

Abstract
Action potential-evoked Ca2+ transients in postganglionic sympathetic axon bundles in mouse vas deferens have been characterized using confocal microscopy and Ca2+ imaging.
Axonal Ca2+ transients were tetrodotoxin sensitive. The amplitude depended on both the frequency of stimulation and the number of stimuli in a train.
Removal of extracellular Ca2+ abolished the Ca2+ transient. Cd2+ (100 μm) inhibited the Ca2+ transient by 78 ± 10 %. The N-type Ca2+ channel blocker ω-conotoxin GVIA (0.1 μm) reduced the amplitude by −35 ± 4 %, whereas nifedipine (10 μm; L-type) and ω-conotoxin MVIIC (0.1 μm; P/Q type) were ineffective.
Caffeine (10 mm), ryanodine (10 μm), cyclopiazonic acid (30 μm) or CCCP (10 μm) had no detectable effects.
Blockade of large and small conductance Ca2+-dependent K+ channels with iberiotoxin (0.1 μm) and apamin (1 μm), respectively, or Ca2+-dependent Cl− channels by niflumic acid (100 μm) did not alter Ca2+ transients.
In contrast, the non-specific K+ channel blockers tetraethylammonium (10 mm) and 4-aminopyridine (10 mm) markedly increased the amplitude of the Ca2+ transient. Blockade of delayed rectifiers and A-like K+ channels, by tityustoxin-K (α) (0.1 μm) and pandinustoxin-K (α) (10 nm), respectively, also increased the Ca2+ transient amplitude.
Thus, Ca2+ transients are evoked by Na+-dependent action potentials in axons. These transients originate mainly from Ca2+ entry through voltage-dependent Ca2+ channels (80 % Cd2+ sensitive of which 40 % was attributable to N-type). Twenty per cent of the Ca2+ transient was not due to Ca2+ entry through voltage-gated Ca2+ channels. Intracellular stores and mitochondria were not involved in the generation of the transient. Ca2+ transients are modulated by A-like K+ channels and delayed rectifiers (possibly KV1.2) but not by Ca2+-activated ion channels.
A sympathetic neurone consists of a well-defined cell body which gives rise to an axon which, after a variable length (1 cm to 1 m), repeatedly branches into a complex network of varicosities separated by thin intervaricose regions. Recently, a technique has been developed to measure action potential-evoked changes in Ca2+ levels in sympathetic nerve terminals (Brain & Bennett, 1997). Using confocal microscopy and Ca2+ imaging, the authors characterized action potential-evoked Ca2+ transients in individual varicosities and in the intervaricose region of postganglionic sympathetic nerve terminals. Ca2+ transients have also been detected in cell bodies of sympathetic neurones (Hua et al. 1993). What is less well known is that Ca2+ transients are elicited by action potentials in the main axonal trunks of neurones. Axonal studies of Ca2+ dynamics have mainly been carried out in the central nervous system i.e. rat cerebellar interneurones (Callewaert et al. 1996; Forti et al. 2000), cortical neurone cultures (Mackenzie et al. 1996), neonatal (Kriegler & Chiu, 1993; Lev-Ram & Grinvald, 1987; Sun & Chiu, 1999) and adult rat optic nerve (Brown et al. 2001). Axonal Ca2+ transients have also been detected in adult rat preganglionic vagus nerve trunks in the peripheral nervous system (Wächtler et al. 1998). However, to date, no action potential-evoked Ca2+ transients have been reported in the main axon trunks of unmyelinated autonomic neurones in regions far removed from the secretory terminals.
The paucity of data on Ca2+ dynamics in axons is due mainly to their small diameter (0.1–1 μm; Cottee et al. 1996) and their inaccessibility to conventional electrophysiological techniques such as the patch clamp. What little is known about axonal ion channel activity in postganglionic sympathetic neurones innervating rodent vas deferens is based on experiments where the action potential was monitored using extracellular recording techniques (Cunnane & Stjärne, 1984; Brock & Cunnane,1987,1988; Stjärne & Stjärne, 1989). It is noteworthy that removal of extracellular Ca2+ significantly reduced the amplitude of the nerve action potential in axons of sympathetic neurones innervating rodent vas deferens (Cunnane & Stjärne, 1984). In addition, tetraethylammonium chloride (TEA), a non-specific blocker of Ca2+-activated K+ channels, alters the configuration of the nerve action potential (Cunnane & Stjärne, 1984). One possible role for Ca2+ influx is to regulate the frequency of action potential firing in these nerves perhaps by activating Ca2+-dependent K+ channels.
The aims of the present study were: (1) to characterize physiologically, action potential-evoked Ca2+ transients in sympathetic axons using confocal microscopy and Ca2+ imaging techniques; (2) to establish the origin of the Ca2+ transient; (3) to determine whether axonal Ca2+ transients are modified by blockers of Ca2+-activated K+ and Cl− channels; and (4) to determine whether axonal Ca2+ transients are modified by other voltage-dependent K+ channels.
METHODS
A schematic illustration of the cell body, main axon and varicose secretory terminals of a postganglionic sympathetic axon is shown in Fig. 1.
Figure 1. Diagram of a postganglionic sympathetic neurone.
