoreflex loop, we maintained the level of the arterial oxygen tension at equal levels before and after acetazolamide administration. In the hypoxic situation, the inhibitor caused a decrease of about 2 kPa in mean value of B, similar to that found during hyperoxia. During hypoxia, the mean CO2 sensitivities of both the peripheral and central chemoreflex loops were considerably reduced by acetazolamide (from 0.16 to 0.07, and from 0.40 to 0.26 l min−1 kPa−1, respectively), and this is illustrated by the individual data in all animals shown in Fig. 2. Peripheral and central time constants and transport delay times were not altered by acetazolamide. Similar to the hyperoxic condition, a small but significant increase in the arterial-end-tidal difference () was seen (see Table 2).
Table 2.
Effects of low-dose acetazolamide on respiratory parameters during hypoxi
| Control | Acetazolamide | P | |
|---|---|---|---|
| B(kPa) | 1.59 ± 1.22 | −0.70 ± 2.99 | 0.030 |
| SC (l min-1 kPa-1) | 0.40 ± 0.16 | 0.26 ± 0.13 | 0.019 |
| Sp (l min-1 kPa-1) | 0.16 ± 0.06 | 0.07 ± 0.05 | 0.002 |
| τp (s) | 4.26 ± 4.92 | 5.12 ± 4.04 | 0.693 |
| τon (s) | 73.68 ± 37.63 | 60.37 ± 22.79 | 0.269 |
| τoff (s) | 104.53 ± 50.35 | 126.82 ± 65.84 | 0.469 |
|
(kPa) |
−0.10 ± 0.57 | 0.39 ± 0.75 | 0.009 |
|
(kPa) |
8.02 ± 0.30 | 8.02 ± 0.66 | 0.986 |
Parameter values obtained from the optimal fits in the analysis of DEF runs performed during hypoxia (32 control runs and 29 runs after acetazolamide). B, the apnoeic threshold or X-intercept of the ventilatory CO2 response curve. SC and Sp, the CO2 sensitivities of the central and peripheral chemoreflex loops, respectively. τp, time constant of the peripheral chemoreflex loop. τon and τoff, central time constants of responses to CO2 on-and off transients, respectively. , arterial–end-tidal difference.
Figure 2. Effect of 4 mg kg−1 acetazolamide (i.v.) on the hypoxic CO2 sensitivities of the peripheral and central chemoreflex loops.
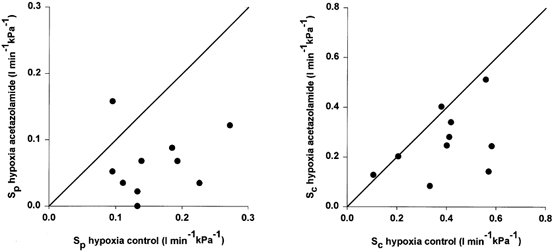
Effect of acetazolamide (4 mg kg−1, i.v.) on the CO2 sensitivities of the peripheral (SP, left panel) and central (SC, right panel) chemoreflex loops during hypoxia. Each animal is represented by one symbol. Diagonal line represents line of identity.
Comparison of the data in Tables 1 and 2 shows that before acetazolamide infusion during hypoxia the CO2 sensitivity of the peripheral chemoreflex loop was considerably larger than during hyperoxia (mean sensitivities 0.16 vs. 0.09 l min−1 kPa−1, P = 0.006). We illustrate this with two DEF runs from one animal in Fig. 3. Figure 4, obtained from the same animal, shows the absence of this CO2–O2 interaction after acetazolamide administration. In all 10 animals studied, a difference between the hypoxic and hyperoxic peripheral CO2 sensitivity was no longer present after acetazolamide administration (0.07 vs. 0.06 l min−1 kPa−1, P = 0.558), indicating the absence of a CO2–O2 stimulus interaction.
Figure 3. CO2–O2 stimulus interaction in the control situation.
