

Abstract
Large-conductance Ca2+- and voltage-activated potassium (BK) channels are important regulators of cellular excitability. Here, we present a patch-clamp electrophysiological analysis of splice-variant-specific regulation by the synthetic glucocorticoid dexamethasone (DEX) of BK channels consisting of cloned STREX or ZERO α-subunit variants expressed in human embryonic kidney (HEK 293) cells.
STREX channels in isolated membrane patches were inhibited by protein kinase A (PKA) and this was blocked on pre-treatment of intact cells with DEX (100 nm) for 2 h.
The effect of DEX required the synthesis of new mRNA and protein. Furthermore, it required protein phosphatase 2A (PP2A)-like activity intimately associated with the channels, as it was blocked by 10 nm okadaic acid but not by the specific protein phosphatase-1 inhibitor peptide PPI−2.
ZERO variant channels that lack the STREX insert were activated by PKA but were not influenced by DEX. ZERO channels containing a mutant STREX domain (S4STREXA) were also activated by PKA. Importantly, DEX blocked PKA activation of S4STREXA channels in a PP2A-dependent manner.
Taken together, the STREX domain is crucial for glucocorticoid regulation of BK channels through a PP2A-type enzyme. Moreover, glucocorticoids appear to induce a generic set of proteins in different types of cells, the actions of which depend on the expression of cell-specific targets.
Adrenal glucocorticoids are pivotal for adaptation to environmental challenge in a wide range of tissues including the central nervous and endocrine systems (Sapolsky et al. 2000). These hormones can potently modify the electrical excitability of target cells through regulation of ion channel activity that requires the rapid induction of new proteins. Increasing evidence suggests that in neurones and endocrine cells, potassium channels are important targets for glucocorticoid action. For example glucocorticoids rapidly induce the expression of potassium channel subunits in some systems (Levitan et al. 1991; Attali et al. 1995), but in both hippocampal CA1 neurones and neuroendocrine anterior pituitary corticotroph (AtT20) cells (Joels & de Kloet, 1989; Shipston et al. 1996; Tian et al. 1998), the activity of Ca2+-activated potassium channels is modulated by glucocorticoid-induced signalling pathways. In both of these systems glucocorticoid action is mediated by type II glucocorticoid receptors, occurs within 2 h, and requires de novo synthesis of mRNA as well as protein. However, the underlying mechanism(s) are poorly understood.
Large-conductance Ca2+-activated potassium channels (BK channels) are pivotal targets of glucocorticoids in AtT20 cells (Shipston et al. 1996; Tian et al. 1998). In these neuroendocrine cells, cAMP-generating agonists inhibit BK channels through cAMP-dependent protein kinase (PKA). Proteins rapidly induced by the synthetic glucocorticoid dexamethasone block the inhibition of BK channels by PKA, and this is crucial for the suppression of stimulated ACTH release by glucocorticoids (Shipston et al. 1996). The effect of glucocorticoids on BK channels is blocked by low nanomolar concentrations of okadaic acid (Tian et al. 1998), suggesting an involvement of serine/threonine protein phosphatases, for example protein phosphatase (PP) 1 or 2A in glucocorticoid action.
AtT20 cells express two major BK channel splice variants, ZERO and STREX (Shipston et al. 1999). Unlike many other voltage-gated potassium channels, the pore-forming BK channel subunits are derived from a single gene that undergoes extensive hormonally regulated alternative splicing (Adelman et al. 1992; Butler et al. 1993; Pallanck & Ganetzky, 1994; Tseng-Crank et al. 1994; Xie & McCobb, 1998; Xie & Black, 2001). The STREX variant is identical to ZERO except for a 59 amino acid insert at site 2, which is one of at least five different splice sites in the C terminal portion of the BK channel α-subunit protein encoded by the mammalian Slo gene (Saito et al. 1997; Shipston et al. 1999). The STREX insert confers enhanced calcium sensitivity to BK channels (Saito et al. 1997; Shipston et al. 1999). Furthermore, STREX channels are inhibited, whereas ZERO channels are activated, by PKA closely associated with the channel complex (Tian et al. 2001).
In this report we have examined whether the pore-forming subunits of BK channels are targets for glucocorticoid regulation and whether such modulation is dependent upon the BK channel splice variant expressed. To address these issues we have investigated the regulation of the cloned BK channel splice variants, STREX and ZERO, heterologously expressed in human embryonic kidney 293 (HEK 293) cells. We demonstrate that the STREX insert confers glucocorticoid sensitivity on BK channels by targeting a glucocorticoid-induced signalling pathway, involving serine/threonine protein phosphatases, to potassium channels. Differential modulation of BK channel splice variants by glucocorticoids would provide a powerful mechanism to tune the excitability of glucocorticoid-sensitive target cells.
METHODS
Construction of BK channel splice variant and mutant cDNA constructs
The cloning and sub-cloning of the mouse BK channel splice variants and respective site directed mutants into the mammalian expression vector pcDNA3 or pcDNA3.1+ (Invitrogen, Leek, The Netherlands) have been described previously (Clark et al. 1999; Shipston et al. 1999; Tian et al. 2001). Briefly, the ZERO variant lacks inserts at mammalian splice sites 1–5 and STREX channels are identical except for a 59 amino acid cysteine-rich insert at mammalian splice site 2 that increase the apparent calcium sensitivity of the channel (Saito et al. 1997; Shipston et al. 1999). The ZERO variant is activated by PKA, requiring a functional serine residue at position 869 within a putative PKA consensus motif present in the C-termini of all mammalian BK channel variants. The STREX variant is inhibited by PKA, dependent upon a conserved serine residue (S4) within the STREX insert itself. The S4STREXA mutant has this serine residue mutated to alanine. The STREX-S869A mutant has a single point mutation of the conserved serine residue at position 869 to alanine. Site-directed mutagenesis was performed using Quik-change (Stratagene) and verified by DNA sequencing. The constructs used in these studies are shown schematically in Fig. 6B.
Figure 6. STREX is required for glucocorticoid regulation of BK channels.