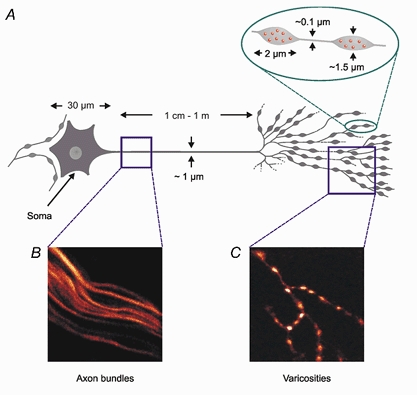
A, schematic diagram of a postganglionic sympathetic neurone, showing the difference in size between cell body, axon and varicosity. B, image of a set of axon bundles loaded with the Ca2+ indicator Oregon Green 488 BAPTA-1 dextran. Note that only a single axon is represented in the corresponding box in A. C, image of part of a sympathetic terminal loaded with the same Ca2+ indicator.
Ca2+ indicator loading
Aganglionic vasa deferentia were removed from 8- to 12-week-old Balb/C mice, which had been humanely killed by cervical fracture. The bathing Krebs solution contained (mm): 118.8 NaCl, 25 NaHCO3, 1.13 NaH2PO4, 4.7 KCl, 1.8 CaCl2, 1.2 MgCl2, 11.1 glucose and was constantly gassed with 95 % O2-5 % CO2 to pH 7.4. The cut prostatic end of each vas deferens was gently sucked into a glass pipette containing a saturated solution of Oregon Green 488 BAPTA-1, dextran linked with a molecular mass of 10 kDa (Molecular Probes Inc., OR, USA), in 2.5 % Triton X-100. Vasa deferentia were loaded for 3 h in the dark at room temperature to allow time for the indicator to travel along axons, and then washed for a further 2 h to remove any extracellular dye.
Stimuli and recording
The prostatic end of a vas deferens was mounted in a 2 ml organ bath and placed on the stage of a Leica TCS NT laser-scanning confocal microscope. The preparation was secured with a pair of parallel platinum electrodes and stimulated using an optically isolated stimulator (Digitimer DS2). The pulse width was set between 0.06 and 0.4 ms and the applied voltage was adjusted to give a reliable change in fluorescence following a single stimulus. The stimulus voltage was then increased by about 20 % to ensure that the stimulus was suprathreshold for that axon bundle thus preventing variation in the Ca2+ transient due to the recruitment of unstimulated axons in the bundle. The stimuli were electronically synchronized with the confocal microscope scans. The 488 nm wavelength of an Argon ion laser was used for exciting fluorescence. A 515 nm long pass emission filter was used. When detecting Ca2+ transients, sets of images were captured for 56 s every 3 min. This protocol prevented excessive photobleaching and phototoxicity. All experiments were carried out at 33 °C in the presence of nifedipine (10 μm; an L-type Ca2+ channel blocker), prazosin (1 μm; a competitive α1-adrenoceptor antagonist) and α,β-methylene ATP (1 μm; a P2X-receptor desensitizing agent) to reduce or abolish contractions elicited by high frequency stimulation.
Stock solutions of tetrodotoxin (TTX), α,β-methylene ATP, ω-conotoxin GVIA, ω-conotoxin MVIIC, tityustoxin-K (α), pandinustoxin-K (α), bepridil and iberiotoxin were dissolved in distilled water. Prazosin, ryanodine, carbonyl cyanide m-chlorophenyl hydrazone (CCCP) and cyclopiazonic acid (CPA) were dissolved in dimethyl sulfoxide, and apamin in 5 % acetic acid. Solutions were prepared and aliquoted before storing at −20 °C; this ensures that the drugs only passed through one freeze-thaw cycle. Stock nifedipine (in ethanol), niflumic acid (in methanol) and cadmium chloride (in distilled water) solutions were diluted as required on the day of the experiment. Caffeine, TEA and 4-aminopyridine (4-AP) were all made up fresh each day and diluted in Krebs solution to the required final bath concentration. The pH of 4-AP was titrated to pH 7.4 with 1 m HCl. CPA was obtained from Calbiochem (Nottingham, UK) and all other compounds were obtained from Sigma (Dorset, UK). Vehicle controls had a final concentration < 0.01 % and had no detectable effect on control axonal Ca2+ transients.
Analysis
Images were analysed using either Scion or NIH Image (available from URL http://www.rsb.info.nih.gov/nih-image) and custom-written macros. As Oregon Green 488 BAPTA-1 is a non-ratiometric dye, changes in intracellular Ca2+ concentration are reported as the change in fluorescence divided by the basal fluorescence (ΔF/F). An example of a typical region analysed is highlighted in yellow in Fig. 2A. Data are presented as means ± standard error of the mean (s.e.m.); n is the number of axon bundles analysed, all of which were obtained from > 3 vas deferens from > 3 animals. Statistical significance was determined using Student's paired t tests. In the figures, *P < 0.05, **P < 0.01 and ***P < 0.005.
Figure 2. Effect of TTX on action potential-evoked axonal Ca2+ transients.