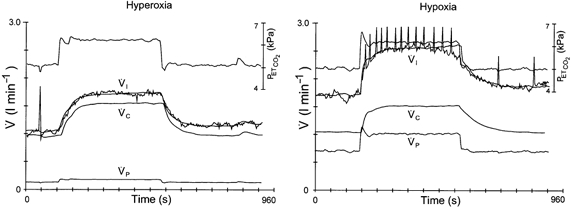
Two DEF runs obtained in the control situation during hyperoxia (left panel) and hypoxia (right panel). Upper traces are end-tidal . Breath-by-breath data points of inspiratory ventilation () are connected; the smooth line represents the optimal model fit.  and
and  are the calculated contributions of the central and peripheral chemoreflex loops, respectively, to the change in ventilation. The sensitivity of the peripheral chemoreflex loop is considerably larger during hypoxia.
are the calculated contributions of the central and peripheral chemoreflex loops, respectively, to the change in ventilation. The sensitivity of the peripheral chemoreflex loop is considerably larger during hypoxia.
Figure 4. Absence of CO2–O2 stimulus interaction after administration of acetazolamide.
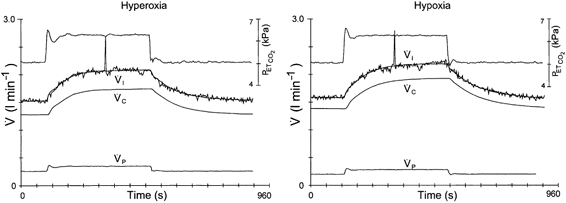
Two DEF runs obtained during hyperoxia (left panel) and hypoxia (right panel) after 4 mg−1 kg−1 acetazolamide. Upper traces are end-tidal . Breath-by-breath data points of inspiratory ventilation () are connected; the smooth line represents the optimal model fit. and are the calculated contributions of the central and peripheral chemoreflex loops, respectively, to the change in ventilation. Going from hyperoxia into hypoxia does not result in an increased contribution of the peripheral chemoreflex loop to the change in ventilation.
Standard bicarbonate (determined for the hypoxic condition only) was not significantly changed by acetazolamide: 20.1 ± 1.5 mmol l−1 (control) vs. 18.5 ± 1.9 mmol l−1 (acetazolamide).
DISCUSSION
The main results obtained in this study can be summarised as follows.
(1) Going from hyperoxia to moderate hypoxia in the control situation (mean from 47.60 to 8.02 kPa) led to an almost doubling of CO2 sensitivity of the peripheral chemoreflex loop. After administration of 4 mg kg−1 acetazolamide, this CO2–O2 interaction was no longer present.
(2) Both during hyperoxia and during moderate hypoxia, acetazolamide reduced the CO2 sensitivity of the peripheral chemoreflex loop, but during hyperoxia this reduction was much smaller and statistically insignificant.
(3) Acetazolamide caused similar decreases in the CO2 sensitivities of the central chemoreflex loop during hyperoxia and moderate hypoxia of 35 and 40 %, respectively, and a decrease in the value of the apnoeic threshold of approximately 2 kPa.
The present studies were performed in anaesthetised animals and we are cautious about applying our results to conscious animals. In cats, inhalational anaesthetics depress respiration due to an effect on the peripheral and central chemoreflex loops (Dahan et al. 1999). For sevoflurane, the effect-site half-life of respiratory changes is about 3.5 min (Dahan et al. 1999). In this study, sevoflurane was discontinued for at least 1 h (> 15 half-lives) before the respiratory studies started. This may also be relevant given the known effect of the inhalational anaesthetic halothane on background potassium channels in carotid body cells from neonatal rats (Buckler et al. 2000). The infusion of α-chloralose–urethane has little effect on respiration and the ventilatory responses to hypercapnia compared with the awake state and does not yield systematic changes in CO2 sensitivity over time (Gautier & Bonora, 1979; Teppema et al. 1997).