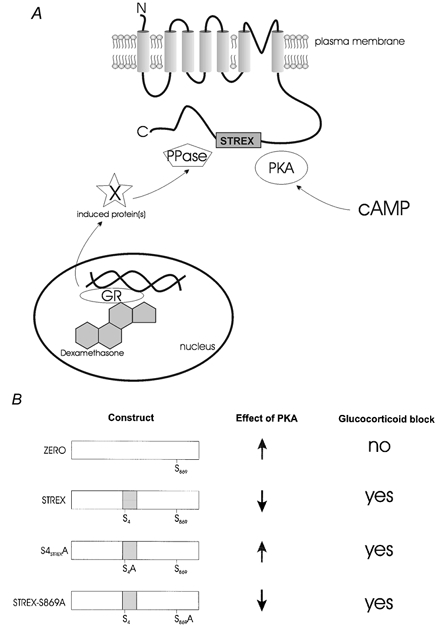
A, schematic illustrating that BK channels containing the STREX insert are targets for both protein kinase A (PKA) and glucocorticoid signalling pathways. PKA closely associated with membrane patches is activated by cyclic 3′,5′-adenosine monophosphate (cAMP). Steroids, such as the synthetic glucocorticoid dexamethasone, activate intracellular glucocorticoid receptors (GR) to regulate gene transcription. GR induced de novo synthesis of unidentified (X) proteins that block PKA-dependent regulation of STREX-containing channels through modification of protein phosphatase 2A-like (PPase) activity closely associated with the membrane patch in dexamethasone-treated cells. B, schematic summarising the effect of PKA and glucocorticoids on the different BK channel splice variant constructs used in this study. The effect of PKA is indicated by activation (up arrow) or inhibition (down arrow) of channel activity. The ability of dexamethasone to block the PKA-induced regulation of BK channel activity is indicated. The STREX insert is essential for glucocorticoid-mediated regulation of BK channel activity irrespective of the functional effect of PKA on channel activity (activation or inhibition) for the different constructs. Serine residues essential for PKA-mediated activation of channel activity (conserved S869 in C-terminal tail of channel) and PKA-mediated inhibition (S4 in the STREX insert) are indicated.
HEK 293 and AtT20 D16:16 cell culture and transfection
HEK 293 and AtT20 D16:16 cells were subcultured as previously described (Shipston et al. 1996, 1999). Briefly, cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10 % fetal calf serum (FCS) in a humidified atmosphere of 95 % air-5 % CO2 at 37 °C. Cells were routinely passaged every 3–7 days using 0.25 % trypsin in Hanks’ buffered salt solution (HBSS) containing 0.1 % EDTA. For immunoblotting studies, cells were grown to 70–80 % confluence in 75 cm2 flasks. For electrophysiological assays, cells were plated on glass coverslips in six-well cluster dishes.
For transient transfections of BK channels, HEK 293 cells were seeded onto glass coverslips in six-well cluster dishes at a density to allow cells to reach 40–60 % confluence after 24 h. Cells were then transfected with 1 μg of the respective cDNA using lipofectamine (Life Technologies) in DMEM, essentially as described by the manufacturer. After 5 h, medium was supplemented with 10 % FCS, replaced after 24 h and electrophysiological recordings made 24–72 h post-transfection. Stable cell lines containing different BK variants were also created from these transfections and the transformants were then selected and maintained for neomycin or zeocin resistance using 0.2 mg ml−1 zeocin (Invitrogen) or 0.8 mg ml−1 geneticin (Life Technologies) as appropriate. The HEK 293 cells do not express endogenous BK channels at the mRNA (by RT-PCR), protein (Western blotting) or functional electrophysiology level.
For luciferase assays, HEK 293 cells were plated directly into the six-well cluster dishes and transfected as above except that transfections and subsequent maintenance of cells was carried out in steroid-free medium (DMEM supplemented with insulin-transferrin-selenium (ITS, Sigma-Aldrich) supplement. The mammary tumour virus long terminal repeat (MMTV-LTR)-luciferase reporter construct (Leckie et al. 1995) was a generous gift from Dr K. Chapman (University of Edinburgh). Similar results were obtained with other artificial serum replacements and supra-maximal stimulation was identical in 10 % FCS. However, the majority of FCS batches contain low to sub-nanomolar concentrations of cortisol that preclude accurate determination of the full dose-response range of MMTV-LTR-luciferase reporter activity to the synthetic glucocorticoid agonist dexamethasone.
Western blot analysis of glucocorticoid receptors
AtT20 and HEK 293 whole cell extracts (from ∼107 cells) were prepared by resuspending cells in lysis buffer containing (mm): 20 Tris-HCl, 2 EDTA, 150 NaCl, 0.5 % Triton X-100, pH 7.4, and supplemented with a protease inhibitor cocktail (Boehringer Mannheim). Cells were sheared through a 26G needle and incubated on ice for approximately 1 h. The lysate was immediately spun for 10 min at 10 000 g and 4 °C and the supernatant used for subsequent SDS-PAGE and Western blot analysis. Total protein content was calculated using the Bradford protein assay (BioRad). Whole cell lysates (10 μg protein from AtT20 cells and 50 μg from HEK 293 cells) were resolved on 8 % SDS-polyacrylamide gels and transferred under semi-dry conditions onto polyvinylidene difluoride (PVDF) membrane. Membranes were blocked using phosphate buffered saline containing 0.05 % Tween 20 (0.05 %) (PBS-T) and 4 % non-fat dry milk (Marvel) for 1.5 h at room temperature (or overnight at 4 °C). All washing stages were carried out using PBS-T. Blots were incubated for 1 h at room temperature with a 1:2000 dilution of anti-GR (M-20) rabbit primary antibody (Autogen BioClear UK, Calne, Wiltshire, UK), and probed with a 1:4000 dilution of donkey anti-rabbit IgG horseradish peroxidase conjugate (Amersham Life Sciences) under the same conditions. Visualisation and processing of the blots was by enhanced chemiluminescence (ECL) essentially as described by the manufacturer.
Luciferase reporter assay
HEK 293 cells transiently transfected with the MMTV-LTR luciferase reporter construct were used 24–72 h post-transfection. Cells were treated with varying concentrations of the synthetic glucocorticoid agonist dexamethasone or vehicle (DMSO) for 2 h at 37 °C. Cells were quickly washed in phosphate buffered saline, lysed by the addition of 0.4 ml lysis buffer mix (25 mm Tris-phosphate, pH 7.8, 2 mm DTT, 1 % Triton X-100 and 10 % glycerol, containing 1.25 mg ml−1 lysozyme and 2.5 mg ml−1 BSA) scraped and briefly centrifuged at 12 000 g. Duplicate (20 μl) aliquots of the resulting supernatants were assayed for luciferase activity using Promega luciferase assay reagent (Promega, Madison, WI, USA) and emitted light detected with a LKB (Wallac) 1250 luminometer. Light emission was averaged over 10 s with a 2 s delay after substrate addition to allow the signal to stabilise. All values were calculated as a percentage of maximum light production with cells treated with 1000 nm of dexamethasone as 100 % after background (vehicle alone) subtraction. The apparent KD was calculated according to the equation:
where Y is luciferase activity at a given dexamethasone concentration [DEX], Ymax is maximal luciferase activity, [DEX] is concentration of dexamethasone and h is the Hill coefficient.