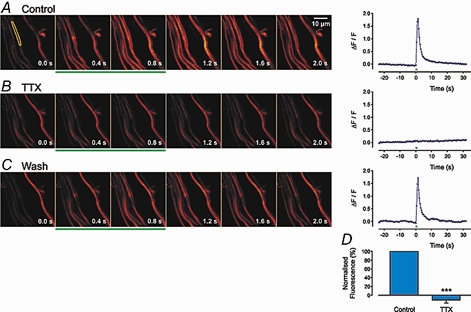
Six example scans are shown; a prestimulus control frame (0.0 s) and frames 0.4, 0.8, 1.2, 1.6 and 2.0 s after the beginning of stimulation (10 stimuli applied at 10 Hz, 0.06 ms, 15 V). The green bars indicate the frames during which the stimuli were applied. The width of each frame is 40 μm. To the right of each panel is a graph showing the change in fluorescence over 50 s, including the period of electrical stimulation, in a single bundle (indicated by the yellow area in the first frame). B as A, 20 min after the bath application of TTX (0.3 μm). C shows a recording 30 min after wash out of TTX. Small differences between bundles may be due to small changes in the focal plane. D, histogram shows the mean data ( ± s.e.m.) from 15 axon bundles.
RESULTS
Characterization of Ca2+ transients
Effects of TTX
Small nerve trunks, which typically contain 2–10 unmyelinated axons (Cottee et al. 1996), were loaded with Oregon Green 488 BAPTA-1 dextran. The Ca2+ transients evoked by a train of 10 stimuli at 10 Hz, in a set of axon bundles, are shown in Fig. 2A. The change in fluorescence was abolished by TTX (0.3 μm; Fig. 2B) and returned after wash out of TTX (Fig. 2C). The mean inhibition of the amplitude of Ca2+ transients evoked by 10 stimuli at 10 Hz in the presence of TTX is shown in Fig. 2D (-106 ± 4 %, n = 15 axon bundles; P < 0.001). Thus the Ca2+ transients detected in axonal bundles are elicited by Na+-dependent, nerve action potentials.
Effects of increasing the number of stimuli in a train
Ca2+ transients could be detected in sympathetic axon bundles in response to even a single action potential (Fig. 3A). The amplitude of the Ca2+ transients could vary markedly from axon bundle to axon bundle, presumably due to either variable dye loading or physiological differences. Increasing the number of stimuli from 1 to 5, 10, 20, 30 or 50 at 10 Hz induced a progressive increase in the magnitude of the evoked Ca2+ transient (Fig. 3A; 1 stimulus, 0.1 ± 0.03; 3 stimuli, 0.2 ± 0.1; 5 stimuli, 0.3 ± 0.1; 10 stimuli, 0.6 ± 0.2; 20 stimuli, 0.7 ± 0.1; 30 stimuli, 0.8 ± 0.2; and 50 stimuli, 0.7 ± 0.2; n = 16 axon bundles). The evoked Ca2+ transient was less than that observed in varicosities (1 stimulus, 0.8 ± 0.2; 3 stimuli, 1.4 ± 0.2; 5 stimuli, 1.5 ± 0.2; 10 stimuli, 1.9 ± 0.3; 20 stimuli, 1.9 ± 0.2; 30 stimuli, 1.9 ± 0.1; and 50 stimuli, 2.1 ± 0.3, n = 19 varicosities). The mean fluorescence change observed with increasing number of action potentials at 10 Hz (n = 16 axon bundles) is shown in Fig. 4A. Experiments were limited to 50 stimuli and/or 50 Hz because of movement artifacts. It was evident that the maximum change in fluorescence was evoked by about 30 stimuli at 10 Hz. When the relative change in fluorescence was plotted against the number of stimuli in a train, a non-linear relationship resulted (Fig. 4A).
Figure 3. Effects of stimulus frequency and train length.

A, Ca2+ transients evoked at a constant frequency of 10 Hz but with a variable number of stimuli (1, 5, 10, 20, 30 and 50). A change in fluorescence was observed even in response to a single stimulus. Note that significant broadening of the Ca2+ transient occurred after 20–50 stimuli. Inspection of the Ca2+ transient evoked by 50 stimuli shows that the signal rose rapidly for about 1200 ms before reaching a plateau that was maintained throughout the period of stimulation. The bars indicate the periods of nerve stimulation. B, Ca2+ transients evoked by trains of 10 stimuli applied at frequencies of 1, 2, 5, 10, 20 and 50 Hz.
Figure 4. Plots of relative changes in fluorescence against stimulus frequency, train length and the effects of ionomycin and 4-AP.
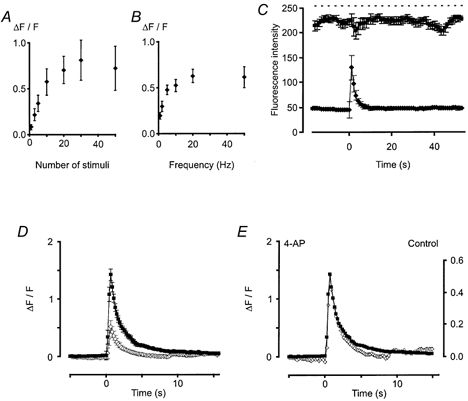
A, plot of relative changes in fluorescence against number of stimuli at a constant frequency of 10 Hz (n = 16). Error bars represent the s.e.m. A non-linear relationship was revealed with a maximum change after 30 stimuli before a plateau was reached. B, plot of relative changes in fluorescence against stimulation frequency, n = 22 (except 2 Hz, n = 15). Again, a non-linear relationship was revealed with a maximum change observed at about 20 Hz before the plateau was reached. C, plot of relative changes in fluorescence after a control period of 10 stimuli at 50 Hz and 90 min after the application of ionomycin (10 μm; top trace). The dashed line represents the maximum output of the photomultiplier tube (i.e. 256, the maximum fluorescence output of an 8-bit detection system). Clearly, the magnitude of the evoked Ca2+ transient is not limited by dye saturation. D, effects of 4-AP on Ca2+ transients evoked by trains of 10 stimuli at 10 Hz (n = 15). 4-AP (10 mm; ▪) produced a threefold increase in the amplitude of the Ca2+ transient compared to control (⋄). Hence, K+ channels regulate the Ca2+ axonal transient and the non-linearities observed in Fig. 3A and B are not a consequence of dye saturation. E, lack of effect of 4-AP on the kinetics of recovery of the Ca2+ transient. Same data as in D, but the error bars have been removed and the control response scaled to match the 4-AP trace and superimposed.