Essential features of the present study included the continuous measurement of arterial blood gases. This is crucial for two reasons. First, with regard to the , we determined the CO2 sensitivity of the peripheral chemoreflex loop, which in the (moderate) hypoxic range may be very sensitive for small variations in . Because end-tidal does not reflect , and, among other factors, depends on the level of ventilation, the experimental design required continuous measurement of the arterial tension. By varying the inspired oxygen concentration when necessary, we were able to perform DEF runs at equal arterial oxygen tensions before and after acetazolamide infusion. Second, regarding the , we wished to use as a measure of erythrocytic carbonic anhydrase inhibition (Maren, 1967). Not only would effective inhibition of the erythrocytic enzyme severely compromise the blood CO2 transport system (resulting in ) differences as large as 2.5–3.5 kPa - Teppema et al. 1988, 1992, 1995; Wagenaar et al. 1996), it would also reduce ventilatory CO2 sensitivity by mechanisms other than inhibition of the endothelial and carotid body enzyme alone (Teppema et al. 1995; Wagenaar et al. 1996). After infusion of 4 mg kg−1 acetazolamide, the animals in this study developed small increases in the arterial-end-tidal gradients (see Tables 1 and 2) only. Our data do not allow us to estimate the extent of the resulting inhibition of the erythrocytic enzyme, but it must have been less than that needed to cause the physiological effect of complete (i.e. > 99.9 %) inhibition (Maren, 1967,1977). In the cat, maximal widening of the arterial-end-tidal gradient occurs after about 30 mg kg−1, a dose substantially higher than is needed in dogs (see Wagenaar et al. 1996, 1998, 2000). Altogether, we have no reasons to assume that the applied dose of acetazolamide caused substantial slowing of the CO2 hydration reaction kinetics in the blood, which in that case could have slowed or even masked a fast (peripheral) component in the ventilatory response upon stepwise changes in end-tidal .
Because, due to its physicochemical properties, acetazolamide crosses biological membranes rather slowly (Holder & Hayes, 1965), the fact that a dose as low as that applied in the present study has such large ventilatory effects is remarkable. The effects on the carbon dioxide sensitivity of the central chemoreflex loop and on the apnoeic threshold B confirm and extend earlier findings obtained in normoxic and moderate hypoxic conditions, respectively (Wagenaar et al. 1996, 1998). Briefly, we attributed the decreases in SC and B to the inhibition by acetazolamide of a membrane-bound carbonic anhydrase on the luminal side of brain capillaries (cf. Hanson et al. 1981a; Ridderstråle & Hanson, 1984). This may influence the relationship between brain tissue and arterial by altering the cerebral blood flow response to changes in arterial (Wagenaar et al. 1996, 1998). In the same studies we ascribed the effect on the peripheral chemoreflex loop to a possible direct action on the peripheral chemoreceptors, and because the interaction between O2 and CO2 is known to occur at the level of the carotid bodies (Lahiri & Delaney, 1975; Fitzgerald & Deghani, 1982), we focused in the present study on the effect of acetazolamide on this interaction.
From our finding that acetazolamide abolished the increase in SP, on going from hyperoxia to moderate hypoxia, we conclude that indeed the low dose must affect the peripheral chemoreflex by a direct action on the carotid bodies.
It is known that chemosensitive (type I) carotid body cells posses carbonic anhydrase but the exact subcellular location(s) and the identity of the isoform(s) are unknown (Ridderstråle & Hanson, 1984; Rigual et al. 1985; Nurse, 1990). Histochemical studies by Ridderstråle & Hanson (1984) and Rigual et al. (1985) showed the existence of both cytosolic and membrane-bound carbonic anhydrase in type I cells from cat. Biochemical assays of rabbit carotid bodies suggested the presence of at least two isoforms (one cytosolic and one membrane-bound) of the enzyme (Botrèet al. 1994). Because we observed such pronounced effects after a low dose of a moderately slow-permeating inhibitor, we should consider the possibility that its effect may be mediated by inhibition of an easily accessible isoform of the enzyme, possibly CA IV, at the extracellular face of the membrane of type I cells. In this context we refer to the finding of Hanson et al. (1981b) that the non-permeant inhibitor benzolamide, at a dose as low as 2 mg kg−1, reduced the overshoot in the carotid body response to fast local applications of CO2-saturated saline. Their in vivo finding in cats, that acetazolamide (50–100 mg kg−1, i.v.) quickly (i.e. within a few minutes) reduced the speed of the carotid body response to a fast hypercapnic stimulus and prevented adaptation, led Torrance and co-workers initially to suggest that an extracellular carbonic anhydrase was mediating the fast hypercapnic response (Black et al. 1971). Because a large dose (25 mg kg−1) of the more lipid-soluble and less ionised agent acetazolamide had more pronounced effects than benzolamide, they suggested later that carotid body responses are triggered by an intracellular pH change rather than by an extracellular one (acidic hypothesis; see Hanson et al. 1981b).