Patch clamp electrophysiology
All experiments were performed in the inside-out configuration of the patch clamp technique at room temperature (20–24 °C) using physiological potassium gradients essentially as described previously (Clark et al. 1999; Shipston et al. 1999; Tian et al. 2001). The pipette solution (extracellular) contained (mm): 140 NaCl, 5 KCl, 0.1 CaCl2, 2 MgCl2, 20 glucose, 10 Hepes, pH 7.4. The bath solution (intracellular) contained (mm): 140 KCl, 5 NaCl, 2 MgCl2, 1 or 5 BAPTA, 30 glucose, 10 Hepes, 1 mm ATP, pH 7.3 with free calcium ([Ca2+]i) buffered to 0.2 μm, unless indicated otherwise. Data acquisition and voltage protocols were controlled by an Axopatch 200A or -B amplifier and pCLAMP 6 software (Axon Instruments, Foster City, USA). All recordings were sampled at 10 kHz and filtered at 2 kHz. Following patch excision, channel activity was allowed to stabilise for at least 10 min (typically 10–15 min after excision) and stability plot experiments demonstrated that BK channel activity was stable for > 1 h under the recording conditions used (data not shown) in the absence of channel modulators. Application of cAMP or other reagents to the intracellular face of patches was by gravity-driven perfusion (10 volumes of the recording bath solution (bath volume, 0.5 ml) at a flow rate of 1–2 ml min−1) or direct application to the bath. In all experiments, 1 mm cAMP was used to activate endogenous PKA; similar results were also observed using 0.1 mm cAMP (not shown). Patches were held at 0 mV and channel activity was determined during voltage steps to +40 mV. To determine mean percentage change in channel activity after a treatment, in patches with low to moderate levels of channel expression, mean NPo (number of functional channels × open probability of channel) was averaged from 60 s of recording at +40 mV immediately before and 10 min after the respective drug treatment. Mean change in activity was expressed as a percentage of the pre-treatment control ± s.e.m. with analysis undertaken using pCLAMP 6 and IGOR Pro 3.1 (Wavemetrics, Lake Oswego, OR, USA). In the respective figure legends and text a positive percentage change in activity reflects activation whereas a negative percentage change reflects channel inhibition.
The single channel slope conductance and half-maximal voltage for activation (V0.5) for each construct under the recording conditions detailed above (except that the assays were performed in the absence of ATP) were as follows: STREX, 126.3 ± 4.1 pS, 26.8 ± 4.2 mV, n = 4; ZERO, 138.0 ± 5.1 pS, 76.1 ± 5.2 mV, n = 4; STREX-S869A, 125.2 ± 3.2 pS, 38.1 ± 7.3 mV, n = 5; S4STREXA, 124.9 ± 3.7 pS, 32.7 ± 6.5 mV, n = 4.
Chemicals and materials
ATP magnesium salt, adenosine 5′-O-(3-thiotriphosphate) (ATPγS) tetralithium salt, adenosine 3′,5′-cyclic monophosphate (cAMP), dexamethasone, puromycin and 5,6-dichloro-1-β-d-ribofluranosylbenzimidazole (DRB) were all purchased from Sigma/Aldrich (Poole, UK). Nucleotides were stored as buffered 1 m or 0.1 m stock solutions at −20 °C prior to use. Dexamethasone was stored at −20 °C at 10 mm in Me2SO. Purified protein phosphatase 2A catalytic subunit was from Promega. Okadaic acid was from LC Laboratories (Alexis Laboratories, Nottingham, UK). The specific protein kinase A inhibitor peptide (PKI5–24) and protein phosphatase 1 inhibitor (PPI-2) were from Calbiochem-Novabiochem (Nottingham, UK). Polyvinylidene difluoride membranes and reagents for SDS-polyacrylamide gel electrophoresis and Western blotting were from Bio-Rad Laboratories (Hemel Hempstead, UK). All other reagents were from Sigma or BDH-Merck (Poole, UK).
Statistical analysis
Data are expressed as means ± s.e.m unless otherwise stated. Statistical significance was defined at P < 0.05 using a non-parametric Kruskal-Wallis or Mann-Whitney U test as appropriate.
RESULTS
HEK 293 cells express functional glucocorticoid receptors
In whole cell lysates of HEK 293 cells, Western blot analysis, using a rabbit polyclonal antibody that recognises both the GRα and GRβ isoforms (Hollenberg et al. 1985; Bamberger et al. 1995), revealed specific immunoreactivity at a molecular mass consistent with that for the GR isoforms (∼95 kDa, Fig. 1A). No immunoreactivity was observed in the absence of primary antibody (not shown) and similar immunoreactive bands were present in whole cell lysates from mouse anterior pituitary AtT20 D16:16 corticotrophs, a cell line that expresses functional GR at a high level (Woods et al. 1992).
Figure 1. HEK 293 cells express functional glucocorticoid receptors.

A, whole cell lysates (10 μg protein from AtT20 cells as positive control and 50 μg from HEK 293 cells) were separated by SDS-PAGE and transferred to PVDF membranes. Blots were probed as described in methods with an anti-GR (M20) rabbit primary antibody that recognises both GRα and GRβ isoforms with detection by ECL; ∼95 kDa immunoreactive bands were detectable in both AtT20 and HEK 293 cell lysates. B, functional assay of glucocorticoid receptor activation in HEK 293 cells using a MMTV-luciferase reporter assay. HEK 293 cells were transiently transfected with 1 μg of MMTV-luc plasmid (○) or pcDNA3 vector alone (•) and treated for 2 h with various concentrations of the synthetic glucocorticoid receptor agonist dexamethasone or vehicle before assay for luciferase activity as described in Methods. Data are expressed as a percentage of maximal (with 1 μm dexamethasone) luciferase activity after vehicle background subtraction. Each data point represents the mean ± s.e.m (n = 4–8).