Effects of increasing the stimulation frequency
The effects of varying the frequency of nerve stimulation but keeping the number of stimuli in a train constant were also investigated. Increasing the frequency of stimulation from 1 to 2, 5, 10, 20 and 50 Hz (10 stimuli), increased the fluorescence signal in all axon bundles studied (Fig. 3B; 10 stimuli at 1 Hz, 0.19 ± 0.04; 2 Hz, 0.30 ± 0.06; 5 Hz, 0.48 ± 0.05; 10 Hz, 0.52 ± 0.07; 20 Hz, 0.63 ± 0.07; 50 Hz, 0.61 ± 0.11, n = 15 axon bundles). The evoked Ca2+ transient was smaller than that observed in varicose regions (10 stimuli at 1 Hz, 1.5 ± 0.7; 2 Hz, 1.2 ± 0.1; 5 Hz, 1.8 ± 0.2; 10 Hz, 2.0 ± 0.2; 20 Hz, 2.5 ± 0.2; 50 Hz, 3 ± 0.2, n = 19 varicosities). A plot of the relative change in fluorescence against frequency (n > 15 axon bundles) is shown in Fig. 4B. A plot of ΔF/F versus the frequency of stimulation revealed a non-linear relationship, the Ca2+ transient reaching a maximum value after a train of 10 stimuli at 20 Hz.
Is the axonal Ca2+ transient amplitude limited by dye saturation?
To investigate whether the peak response was limited by dye saturation, the Ca2+ ionophore ionomycin (10 μm) was used. Ionomycin produced a peak fluorescence that was greater than that produced by nerve stimulation (Fig. 4C; control 10 stimuli at 50 Hz, 129 ± 16; ionomycin, 225 ± 6, n = 3 axon bundles). Furthermore, the application of the K+ channel blocker 4-AP (10 mm) also increased the action potential-evoked Ca2+ transient about threefold (Fig. 4D; 10 stimuli at 10 Hz, n = 15 axon bundles; P < 0.001) with no change in the time course of the signal (Fig. 4E). Taken together, these data strongly suggest that the non-linearity of the Ca2+ transient, observed during repetitive stimulation (Fig. 4), is unlikely to be due to dye saturation, and indicate that K+ channel activation might limit the magnitude of axonal Ca2+ transients.
Origin of Ca2+ transient
Evidence for Ca2+ influx
The potential sources of Ca2+ involved in the genesis of the axonal Ca2+ transients were investigated. When tissues were perfused with Krebs solution containing no added Ca2+ and with the Ca2+ chelator EGTA (1 mm) present, the evoked axonal Ca2+ transient was abolished. Thus, the presence of extracellular Ca2+ is obligatory for the generation of the Ca2+ transient. A histogram summarizing the effects of Ca2+-free solution on axonal Ca2+ transients evoked by trains of 10 stimuli at 10 Hz (-98 ± 4 %, n = 9 axon bundles; P < 0.001) is shown in Fig. 5A. It is noteworthy that in addition to abolishing the evoked axonal Ca2+ transient, removal of extracellular Ca2+ also reduced the basal fluorescence (-28 ± 5 %, n = 10 axon bundles; P < 0.005).
Figure 5. Effects of Ca2+-free Krebs solution and of Cd2+ on action potential-evoked axonal Ca2+ transients.

A, effects of Ca2+-free Krebs solution (EGTA 1 mm) on the Ca2+ transient evoked by 10 stimuli at 10 Hz in a single axon bundle. The histogram shows the mean data ( ± s.e.m.) from 9 axon bundles in different preparations. B, effects of the non-specific Ca2+ channel blocker Cd2+ (100 μm) on the Ca2+ transient evoked by 10 stimuli at 10 Hz in a single axon bundle. The histogram shows the mean inhibition of the Ca2+ transient produced by Cd2+ in 8 axon bundles ( ± s.e.m.).
Are voltage-gated calcium channels the route of Ca2+ entry into the axon?
The most likely route of Ca2+ entry into axons is through voltage-gated Ca2+ channels (VGCCs). This potential route of Ca2+ entry was investigated using the non-specific VGCC blocker Cd2+ (100 μm). A sample trace showing the inhibitory effect of Cd2+ on the Ca2+ transient evoked in a single axonal bundle (10 stimuli at 10 Hz) is shown in Fig. 5B. The inhibitory effect of Cd2+ was not readily reversible upon wash. A histogram summarizing the mean effect of Cd2+ on action potential-evoked Ca2+ transients (10 stimuli at 10 Hz, −78 ± 10 %, n = 8 axon bundles; P < 0.01) is shown in Fig. 5B. Interestingly, about 20 % of the fluorescence signal remained 1 h after the application of Cd2+. It should be pointed out that the Na+–Ca2+ exchanger may be blocked by Cd2+ (Trosper & Philipson, 1983; Hobai et al. 1997) so these results by themselves cannot distinguish between Ca2+ influx through axonal VGCCs and reverse mode Na+–Ca2+ exchange. However, the Na+–Ca2+ exchange blocker bepridil (100 μm) had no detectable effect on the axonal Ca2+ transient indicating that Ca2+ entry into the axons was mainly through VGCCs (10 stimuli at 10 Hz, 2 ± 14 %, n = 5 axon bundles; P > 0.05).