At present, it is generally accepted that intracellular acidosis is a crucial factor in the cascade of events from a rise in to an increase in carotid body discharge. The activity of local carbonic anhydrase is necessary to obtain the normal biphasic response of the afferent nerve upon a fast change in , i.e. an over- or undershoot followed by a slower adaptation to the new steady-state level: apart from a decrease in resting activity, inhibitors of the enzyme cause a reduction in the peak and steady-state carotid sinus nerve responses (Hanson et al. 1981b; Iturriaga et al. 1991; Iturriaga, 1993; Lahiri et al. 1996). In vitro, the predominant effect of complete carbonic anhydrase inhibition is to reduce the fast initial rather than the steady-state response (Itturiaga et al. 1993; Lahiri et al. 1996). This, however, may depend on the degree of inhibition, since during partial inhibition the in vitro steady-state response may be reduced (Itturiaga et al. 1991). The effects of carbonic anhydrase inhibitors may be due to a rise in intracellular pH. Not only has intracellular alkalosis by acetazolamide been shown to occur in type I carotid body cells from neonatal rats (Buckler et al. 1991), it has also been observed in other tissues such as smooth muscle cells from guinea-pig mesentery arteries (Pickkers et al. 1999). So if the effects of acetazolamide on SP and on the CO2–O2 interaction that we report are also due to intracellular alkalosis in type I cells, normal activity of the uninhibited enzyme will have an acidifying effect. How then could a carbonic anhydrase iso-enzyme, possibly located at the extracellular face of the membrane (see above), acidify the cell interior? One possibility is that normal enzyme activity is necessary for the operation of a bicarbonate-chloride (bicarbonate out, chloride in) exchanger in the membrane of type I cells, which indeed appears to exist (Buckler et al. 1991). Inhibition of the exchanger results in intracellular alkalosis (Buckler et al. 1991; Wilding et al. 1992), and in a reduced steady-state response of the carotid body to hypoxia and hypercapnia (Itturiaga et al. 1998). In several tissues, intracellular alkalosis by inhibition of a membrane-bound carbonic anhydrase may be due to inhibition of a chloride-bicarbonate exchanger (Pickkers et al. 1999).
In guinea-pig mesenteric smooth muscle cells, benzolamide and acetazolamide, apart from causing intracellular alkalosis, stimulate large-conductance (Ca2+- dependent) potassium channels, resulting in hyperpolarisation, reduced Ca2+ influx and relaxation (Pickkers et al. 1999). Conversely, Peers & Green (1991) showed that weak-acid-induced intracellular acidosis in type I cells from neonatal rats inhibits large-conductance potassium channels.
A reduced influx of calcium ions secondary to acetazolamide-induced hyperpolarisation of glomus cells could well result in a substantial reduction in CO2–O2 interaction: Dasso et al. (2000) recently showed that within individual type I cells from neonatal rats a combined hypoxic-hypercapnic stimulus invariably led to a greater rise in intracellular rise [Ca2+] than either stimulus alone.
In summary, considering the data available in the literature we suggest that the reduction in SP and the loss of CO2–O2 interaction induced by low-dose acetazolamide may be due to an alkalising effect on carotid body cells, followed by an increased opening probability of large conductance potassium channels, hyperpolarisation and reduced calcium influx. Obviously, such a scenario warrants electrophysiological verification. In addition, the mechanism(s) by which carbonic anhydrase influences the pH in glomus cells will remain incompletely understood until more detailed information on both the identity and subcellular location(s) of the isoforms becomes available.