To examine whether HEK 293 cells contain functional GRα receptors, the mouse mammary tumour virus long terminal repeat (MMTV-LTR)-luciferase reporter plasmid, which responds to activated GR by enhancing luciferase expression (Leckie et al. 1995), was transiently transfected into the cells. Indeed, in this system DEX produced a concentration-dependent stimulation of luciferase activity with a KD of 4.5 ± 3.6 nm with maximal activity at 100 nm.
Dexamethasone pre-treatment blocks PKA-mediated inhibition of STREX BK channels
In inside-out patches from HEK 293 cells expressing cloned BK channel splice variants, single BK channel events were characterised by their unitary slope conductance and sensitivity to voltage and calcium as previously described (data not shown; Shipston et al. 1999). Pre-treatment of cells with a maximally effective concentration (0.1–1 μm) of the synthetic glucocorticoid agonist dexamethasone (Woods et al. 1992; Shipston et al. 1996) had no significant effects on intrinsic BK channel properties.
Application of cAMP (0.1–1 mm) to the intracellular face of isolated inside-out patches from HEK 293 cells expressing STREX or the site directed mutant STREX-S869A in the presence of 1 mm Mg-ATP and 0.2 μm[Ca2+]i resulted in a significant inhibition of BK channel activity (expressed as the percentage change of pre-treatment BK channel activity: for STREX, −57.5 ± 8.7 %, n = 13, P < 0.01; for STREX-S869A, −58.1 ± 12.6 %, n = 11, P < 0.01; determined 10 min after cAMP application compared with pre-treatment BK channel activity, Fig. 2A and B). As the effect of cAMP was identical in STREX and STREX-S869A channels, further analysis was largely carried out on STREX-S869A channels with results identical to those in STREX channels (not shown). The cAMP inhibition was dependent upon protein kinase A associated with the channels as the specific protein kinase A inhibitor peptide PKI5–24 blocked the inhibitory effect of cAMP (the change was −2.2 ± 7.8 %, P < 0.01, compared to −58.1 ± 12.6 % with cAMP alone, Fig. 2B). In parallel experiments, pre-treatment of HEK 293 cells with maximally effective concentrations of dexamethasone (0.1–1 μm for 2 h) abolished the inhibitory action of cAMP in excised inside-out patches containing STREX or STREX-S869A channels compared to the inhibition observed in patches from control cells (Fig. 2B). The percentage change in BK channel activity was 0.24 ± 7.7 % in DEX-treated cells (n = 11).
Figure 2. Dexamethasone blocks PKA-mediated inhibition of STREX BK channels via a genomic mechanism.
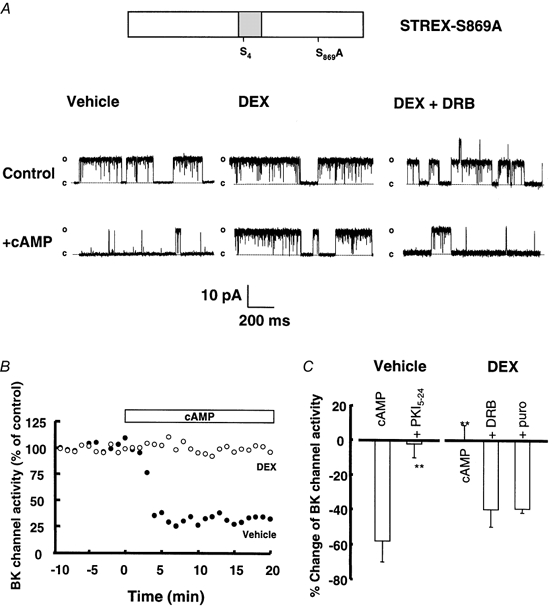
A, representative single channel traces from isolated inside-out patches from vehicle, dexamethasone and dexamethasone + DRB pre-treated HEK 293 cells expressing STREX-S869A channels before and 10 min after application of 1 mm cAMP to the intracellular face of the patch. Patches containing a single STREX BK channel were exposed to 0.2 μm[Ca2+]i and 1 mm ATP in physiological potassium gradients and depolarised to +40 mV as described in Methods. B, representative time course of the effect cAMP on BK channel activity in patches from vehicle-treated (Vehicle, •) and dexamethasone-treated (DEX, ○) cells. Activity is expressed as a percentage of the activity at time −10 min before cAMP application and averaged over 1 min for each data point. The open horizontal bar indicates the period of cAMP application. C, summary of the effect of cAMP application to the intracellular face of patches from STREX-S869A-expressing HEK 293 cells pre-treated for 2 h with vehicle (n = 11) or 1 μm dexamethasone (n = 11) prior to patch excision. Inhibitors of transcription (DRB, 0.1 mm, n = 4) and translation (puromycin, 0.1 mm, n = 4) were applied 15 min before and during dexamethasone treatment of intact HEK 293 cells. The specific PKA inhibitor peptide PKI5–24 (n = 4) was applied to the intracellular face of inside-out patches 5 min before application of cAMP. All data are expressed as the percentage change of pre-treatment BK channel activity measured at +40 mV in the presence of 0.2 μm[Ca2+]i and 1 mm ATP as described in Methods. The bars show means and s.e.m. **P < 0.01 compared with vehicle-treated cAMP group.
Dexamethasone action is dependent upon de novo mRNA and protein synthesis
Early (within 2 h) glucocorticoid action on BK channel regulation and hormone secretion in corticotrophs is dependent upon activation of intracellular GR and induction of new mRNA and protein synthesis (Woods et al. 1992; Shipston, 1995; Shipston et al. 1996). To address whether such a genomic mechanism is responsible for the action of dexamethasone, HEK 293 cells were pre-treated with inhibitors of mRNA transcription (5,6-dichloro-1-β-d-ribofluranosylbenzimidazole (DRB), 50 nm, 15 min before and during DEX treatment) or protein translation (puromycin, 100 μm (Woods et al. 1992; Tian et al. 1999), 20 min before and during DEX treatment). Treatment of HEK 293 cells expressing STREX-S869A channels with either DRB or puromycin prior to and during exposure to dexamethasone blocked the action of dexamethasone, i.e. cAMP-mediated inhibition of BK channel activity returned to near control (non-dexamethasone-treated) levels (Fig. 2A and B). The cAMP-mediated percentage inhibition of BK channel activity in DRB and puromycin pre-treated cells was −39.8 ± 11.4 % (P < 0.01, n = 3) and −39.7 ± 2.0 % (P < 0.01, n = 4), respectively. These data suggest that dexamethasone attenuates PKA-mediated inhibition of BK channel activity via a genomic mechanism. Dexamethasone had no effect on BK channel transcript or protein expression levels as determined by RT-PCR and Western blotting, respectively, over the time course (2 h) of these studies (not shown).