Pharmacological characterization of VGCCs
The L-type Ca2+ entry blocker nifedipine (10 μm) had no detectable effects on axonal Ca2+ transients evoked by a single stimulus (20 ± 18 %, n = 5 axon bundles; P > 0.05). The relatively high concentration of nifedipine employed here may also inhibit low-threshold T-type VGCCs (Richard et al. 1991; Takahashi & Akaike, 1991). All subsequent experiments were carried out with nifedipine present in the Krebs solution, to minimize smooth muscle contraction during high frequency stimulation. The most common VGCCs expressed in sympathetic nerve terminals, and shown to be involved in transmitter release, are the N-, P- and Q-types (Smith & Cunnane, 1997; Brain & Bennett 1997). The N-type Ca2+ channel blocker ω-conotoxin GVIA (0.1 μm) inhibited, but did not abolish, the axonal Ca2+ transient (Fig. 6A). Alternative routes of Ca2+ entry were investigated by using relatively high concentrations of ω-conotoxin MVIIC (0.1 μm), which blocks P/Q-type VGCCs. ω-Conotoxin MVIIC had no detectable effect on the axonal Ca2+ transient even when applied for 1 h or more (Fig. 6B; 10 stimuli at 10 Hz, 10 ± 12 %, n = 14 axon bundles; P > 0.05). To establish whether different types/populations of VGCCs were recruited when the frequency of stimulation was varied, the effects of N-, P- and Q-type VGCC blockers were investigated over a wide range of stimulation frequencies (1, 10 and 50 Hz). ω-Conotoxin GVIA produced the same degree of inhibition at each stimulation frequency employed (Fig. 6C; 1 Hz, −43 ± 14 %; 10 Hz, −35 ± 4 %; 50 Hz, −42 ± 6 %; n = 10 axon bundles; P < 0.005). Similarly, ω-conotoxin MVIIC was ineffective at all stimulation frequencies employed (Fig. 6C; 1 Hz, −2 ± 21 %; 10 Hz, 26 ± 14 %; 50 Hz, 17 ± 13 %; n = 9 axon bundles; P > 0.05).
Figure 6. Effects of ω-conotoxin GVIA and ω-conotoxin MVIIC on axonal Ca2+ transients.
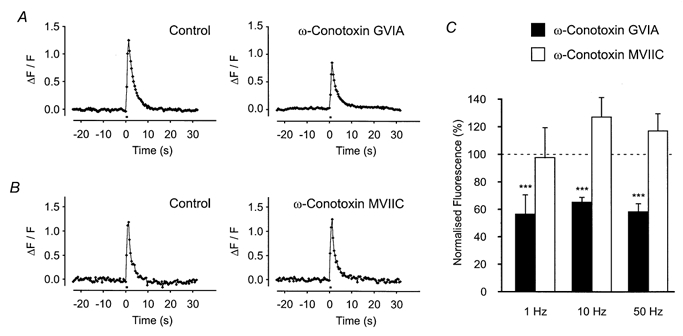
A, effects of the selective N-type VGCC blocker ω-conotoxin GVIA (0.1 μm) on the Ca2+ transient evoked by 10 stimuli at 10 Hz in a single axon bundle. The toxin inhibited the transient by ∼40 %, even after 1 h of exposure. B, effects of the P/Q-type VGCC blocker ω-conotoxin MVIIC (0.1 μm) on the Ca2+ transient evoked by 10 stimuli at 10 Hz in a single axon bundle. The toxin had no significant effect even after 1 h of exposure. C, histogram showing the effects of VGCC blockers on Ca2+ transients evoked at different stimulation frequencies. The inhibitory effects of ω-conotoxin GVIA (0.1 μm) were independent of stimulation frequency (1 Hz, −43 ± 14 %; 10 Hz, −35 ± 4 %; 50 Hz, −42 ± 6 %; n = 10 axon bundles). ω-Conotoxin MVIIC was ineffective at all stimulation frequencies employed (1 Hz, −2 ± 21 %; 10 Hz, 26 ± 14 %; 50 Hz, 17 ± 13 %; n = 9 axon bundles).