An inhibiting effect of acetazolamide has also been shown in human subjects. After an intravenous infusion of 500 mg, Swenson & Hughes (1993) reported that the eucapnic hypoxic ventilatory response was abolished. Scheuermann et al. (1999a,b) showed that an intravenous dose of 10 mg kg−1 slowed the  and
and 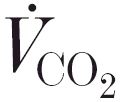 kinetics and prevented a decrease in ventilation upon the transition from normoxia to hyperoxia during steady-state exercise, and they attributed this to a decrease in carotid body gain. However, after a standard clinical oral dosage of 250 mg every 8 h (for 3 days), we did not find any change in the CO2 sensitivity of the peripheral chemoreflex loop in healthy volunteers (Teppema and Dahan, 1999). It seems therefore that the effect of the inhibitor on the carotid bodies depends critically on the dose and route of administration. In most clinical applications of acetazolamide, an oral dose is given. Usually this results in metabolic acidosis, frequently (but not always) followed by a rise in ventilation (Teppema and Dahan, 1999). Acute intravenous administration, however, does not lead to metabolic acidosis but may be followed by inhibiting effects on the control of breathing (reviewed by Swenson, 1998).
kinetics and prevented a decrease in ventilation upon the transition from normoxia to hyperoxia during steady-state exercise, and they attributed this to a decrease in carotid body gain. However, after a standard clinical oral dosage of 250 mg every 8 h (for 3 days), we did not find any change in the CO2 sensitivity of the peripheral chemoreflex loop in healthy volunteers (Teppema and Dahan, 1999). It seems therefore that the effect of the inhibitor on the carotid bodies depends critically on the dose and route of administration. In most clinical applications of acetazolamide, an oral dose is given. Usually this results in metabolic acidosis, frequently (but not always) followed by a rise in ventilation (Teppema and Dahan, 1999). Acute intravenous administration, however, does not lead to metabolic acidosis but may be followed by inhibiting effects on the control of breathing (reviewed by Swenson, 1998).
In this and previous studies in cat and rabbit, we have shown that an intravenous dose as low as 4 mg kg−1 may have both stimulating (decrease in apnoeic threshold) and inhibiting (decrease in CO2 sensitivity) effects (Wagenaar et al. 1995, 1996, 2000). Although we cannot exclude effects at the (neuro)muscular level as shown in rabbit (see Kiwull-Schöne et al. 2001), the fact that in the present study the agent abolished or at least reduced the CO2–O2 interaction suggests a direct action on the carotid bodies. Preliminary data from our laboratory show that in the cat acetazolamide, at the same dose, also reduces the isocapnic hypoxic response; larger doses, sufficient to cause effective inhibition of intracellular (e.g. red cell) carbonic anhydrase, completely abolish it (Teppema et al. 1988, 1992). Again this indicates that the effect is dose dependent. If carbonic anhydrase inhibitors cause physiological effects only when inhibition exceeds 99 % (Maren, 1967,1977), this dose dependency could result from enzyme activity in different compartments with variable accessibility. Thus a relatively high dose of a moderately slow-permeating agent would be needed to inhibit the enzyme at intracellular locations.
Our data do not provide a direct clue as to the specific site of action of acetazolamide within the carotid bodies. As mentioned above, data obtained from cat and rabbit indicate the existence of at least two iso-enzymes in type-I cells. Biochemical analysis of rabbit carotid bodies showed that during hyperoxia enzyme activity was lower than during hypoxia (Botrèet al. 1994). This may explain our present finding that during hyperoxia the effect of acetazolamide on SP was smaller than during hypoxia (see also previous data from hypoxic animals; Wagenaar et al. 1998). Carbonic anhydrase may thus be an oxygen-sensitive element within the carotid bodies.
In conclusion, the data presented here show that low-dose acetazolamide lowers the CO2 sensitivity of the peripheral chemoreflex loop (SP) particularly during hypoxia. Furthermore, the agent prevented the increase in SP on going from hyperoxia to moderate hypoxia. Because the interaction between O2 and CO2 resides within the carotid bodies, it was concluded that the effect of acetazolamide is probably mediated by a direct action on the carotid bodies. The precise role of carbonic anhydrase within the carotid bodies is not completely known at this stage, but our data show that normal activity of the enzyme is crucial for the carotid bodies to function normally, even in steady-state conditions.