Glucocorticoid action is mediated via protein phosphatase activity intimately associated with the channel complex
Using ATPγS as the phosphate donor in place of ATP in excised inside-out patches from dexamethasone-treated STREX-S869A cells restored cAMP-mediated inhibition of BK channel activity to almost the same extent as that observed in control cells (percentage change of BK channel activity: −33.9 ± 5.2 %, n = 5, Fig. 3B). This effect required PKA activity as it was abolished by PKI5–24 (not shown). ATPγS can be used by protein kinases to phosphorylate target proteins. However, thio-phosphorylated amino acid residues are largely resistant to dephosphorylation by protein phosphatases. Thus these data suggest a closely associated protein phosphatase is responsible for the attenuation of PKA-mediated inhibition of BK channel activity in isolated patches from dexamethasone-treated STREX-S869A cells. Identical results were obtained using the STREX channel under the same experimental conditions (n = 3, data not shown).
Figure 3. Glucocorticoid action is mediated via protein phosphatase activity associated with the patch.

A, representative single channel traces from isolated inside-out patches from a vehicle-treated cell with patch exposed to exogenous PP2A (left) and a dexamethasone pre-treated cell with patch exposed to 10 nm okadaic acid before (control) and after (cAMP) application of cAMP to the intracellular face of the patch. Patches containing STREX-S869A BK channels were exposed to 0.2 μm[Ca2+]i and 1 mm ATP in physiological potassium gradients and depolarised to +40 mV as described in Methods. B, summary of the effect of manipulating serine/threonine protein phosphatase activity in isolated inside-out patches. Vehicle: effect of 1 mm cAMP on BK channel activity in the absence (n = 11) and presence (PP2A, n = 4) of exogenous catalytic subunit of PP2A (1 unit ml−1) applied to the intracellular face of isolated patches from vehicle-treated HEK 293 cells expressing STREX-S869A channels. DEX: effect of 1 mm cAMP on STREX-S869A channel activity in the presence of ATP (n = 11) or ATPγS (0.1 mm, n = 5) as phosphate donor or on co-application with the serine/threonine phosphatase inhibitor, okadaic acid (Ok, 10–100 nm, n = 5) or the specific protein phosphatase 1 (PP1) inhibitor peptide (PPI-2, 20 nm) in the presence of ATP. The PKA inhibitor peptide PKI5–24 (0.45 μm) blocked the effects of cAMP in the presence of okadaic acid (n = 6). All data are expressed as the percentage change of pre-treatment BK channel activity measured at +40 mV in the presence of 0.2 μm[Ca2+]i and 1 mm ATP as described in Methods. The bars show means and s.e.m. **P < 0.01 compared with vehicle-treated cAMP group.
In the presence of 10–100 nm okadaic acid, a blocker of serine/threonine phosphatases, cAMP significantly inhibited BK channel activity in isolated patches from dexamethasone-treated HEK 293 cells expressing STREX-S869A channels (Fig. 3A and B). The percentage change of BK channel activity in the presence of okadaic acid was −37.3 ± 12.9 % (n = 5), similar to that seen in control (non-dexamethasone-treated cells). Importantly, the effect of cAMP was blocked by PKI5–24 (Fig. 3B). The percentage change of BK channel activity in the presence of okadaic acid and PKI5–24 was −6.1 ± 12.6 % (P < 0.01, n = 6). In contrast, application of a specific peptide inhibitor of protein phosphatase 1 (PP1), PPI-2 (20 nm), failed to restore cAMP-mediated inhibition of BK channels in patches from dexamethasone-treated HEK 293 cells expressing STREX-S869A channels (percentage change of BK channel activity after cAMP application in the presence of PPI-2 was 3.8 ± 7.8 %, n = 5, Fig. 3B). The PPI-2 was functionally active in other electrophysiological and biochemical assays of PP1 action (not shown). Thus the action of glucocorticoids on BK channel function does not involve PP1 activity and is likely to be mediated via PP2A-like phosphatase activity associated with the channel.
To test this hypothesis the purified catalytic subunit of PP2A was applied to the intracellular face of patches containing STREX-S869A channels from vehicle-treated HEK 293 cells. In the presence of 1 U ml−1 PP2A, cAMP failed to inhibit BK channel activity (Fig. 3A and B): the percentage change of BK channel activity 10 min after application of cAMP in the presence of PP2A was 15.3 ± 15.0 % (P < 0.01, n = 4).
Overall, these results suggest that the glucocorticoid blockade of PKA-mediated inhibition of BK channel activity is mediated via a PP2A-like phosphatase closely associated with the STREX variant channel complex.
ZERO channels are stimulated by PKA and are not modulated by dexamethasone
Application of cAMP (1 mm) to the intracellular face of isolated inside-out patches from vehicle-treated HEK 293 cells expressing ZERO BK channels (identical to STREX except they lack the STREX insert) results in significant stimulation of ZERO BK channel activity (Fig. 4A and B (see also Nara et al. 1998; Tian et al. 2001); the mean percentage activation was 43.0 ± 10.0 %, n = 8, P < 0.01). This stimulation is entirely dependent upon activation of PKA associated with the patch as the protein kinase A-specific inhibitor peptide, PKI5–24, prevents channel activation (the mean percentage change in activity after cAMP in the presence of PKI5–24 was −9.3 ± 8.2 %, n = 6).
Figure 4. The ZERO variant is not regulated by glucocorticoids.