Possible role of intracellular Ca2+ stores
Although removal of extracellular Ca2+ abolished the axonal Ca2+ transient, these experiments did not rule out the potential involvement of Ca2+-induced Ca2+ release (CICR). Ryanodine acts in a use- and time-dependent manner at the ryanodine receptor (Nagasaki & Fleischer, 1988) to either lock the channel in a sub-conductance state or to block it (Rousseau et al. 1987; McPherson et al. 1991). Nerves were therefore stimulated continually for 60–90 min in the presence of ryanodine to ensure that any potential ryanodine receptors present were activated by Ca2+ entry and therefore blockable by ryanodine (Smith & Cunnane, 1996). Under these experimental conditions, ryanodine (10 μm) did not alter the axonal Ca2+ transient even when long trains of stimuli were applied. The mean effects of ryanodine on the amplitude of the axonal Ca2+ transient elicited by several stimulation protocols were: 1 stimulus, −8 ± 25 %, n = 5 axon bundles; 10 stimuli at 10 Hz, 9 ± 47 %, n = 5 axon bundles; 50 stimuli at 10 Hz, −1 ± 37 %, n = 6 axon bundles, P > 0.05. Caffeine was also employed to deplete putative axonal Ca2+ stores. Caffeine (10 mm) had no significant effects on evoked axonal Ca2+ transients (1 stimulus, −16 ± 13 %; 10 stimuli at 10 Hz, 3 ± 16 %; 50 stimuli at 10 Hz, 2 ± 12 %; n = 9 axon bundles; P > 0.05). Ryanodine and caffeine had no detectable effect on basal fluorescence (8 ± 16 %, n = 9 axon bundles and 4 ± 15 %, n = 6 axon bundles, respectively; P > 0.05). Similarly, high concentrations of the Ca2+-ATPase inhibitor CPA (30 μm) had no detectable effect on the axonal Ca2+ transient (-7 ± 9 %, n = 8 axon bundles; P > 0.05) or on the basal fluorescence (10 ± 10.5 %, n = 9 axon bundles; P > 0.05).
To investigate whether mitochondria play a role in modulating the axonal Ca2+ transient, the effects of CCCP were investigated. CCCP (10 μm) had no effect on the axonal Ca2+ transient even after 30 min exposure (10 stimuli at 1 Hz, −12 ± 12 %; 10 Hz, −10 ± 13 %; 50 Hz, −10 ± 17 %; n = 11 axon bundles; P > 0.05), nor on the basal fluorescence (12 ± 29 %, n = 11 axon bundles; P > 0.05).
Do Ca2+-activated K+ and/or Cl− channel blockers modulate axonal Ca2+ transients?
To determine whether Ca2+-activated K+ channel blockers could affect the Ca2+ transient, the non-selective K+ channel blocker TEA was used. TEA (10 mm) produced a large potentiation of the action potential-evoked axonal Ca2+ transient as shown in Fig. 7. As it has been suggested that activation of K+ currents in axons might depend on the stimulation frequency employed (see Hille, 1992), the effects of TEA (10 mm) were tested over a wide range of stimulation frequencies (10 stimuli at 1, 10 and 50 Hz). TEA increased the amplitude of the evoked Ca2+ transient at all frequencies of stimulation studied (Fig. 7; 10 stimuli at 1 Hz, 116 ± 32 %, n = 16 axon bundles; 10 Hz, 89 ± 38 %, n = 18 axon bundles; 50 Hz, 39 ± 17 %, n = 10 axon bundles; P < 0.005). TEA had no effect on the basal fluorescence (-1 ± 7 %, n = 13 axon bundles; P > 0.05).
Figure 7. Effect of TEA on axonal Ca2+ transients.

Effects of the K+ channel blocker TEA (10 mm) on the Ca2+ transient evoked by 10 stimuli at 10 Hz in a single axon bundle. Histogram showing the mean ( ± s.e.m.) increase in amplitude of the Ca2+ transient produced by TEA following trains of 10 stimuli applied at 1, 10 and 50 Hz from n = 16, 18 and 10 bundles, respectively.
As TEA is believed to block large conductance Ca2+-activated K+ channels, the specific blocker iberiotoxin was used to characterize further the nature of the K+ channel involved. Iberiotoxin (0.1 μm) had no effect on the action potential-evoked Ca2+ transients, at any stimulation frequency employed (Fig. 8; 10 stimuli at 1 Hz, 7 ± 9 %; 10 Hz, −2 ± 6 %; 50 Hz, 9 ± 10 %; n = 11 axon bundles; P > 0.05). Iberiotoxin had no detectable effect on the basal fluorescence (2 ± 7 %, n = 11 axon bundles; P > 0.05).
Figure 8. Effect of iberiotoxin on axonal Ca2+ transients.
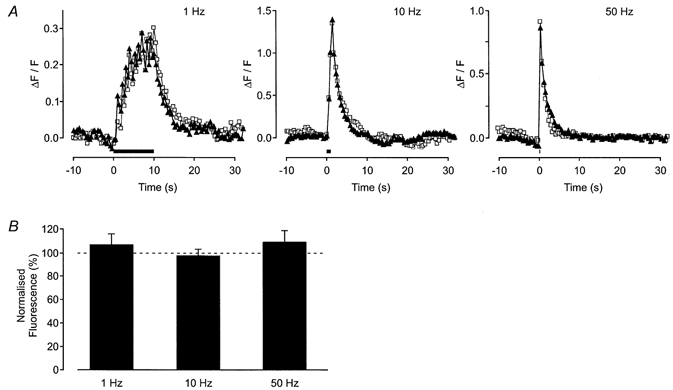
A, graphs showing the change in fluorescence in a single axon bundle when a stimulus was applied (trains of 10 stimuli) at 1, 10 and 50 Hz (from left to right in the figure). The period of electrical stimulation is shown by the bar. The response in the absence (▴) and presence (□) of iberiotoxin (0.1 μm) is shown. B, histogram shows the mean ( ± s.e.m.) for trains of 10 stimuli applied at 1, 10 and 50 Hz from 11 axon bundles. Iberiotoxin had no significant effect.