References
- Black AMS, McCloskey DI, Torrance RW. The responses of carotid body chemoreceptors in the cat to sudden changes of hypercapnic and hypoxic stimuli. Respiration Physiology. 1971;13:36–49. doi: 10.1016/0034-5687(71)90063-6. [DOI] [PubMed] [Google Scholar]
- Botrè F, Botrè C, Greco A, Data PG, Di Giulio C, Morelli L. Potentiometric determination of carbonic anhydrase activity in rabbit carotid bodies: comparison among normoxic, hyperoxic and hypoxic animals. Neuroscience Letters. 1994;166:126–130. doi: 10.1016/0304-3940(94)90467-7. [DOI] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD, Peers C, Nye PC. Intracellular pH and its regulation in isolated type I carotid body cells of the neonatal rat. Journal of Physiology. 1991;436:107–129. doi: 10.1113/jphysiol.1991.sp018542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Williams BA, Honore E. An oxygen, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemoreceptor cells. Journal of Physiology. 2000;525:135–142. doi: 10.1111/j.1469-7793.2000.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahan A, Olofsen E, Teppema L, Sarton E, Olievier C. Speed of onset and offset and mechanisms of ventilatory depression from sevoflurane. Anesthesiology. 1999;90:1119–1128. doi: 10.1097/00000542-199904000-00027. [DOI] [PubMed] [Google Scholar]
- Dasso LLT, Buckler KJ, Vaughan-Jones R. Interactions between hypoxia and hypercapnic acidosis on calcium signalling in carotid body type I cells. American Journal of Physiology. 2000;279:L36–42. doi: 10.1152/ajplung.2000.279.1.L36. [DOI] [PubMed] [Google Scholar]
- Degoede J, Berkenbosch A, Ward DS, Bellville JW, Olievier CN. Comparison of chemoreflex gains obtained with two different methods in cats. Journal of Applied Physiology. 1985;59:170–179. doi: 10.1152/jappl.1985.59.1.170. [DOI] [PubMed] [Google Scholar]
- Fitzgerald RS, Deghani GA. Neural responses of the cat carotid and aortic bodies to hypercapnia and hypoxia. Journal of Applied Physiology. 1982;52:596–601. doi: 10.1152/jappl.1982.52.3.596. [DOI] [PubMed] [Google Scholar]
- Gautier H, Bonora M. Effects of carotid chemodenervation on respiratory pattern of awake cats. Journal of Applied Physiology. 1979;46:1127–1131. doi: 10.1152/jappl.1979.46.6.1127. [DOI] [PubMed] [Google Scholar]
- Hanson MA, Nye PCG, Torrance RW. The location of carbonic anhydrase in relation to the blood-brain barrier at the medullary chemoreceptors of the cat. Journal of Physiology. 1981a;320:113–125. doi: 10.1113/jphysiol.1981.sp013938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MA, Nye PCG, Torrance RW. The exodus of an extracellular bicarbonate theory of chemoreception and the genesis of an intracellular one. In: Belmonte C, Pallot D, Acker H, Fidone S, editors. Arterial Chemoreceptors. Leicester UK: Leicester University Press; 1981b. pp. 403–416. [Google Scholar]
- Holder LB, Hayes SL. Diffusion of sulfonamides in aqueous buffers and into red cells. Molecular Pharmacology. 1965;1:266–279. [PubMed] [Google Scholar]
- Iturriaga R. Carotid body chemoreception: The importance of CO2-HCO3− and carbonic anhydrase (review) Biological Research. 1993;26:319–329. [PubMed] [Google Scholar]
- Itturiaga R, Lahiri S, Mokashi A. Carbonic anhydrase and chemoreception in the carotid body. American Journal of Physiology. 1991;265:C565–573. doi: 10.1152/ajpcell.1991.261.4.C565. [DOI] [PubMed] [Google Scholar]