A, representative single channel traces from isolated inside-out patches from dexamethasone pre-treated HEK 293 cells expressing ZERO variant channels before and 10 min after application of 1 mm cAMP to the intracellular face of the patch. Patches containing ZERO channels were exposed to 0.2 μm[Ca2+]i and 1 mm ATP in physiological potassium gradients and depolarised to +40 mV as described in Methods. B, representative time course of ZERO channel activity from vehicle-treated cells in the presence (+cAMP, ▪) or absence (-cAMP, □) of cAMP. Activity is expressed as a percentage of the activity at time −10 min before cAMP application and averaged over 1 min for each data point. Open horizontal bar indicates the period of cAMP application for the +cAMP patch. C, summary of the effect of cAMP application to the intracellular face of patches from ZERO-expressing HEK 293 cells pre-treated for 2 h with vehicle (n = 8) or 1 μm dexamethasone (n = 8) prior to patch excision. The specific PKA inhibitor peptide PKI5–24 was applied to the intracellular face of inside-out patches 5 min before application of cAMP in vehicle (n = 4) or dexamethasone-treated (n = 6) cells. All data are expressed as the percentage change of pre-treatment BK channel activity measured at +40 mV in the presence of 0.2 μm[Ca2+]i and 1 mm ATP as described in Methods. The bars show means and s.e.m. **P < 0.01 compared with vehicle-treated cAMP group.
In direct contrast to the effect of dexamethasone on PKA-mediated regulation of BK channels containing the STREX insert, dexamethasone pre-treatment did not significantly modify PKA-mediated stimulation of ZERO BK channels. The percentage change of BK channel activity 10 min after cAMP application to the intracellular face of patches from dexamethasone-treated HEK 293 cells expressing ZERO channels was 49.8 ± 13.0 %, n = 8 (Fig. 4B). Importantly, this activation was dependent upon PKA activation as the specific protein kinase A inhibitor peptide (PKI5–24, Fig. 4B) blocked activation by cAMP (mean percentage change in activity was 3.6 ± 4.8 %, n = 4).
S4STREX domain confers glucocorticoid sensitivity to ZERO channels
To examine whether the lack of glucocorticoid regulation of ZERO channels is a result of the absence of the STREX insert per se or a consequence of ZERO channels being activated by PKA (dependent upon a functional S869; Nara et al. 1998; Tian et al. 2001) compared to the inhibition observed with channels containing the STREX insert, we examined the glucocorticoid regulation of the site directed mutant channel S4STREXA. This construct is identical to STREX channels except that the serine residue (S4STREX) within a PKA consensus site of the STREX insert (required for PKA-inhibition; Tian et al. 2001) is mutated to alanine. This channel is activated by PKA as for ZERO and this activation is dependent upon a functional S869 residue within the C-terminal consensus PKA site (Fig. 5; see also Tian et al. 2001). S4STREXA channel activity was stimulated to the same extent as ZERO channels (by 45.9 ± 8.9 %, P < 0.01, n = 8) upon cAMP application to the intracellular face of the channel and this effect was abolished by PKI5–24 (mean percentage change in activity was −0.9 ± 11.9 %, n = 4, P < 0.01).
Figure 5. S4STREXA is activated by PKA and is modulated by DEX.
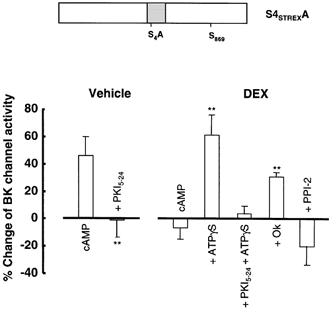
Summary of the effect of cAMP application to the intracellular face of patches from HEK 293 cells expressing S4STREXA channels pre-treated for 2 h with vehicle (n = 8) or 1 μm, dexamethasone (n = 8) prior to patch excision. The specific PKA inhibitor peptide PKI5–24 was applied to the intracellular face of inside-out patches 5 min before application of cAMP in vehicle (n = 4) or dexamethasone-treated (n = 4) cells. ATPγS, in the presence (n = 4) or absence (n = 6) of PKI5–24, and okadaic acid (Ok, 2 nm) (n = 3) were applied as in Fig. 3. All data are expressed as the percentage change of pre-treatment BK channel activity measured at +40 mV in the presence of 0.2 μm[Ca2+]i and 1 mm ATP as described in Methods. The bars show means and s.e.m. **P < 0.01 compared with vehicle-treated cAMP group.
However, in contrast to ZERO channels, dexamethasone pre-treatment abolished PKA-mediated activation of S4STREXA channels. The percentage change in BK channel activity after cAMP application to patches from dexamethasone-treated S4STREXA was −6.8 ± 8.6 % (n = 8, P < 0.01, Fig. 5). Importantly, PKA-mediated activation of S4STREXA channels could be restored in patches from dexamethasone-treated HEK 293 cells when ATPγS was used as the phosphate donor (mean percentage PKA activation of BK channel activity was 60.9 ± 14.9 %, n = 6, Fig. 5) and this cAMP-dependent stimulation was blocked by PKI5–24 (mean percentage PKA activation of BK channel activity was 3.9 ± 5.3 %, n = 4, P < 0.01 Fig. 5). This suggested protein phosphatase activity is required for the action of glucocorticoid. Indeed, inhibition of PP2A-like activity with 2 nm okadaic acid, but not PP1 activity when inhibited by PPI-2 (Fig. 5), prevented the glucocorticoid block of S4STREXA channel regulation by PKA (mean percentage PKA activation of BK channel activity in the presence of okadaic acid was 30.7 ± 3.1 %, n = 3, Fig. 5).
DISCUSSION
We have established in HEK 293 cells a model for the analysis of the modulation of ion channels by glucocorticoids. In this system, novel proteins rapidly induced by dexamethasone differentially modulate the regulation of BK channels by PKA. The presence of the 59 amino acid STREX insert at splice site 2 of the BK channel α-subunit is sufficient for modulation by glucocorticoid-induced proteins through a serine/threonine protein phosphatase, irrespective of the direction of the PKA effect on the channel (Fig. 6).
Glucocorticoids differentially modulate the activity of BK channel splice variants
In order to address whether the pore-forming subunits of BK channels are the targets for glucocorticoids, the regulation of cloned BK channel splice variants was analysed in HEK 293 cells as a heterologous mammalian expression system. HEK 293 cells do not endogenously express BK channel subunits and show the pivotal features of the glucocorticoid response previously reported in anterior pituitary corticotroph cells and CA1 pyramidal neurones (Joels & de Kloet, 1989; Shipston et al. 1996). Thus HEK 293 cells are a suitable model for the analysis of glucocorticoid regulation of BK, and possibly other, ion channels.