To test whether the action potential-evoked Ca2+ transient was affected by small conductance Ca2+-activated K+ channels, the specific blocker apamin was applied. High concentrations of apamin (1 μm) failed to affect the evoked Ca2+ transient (10 stimuli at 1 Hz, 25 ± 22 %, n = 6 axon bundles; 10 Hz, −3 ± 10 %, n = 6 axon bundles; 50 Hz, −5 ± 7 %, n = 4 axon bundles; P > 0.05). Apamin had no effect on the basal fluorescence (7 ± 7 %, n = 9 axon bundles; P > 0.05).
As specific Ca2+-activated K+ channel blockers did not modify the action potential-evoked axonal Ca2+ transients, the possible involvement of Ca2+-activated Cl− channels was investigated using niflumic acid. Niflumic acid (100 μm) had no detectable effect on the Ca2+ transient evoked by 10 stimuli at 10 Hz (-9 ± 13 %, n = 6 axon bundles; P > 0.05). Niflumic acid did not detectably alter the basal fluorescence (6 ± 8 %, n = 6 axon bundles; P > 0.05).
Pharmacological characterization of the K+ channels modulating axonal Ca2+ transients
The data obtained with iberiotoxin suggest that TEA does not potentiate the axonal Ca2+ transient by blocking large conductance Ca2+-activated K+ channels. One possibility is that TEA exerts its effects on K+ channels that are also sensitive to 4-AP. It is interesting to note that 4-AP (10 mm) also potentiated Ca2+ transients evoked by 10 stimuli at 10 Hz (Fig. 9). Like TEA, the effects of 4-AP were observed at all stimulation frequencies studied (Fig. 9; 1 stimulus, 662 ± 126 %, n = 13 axon bundles, P < 0.005; 10 stimuli at 1 Hz, 430 ± 217 %, n = 8 axon bundles, P < 0.01; 10 Hz, 238 ± 45 %, n = 18 axon bundles, P < 0.005; 50 Hz, 106 ± 50 %, n = 8 axon bundles, P < 0.01). 4-AP had no effect on the basal fluorescence (4 ± 6 %, n = 18 axon bundles; P > 0.05).
Figure 9. Effects of 4-AP on action potential-evoked Ca2+ transients.
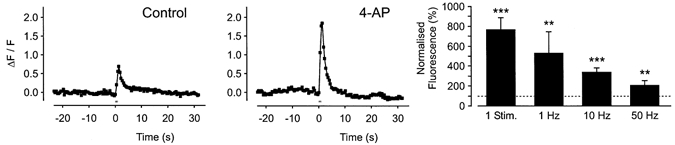
Effects of the K+ channel blocker 4-AP (10 mm) on the Ca2+ transient evoked by 10 stimuli at 10 Hz in a single axon bundle. Histogram showing the mean ( ± s.e.m.) increase in amplitude of the Ca2+ transient produced by 4-AP following a single stimulus, or trains of 10 stimuli applied at 1, 10 and 50 Hz from n = 8–18 bundles.
4-AP is known to block both the transient A-like K+ current (IA) and delayed rectifiers (IV). Therefore, the selective antagonist for IA, pandinustoxin-K (α), and the selective antagonist for the slow inactivating IV, tityustoxin-K (α), were studied (Juhng et al. 1999). Pandinustoxin-K (α) (10 nm) potentiated the action potential-evoked axonal Ca2+ transients (Fig. 10). The mean effect of pandinustoxin-K (α) (10 stimuli at 1 Hz, 54 ± 13 %, n = 7 axon bundles, P < 0.005; 10 Hz, 48 ± 5 %, n = 6 axon bundles, P < 0.005; 50 Hz, 58 ± 23 %, n = 6 axon bundles, P < 0.01) is shown in Fig. 10B. Pandinustoxin-K (α) had no effect on the basal fluorescence (2 ± 2 %, n = 6 axon bundles; P > 0.05).
Figure 10. Effect of pandinustoxin-K (α) on action potential-evoked Ca2+ transients.
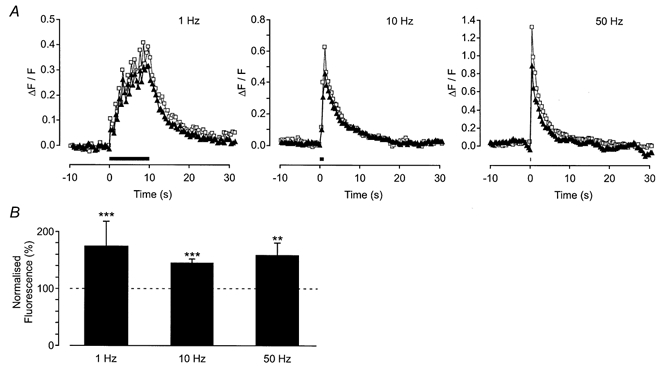
A, change in fluorescence in a single axon bundle when a stimulus was applied (trains of 10 stimuli) at 1, 10 and 50 Hz (from left to right in the figure). The period of electrical stimulation is shown by the bar. The response in the absence (▴) and presence (□) of pandinustoxin-K (α) (10 nm) is shown. B, histogram shows the mean ( ± s.e.m.) for trains of 10 stimuli applied at 1, 10 and 50 Hz from 7, 6 and 6 axon bundles, respectively. Pandinustoxin-K (α) significantly increased the amplitude of the evoked response.