- Iturriaga R, Mokashi A, Lahiri S. Dynamics of carotid body responses in vitro in the presence of CO2-HCO3−: role of carbonic anhydrase. Journal of Applied Physiology. 1993;75:1587–1594. doi: 10.1152/jappl.1993.75.4.1587. [DOI] [PubMed] [Google Scholar]
- Iturriaga R, Mokashi A, Lahiri S. Anion exchanger and chloride channel in cat carotid body chemotransduction. Journal of the Autonomic Nervous System. 1998;70:23–31. doi: 10.1016/s0165-1838(98)00019-8. [DOI] [PubMed] [Google Scholar]
- Kiwull-Schöne H, Teppema LJ, Kiwull P. Low-dose acetazolamide does affect respiratory muscle function in spontaneously breathing anesthetized rabbits. American Journal of Respiratory and Critical Care Medicine. 2001;163:478–483. doi: 10.1164/ajrccm.163.2.9911075. [DOI] [PubMed] [Google Scholar]
- Lahiri S, Delaney R. Stimulus interaction in the responses of carotid body single afferent fibers. Respiration Physiology. 1975;24:249–266. doi: 10.1016/0034-5687(75)90017-1. [DOI] [PubMed] [Google Scholar]
- Lahiri S, Iturriaga R, Mokashi A, Botrè F, Chugh D, Osanai S. Adaptation to hypercapnia vs. intracellular pH in cat carotid body: responses in vitro. Journal of Applied Physiology. 1996;80:1090–1099. doi: 10.1152/jappl.1996.80.4.1090. [DOI] [PubMed] [Google Scholar]
- Lee KD, Mattenheimer H. The biochemistry of the carotid body. Enzymologia Biologica et Clinica. 1964;4:199–216. doi: 10.1159/000458029. [DOI] [PubMed] [Google Scholar]
- Maren TH. Carbonic anhydrase: Chemistry, physiology and inhibition. Physiological Reviews. 1967;47:595–761. doi: 10.1152/physrev.1967.47.4.595. [DOI] [PubMed] [Google Scholar]
- Maren TH. Use of inhibitors in physiological studies of carbonic anhydrase. American Journal of Physiology. 1977;232:F291–297. doi: 10.1152/ajprenal.1977.232.4.F291. [DOI] [PubMed] [Google Scholar]
- Nurse CA. Carbonic anhydrase and neuronal enzymes in cultured glomus cells of the carotid body of the rat. Cell and Tissue Research. 1990;261:65–71. doi: 10.1007/BF00329439. [DOI] [PubMed] [Google Scholar]
- Peers C, Green FK. Inhibition of Ca2+-activated K+ currents by intracellular acidosis in isolated type I cells of the neonatal rat carotid body. Journal of Physiology. 1991;437:589–602. doi: 10.1113/jphysiol.1991.sp018613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickkers P, Garcha RS, Schachter M, Smits P, Hughes AD. Inhibition of carbonic anhydrase accounts for the direct vascular effects of hydrochlorothiazide. Hypertension. 1999;33:1043–1048. doi: 10.1161/01.hyp.33.4.1043. [DOI] [PubMed] [Google Scholar]
- Ridderstråle Y, Hanson MA. Histochemical localization of carbonic anhydrase in the cat carotid body. Proceedings of the New York Academy of Sciences. 1984;429:398–400. doi: 10.1111/j.1749-6632.1984.tb12363.x. [DOI] [PubMed] [Google Scholar]
- Rigual C, Iñiguez C, Carreres J, Gonzalez C. Carbonic anhydrase in the carotid body and the carotid sinus nerve. Histochemistry. 1985;82:577–580. doi: 10.1007/BF00489979. [DOI] [PubMed] [Google Scholar]
-
Scheuermann BW, Kowalchuk JM, Paterson DH, Cunningham D. and kinetics during moderate- and heavy-intensity exercise after acetazolamide administration. Journal of Applied Physiology. 1999a;86:1534–1543. doi: 10.1152/jappl.1999.86.5.1534. [DOI] [PubMed] [Google Scholar]