BK channels that express the wild-type STREX insert at site 2 (STREX and the site directed mutant STREX-S869A) are potently inhibited by cAMP-dependent protein kinase (PKA) activity intimately associated with the channel complex in HEK 293 cells (Shipston et al. 1999; Tian et al. 2001). This inhibition requires a PKA consensus site in the STREX insert (S4) but is not dependent upon the conserved PKA site at serine residue 869 of the C-terminus (Tian et al. 2001). Glucocorticoid pre-treatment blocked the PKA-mediated inhibition of STREX, or STREX-S869A, BK channels expressed in HEK 293 cells. Glucocorticoid action was manifest within 2 h of exposure, was dependent upon de novo mRNA and protein synthesis and was mediated by okadaic acid-sensitive (PP2A-like) protein phosphatase activity intimately associated with the channel.
However, glucocorticoid regulation was not evident in the ZERO BK channel splice variant that does not contain the STREX insert. ZERO channels are activated by PKA, dependent upon a functional conserved C-terminal PKA consensus phosphorylation site serine residue, S869 (Nara et al. 1998; Tian et al. 2001). The difference in responsiveness of the STREX and ZERO variants to glucocorticoids could be a reflection of the opposite action of PKA on these channels. To address this issue, we used a STREX site directed mutant (S4STREXA) that lacks the STREX PKA consensus site but retains a functional C-terminal site S869. S4STREXA is activated by PKA and this activation was blocked by glucocorticoids, which indicates that the STREX domain confers glucocorticoid modulation. The exact mode of the STREX-dependent modulation remains to be clarified.
Evidence for short- and long-term regulation of cellular excitability through glucocorticoid modulation of BK channels
Previous studies have shown that endogenously expressed BK channels from a variety of tissues may be activated or inhibited by PKA-dependent phosphorylation (Reinhart et al. 1991, 1995; White et al. 1991) and that the direction of this regulation is influenced by circulating hormones such as adrenal corticosteroids (Perez & Toro, 1994; Shipston et al. 1996; Zhou et al. 2000). The diversity and plasticity of modulation is, at least in part, a result of the opposing effects of PKA phosphorylation on distinct BK channel splice variants. Yet a further level of control is the ability of proteins rapidly induced by glucocorticoids to switch the functional outcome of PKA activation targeted to BK channels. Importantly, STREX subunits are widely expressed in glucocorticoid-sensitive endocrine cells and neurones (Tseng-Crank et al. 1994; Ferrer et al. 1996; Xie & McCobb, 1998; Shipston et al. 1999) where cAMP signalling is pivotal for functional activity. Thus glucocorticoid modulation of the phosphorylation of BK channels could provide a short-term switch of cellular excitability in these systems.
In a longer time domain (> 6 h) the regulation of the expression of STREX variant subunits, along with other BK channel splice variants, could also contribute to the specification of the control of cellular excitability. Indeed, the expression of STREX channels is regulated by stress hormones in adrenal medullary chromaffin cells (Xie & McCobb, 1998) and by depolarisation in GH3 pituitary tumour cells (Xie & Black, 2001), and the STREX mRNA profile changes during pregnancy in adult myometrium (Benkusky et al. 2000). Although the impact of STREX α-subunit expression on heteromultimeric BK channel properties has not been explored, previous evidence suggests that the resultant channel properties would reflect the functional characteristics of the STREX subunit (Shen et al. 1994; Lovell & McCobb, 2001).
Acknowledgments
We thank members of the Membrane Biology Group and MRC Membrane and Adapter Protein COOP for advice and useful discussions. This work was supported by The Wellcome Trust and the Biotechnology and Biologicial Sciences Research Council.
References
- Adelman JP, Shen KZ, Kavanaugh MP, Warren RA, Wu YN, Lagrutta A, Bond CT, North RA. Calcium-activated potassium channels expressed from cloned complimentary DNAs. Neuron. 1992;9:209–216. doi: 10.1016/0896-6273(92)90160-f. [DOI] [PubMed] [Google Scholar]
- Attali B, Latter H, Rachamim N, Garty H. A corticosteroid-induced gene expressing the ‘Isk-like’ K+ channel activity in Xenopus oocytes. Proceedings of the National Academy of Sciences of the USA. 1995;92:6092–6096. doi: 10.1073/pnas.92.13.6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamberger CM, Bamberger AM, de Castro M, Chrousos GP. Glucocorticoid receptor-β, a potential endogenous inhibitor of glucocorticoid action in humans. Journal of Clinical Investigation. 1995;95:2435–2441. doi: 10.1172/JCI117943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkusky NA, Fergus DJ, Zucchero TM, England SK. Regulation of the Ca2+-sensitive domains of the maxi-K channel in the mouse myometrium during gestation. Journal of Biological Chemistry. 2000;275:27712–27719. doi: 10.1074/jbc.M000974200. [DOI] [PubMed] [Google Scholar]
- Butler A, Tsunoda S, McCobb DP, Wei A, Salkoff L. mSlo, a complex mouse gene encoding ‘maxi’ calcium-activated potassium channels. Science. 1993;261:221–224. doi: 10.1126/science.7687074. [DOI] [PubMed] [Google Scholar]
- Clark AG, Hall SK, Shipston MJ. ATP inhibition of a mouse brain large-conductance K+ (mslo) channel variant by a mechanism independent of protein phosphorylation. Journal of Physiology. 1999;516:45–53. doi: 10.1111/j.1469-7793.1999.045aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer J, Wasson J, Salkoff L, Permutt MA. Cloning of human pancreatic islet large conductance Ca2+-activated K+ channel (hslo) cDNAs: evidence for high levels of expression in pancreatic islets and identification of a flanking genetic marker. Diabetologia. 1996;39:891–898. doi: 10.1007/BF00403907. [DOI] [PubMed] [Google Scholar]
- Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, Thompson EB, Rosenfeld MG, Evans RM. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature. 1985;318:635–641. doi: 10.1038/318635a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joels M, de Kloet ER. Effects of glucocorticoids and norepinephrine on the excitability in the hippocampus. Science. 1989;245:1502–1505. doi: 10.1126/science.2781292. [DOI] [PubMed] [Google Scholar]
- Leckie C, Chapman KE, Edwards CR, Seckl JR. LLC-PK1 cells model 11 β-hydroxysteroid dehydrogenase type 2 regulation of glucocorticoid access to renal mineralocorticoid receptors. Endocrinology. 1995;136:5561–5569. doi: 10.1210/endo.136.12.7588309. [DOI] [PubMed] [Google Scholar]
- Levitan ES, Hemmick LM, Birnberg NC, Kaczmarek LK. Dexamethasone increases potassium channel messenger RNA and activity in clonal pituitary cells. Molecular Endocrinology. 1991;5:1903–1908. doi: 10.1210/mend-5-12-1903. [DOI] [PubMed] [Google Scholar]
- Lovell PV, McCobb DP. Pituitary control of BK potassium channel function and intrinsic firing properties of adrenal chromaffin cells. Journal of Neuroscience. 2001;21:3429–3442. doi: 10.1523/JNEUROSCI.21-10-03429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nara M, Dhulipala PD, Wang YX, Kotlikoff MI. Reconstitution of β-adrenergic modulation of large conductance, calcium-activated potassium (maxi-K) channels in Xenopus oocytes. Identification of the cAMP-dependent protein kinase phosphorylation site. Journal of Biological Chemistry. 1998;273:14920–14924. doi: 10.1074/jbc.273.24.14920. [DOI] [PubMed] [Google Scholar]
- Pallanck L, Ganetzky B. Cloning and characterization of human and mouse homologs of the Drosophila calcium-activated potassium channel gene, slowpoke. Human Molecular Genetics. 1994;3:1239–1243. doi: 10.1093/hmg/3.8.1239. [DOI] [PubMed] [Google Scholar]
- Perez G, Toro L. Differential modulation of large-conductance KCa channels by PKA in pregnant and nonpregnant myometrium. American Journal of Physiology. 1994;266:C1459–1463. doi: 10.1152/ajpcell.1994.266.5.C1459. [DOI] [PubMed] [Google Scholar]
- Reinhart PH, Chung SK, Martin BL, Brautigan DL, Levitan IB. Modulation of calcium-activated potassium channels from rat brain by protein kinase A and phosphatase 2A. Journal of Neuroscience. 1991;11:1627–1635. doi: 10.1523/JNEUROSCI.11-06-01627.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart PH, Levitan IB. Kinase and phosphatase activities intimately associated with a reconstituted calcium-dependent potassium channel. Journal of Neuroscience. 1995;15:4572–4579. doi: 10.1523/JNEUROSCI.15-06-04572.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Nelson C, Salkoff L, Lingle CJ. A cysteine-rich domain defined by a novel exon in a Slo variant in rat adrenal chromaffin cells and PC12 cells. Journal of Biological Chemistry. 1997;272:11710–11717. doi: 10.1074/jbc.272.18.11710. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocrine Reviews. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- Shen KZ, Lagrutta A, Davies NW, Standen NB, Adelman JP, North RA. Tetraethylammonium block of Slowpoke calcium-activated potassium channels expressed in Xenopus oocytes: evidence for tetrameric channel formation. Pflügers Archiv. 1994;426:440–445. doi: 10.1007/BF00388308. [DOI] [PubMed] [Google Scholar]
- Shipston MJ. Mechanism(s) of early glucocorticoid inhibition of adrenocorticotropin secretion from anterior pituitary corticotropes. Trends in Endocrinology and Metabolism. 1995;6:261–266. doi: 10.1016/1043-2760(95)00149-2. [DOI] [PubMed] [Google Scholar]
- Shipston MJ, Duncan RR, Clark AG, Antoni FA, Tian L. Molecular components of large conductance calcium-activated potassium (BK) channels in mouse pituitary corticotropes. Molecular Endocrinology. 1999;13:1728–1737. doi: 10.1210/mend.13.10.0355. [DOI] [PubMed] [Google Scholar]
- Shipston MJ, Kelly JS, Antoni FA. Glucocorticoids prevent protein kinase A inhibition of calcium-activated potassium channels. Journal of Biological Chemistry. 1996;271:9197–9200. doi: 10.1074/jbc.271.16.9197. [DOI] [PubMed] [Google Scholar]
- Tian L, Duncan RR, Hammond SL, Coghill LC, Wen H, Rusinova R, Clark AG, Levitan IB, Shipston MJ. Alternative splicing switches potassium channel sensitivity to protein phosphorylation. Journal of Biological Chemistry. 2001;276:1717–1720. doi: 10.1074/jbc.C000741200. [DOI] [PubMed] [Google Scholar]
- Tian L, Knaus H-G, Shipston MJ. Glucocorticoid regulation of calcium-activated potassium channels mediated by serine/threonine protein phosphatase. Journal of Biological Chemistry. 1998;273:13531–13536. doi: 10.1074/jbc.273.22.13531. [DOI] [PubMed] [Google Scholar]
- Tian L, Philp JAC, Shipston MJ. Glucocorticoid block of protein kinase C signalling in mouse pituitary corticotroph AtT20 D16:16 cells. Journal of Physiology. 1999;516:757–768. doi: 10.1111/j.1469-7793.1999.0757u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng-Crank J, Foster CD, Krause JD, Mertz R, Godinot N, Dichiara TJ, Reinhart PH. Cloning, expression, and distribution of functionally distinct Ca2+-activated K+ channel isoforms from human brain. Neuron. 1994;13:1315–1330. doi: 10.1016/0896-6273(94)90418-9. [DOI] [PubMed] [Google Scholar]
- White RE, Schonbrunn A, Armstrong DL. Somatostatin stimulates Ca2+-activated K+ channels through protein dephosphorylation. Nature. 1991;351:570–573. doi: 10.1038/351570a0. [DOI] [PubMed] [Google Scholar]
- Woods MD, Shipston MJ, Mullens EL, Antoni FA. Pituitary corticotroph tumour (AtT-20) cells as a model system for the study of early inhibition by glucocorticoids. Endocrinology. 1992;131:2873–2880. doi: 10.1210/endo.131.6.1332850. [DOI] [PubMed] [Google Scholar]
- Xie J, Black DL. A CaMK IV responsive RNA element mediates depolarization-induced alternative splicing of ion channels. Nature. 2001;410:936–939. doi: 10.1038/35073593. [DOI] [PubMed] [Google Scholar]
- Xie J, McCobb DP. Control of alternative splicing of potassium channels by stress hormones. Science. 1998;280:443–446. doi: 10.1126/science.280.5362.443. [DOI] [PubMed] [Google Scholar]
- Zhou X-B, Wang G-X, Huneke B, Wieland T, Korth M. Pregnancy switches adrenergic signal transduction in rat and human uterine myocytes as probed by BKCa channel activity. Journal of Physiology. 2000;524:339–352. doi: 10.1111/j.1469-7793.2000.t01-1-00339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]