- Scheuermann BW, Kowalchuk JM, Paterson DH, Cunningham D. Peripheral chemoreceptor function after carbonic anhydrase inhibition during moderate-intensity exercise. Journal of Applied Physiology. 1999b;86:1544–1551. doi: 10.1152/jappl.1999.86.5.1544. [DOI] [PubMed] [Google Scholar]
- Sly WS, Hu PY. Human carbonic anhydrases and carbonic anhydrase deficiencies. Annual Review of Biochemistry. 1995;64:375–401. doi: 10.1146/annurev.bi.64.070195.002111. [DOI] [PubMed] [Google Scholar]
- Swenson ER. Carbonic anhydrase inhibition and ventilation: a complex interplay of stimulation and suppression. European Respiratory Journal. 1998;12:1242–1247. doi: 10.1183/09031936.98.12061242. [DOI] [PubMed] [Google Scholar]
- Swenson ER, Hughes JMB. Effects of acute and chronic acetazolamide on resting ventilation and ventilatory responses in man. Journal of Applied Physiology. 1993;73:230–237. doi: 10.1152/jappl.1993.74.1.230. [DOI] [PubMed] [Google Scholar]
- Teppema L, Berkenbosch A, Degoede J, Olievier C. Carbonic anhydrase and control of breathing: different effects of benzolamide and methazolamide in the anaesthetized cat. Journal of Physiology. 1995;488:767–777. doi: 10.1113/jphysiol.1995.sp021008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teppema L, Berkenbosch A, Olievier C. Effect of Nω-nitro-L-arginine on ventilatory response to hypercapnia in anesthetized cats. Journal of Applied Physiology. 1997;82:292–297. doi: 10.1152/jappl.1997.82.1.292. [DOI] [PubMed] [Google Scholar]
- Teppema LJ, Dahan A. Acetazolamide and breathing. Does a clinical dose alter peripheral and central CO2 sensitivity? American Journal of Respiratory and Critical Care Medicine. 1999;160:1592–1597. doi: 10.1164/ajrccm.160.5.9903088. [DOI] [PubMed] [Google Scholar]
- Teppema LJ, Rochette F, Demedts M. Ventilatory response to carbonic anhydrase inhibition in cats: effects of acetazolamide in intact vs. peripherally denervated animals. Respiration Physiology. 1988;74:373–382. doi: 10.1016/0034-5687(88)90044-8. [DOI] [PubMed] [Google Scholar]
- Teppema LJ, Rochette F, Demedts M. Ventilatory effects of acetazolamide in cats during hypoxemia. Journal of Applied Physiology. 1992;72:1717–1723. doi: 10.1152/jappl.1992.72.5.1717. [DOI] [PubMed] [Google Scholar]
- Torrance RW. Prolegomena. Chemoreception upstream of transmitters. Advances in Experimental Biology and Medicine. 1996;410:13–38. [PubMed] [Google Scholar]
- Wagenaar M, Teppema L, Berkenbosch A, Olievier C, Folgering H. The effect of low-dose acetazolamide on the ventilatory CO2 response curve in the anaesthetized cat. Journal of Physiology. 1996;495:227–237. doi: 10.1113/jphysiol.1996.sp021587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenaar M, Teppema L, Berkenbosch A, Olievier C, Folgering H. Effect of low-dose acetazolamide on the ventilatory CO2 response during hypoxia in the anaesthetized cat. European Respiratory Journal. 1998;12:1271–1277. doi: 10.1183/09031936.98.12061271. [DOI] [PubMed] [Google Scholar]
- Wagenaar M, Teppema L, Berkenbosch A, Olievier C, Folgering H. Medroxyprogesterone acetate with acetazolamide stimulates breathing in cats. Respiration Physiology. 2000;119:19–29. doi: 10.1016/s0034-5687(99)00098-5. [DOI] [PubMed] [Google Scholar]
- Wilding TJ, Cheng B, Roos A. pH regulation in adult rat carotid body glomus cells. Journal of General Physiology. 1992;100:593–608. doi: 10.1085/jgp.100.4.593. [DOI] [PMC free article] [PubMed] [Google Scholar]