Like pandinustoxin-K (α), tityustoxin-K (α) (100 nm) also potentiated the action potential-evoked axonal Ca2+ transients evoked by 10 stimuli at 1, 10 and 50 Hz (Fig. 11). Inspection of the histogram in Fig. 11B shows that the effect of tityustoxin-K (α) was independent of stimulation frequency (10 stimuli at 1 Hz, 75 ± 43 %, n = 11 axon bundles, P < 0.01; 10 Hz, 72 ± 31 %, n = 16 axon bundles, P < 0.005; 50 Hz, 42 ± 12 %, n = 13 axon bundles, P < 0.005). Tityustoxin-K (α) had no effect on the basal fluorescence (1 ± 3 %, n = 11 axon bundles; P > 0.05).
Figure 11. Effect of tityustoxin-K (α) on action potential-evoked Ca2+ transients.
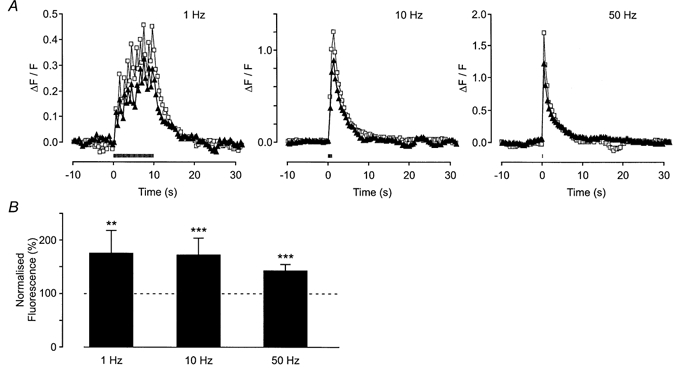
A, change in fluorescence in a single axon bundle in response to trains of 10 stimuli at 1, 10 and 50 Hz (from left to right in the figure). The period of electrical stimulation is shown by the bar. The response in the absence (▴) and presence (□) of tityustoxin-K (α) (100 nm) is shown. B, histogram shows the mean data ( ± s.e.m.) for trains of 10 stimuli applied at 1, 10 and 50 Hz from 11, 16 and 13 axon bundles, respectively. Tityustoxin-K (α) significantly increased the amplitude of the evoked response.
DISCUSSION
The present study shows that Ca2+ transients are evoked in the main axon of postganglionic sympathetic nerve fibres (see Fig. 1) by single or brief trains of action potentials. Axonal Ca2+ transients may well be a general phenomenon of mammalian axons, having been detected in axons in the central nervous system (Lev-Ram & Grinvald, 1987; Kriegler & Chiu, 1993; Callewaert et al. 1996; Mackenzie et al. 1996; Sun & Chiu, 1999; Forti et al. 2000; Brown et al. 2001) and in preganglionic parasympathetic axons (Wächtler et al. 1998).
The first question to address is whether action potential-evoked Ca2+ transients occur in axon bundles or Schwann cells. Unlike previous studies with Ca2+ indicators in axons (for example, Lev-Ram & Grinvald, 1987; Kriegler & Chiu, 1993; Sun & Chiu, 1999), the present experiments involved the use of a dextran-conjugated dye. This membrane-impermeable dye cannot penetrate a series of sequential Schwann cells and therefore the evoked fluorescence changes report Ca2+ transients within axons rather than in Schwann cells.
Characterization of axonal Ca2+ transients
When the number of stimuli in a train was increased, the axonal Ca2+ transient reached a stable plateau after about 30 stimuli. Our findings differ from those obtained in single axons of rat cerebellar Purkinje neurones (Callewaert et al. 1996), where an increase in the number of evoked-action potentials produced an increase in the amplitude of the axonal Ca2+ transient that was directly proportional to the number of stimuli in a train (although only the first 20 stimuli were tested). A hyperbolic relationship was found in cell bodies of rabbit vagal afferent neurones by Cohen et al. (1997) who concluded that the plateau phase was due to CICR. However, this is unlikely to be the case in the present experiments as ryanodine, caffeine and CPA failed to affect the axonal Ca2+ transient.
The plateau phase does not appear to result from dye saturation as the Ca2+ ionophore ionomycin produced about a fivefold greater increase in fluorescence. This is more than twice that evoked by maximal stimulation. Furthermore, the K+ channel blocker 4-AP markedly increased the amplitude of the Ca2+ transient without altering its kinetics of recovery. Taken together, these findings suggest that the plateau phase of the Ca2+ transient is determined by biological mechanisms. It is possible that this plateau is where Ca2+ influx is in equilibrium with Ca2+ extrusion mechanisms.
What is the origin of the action potential-evoked axonal Ca2+ transient?
Removal of extracellular Ca2+ has previously been shown to reduce the amplitude of the extracellularly recorded nerve action potential in axon bundles innervating rodent vas deferens (Cunnane & Stjärne, 1984). In the present study, removal of extracellular Ca2+ abolished the axonal Ca2+ transient, showing that the genesis of the Ca2+ transient requires extracellular Ca2+ and is wholly dependent on Ca2+ influx. It must also be noted that the axonal basal [Ca2+]i was reduced in Ca2+-free conditions, as there is now a change in the electrochemical gradient for Ca2+.
How does Ca2+ enter the neurone?
There are three main potential routes of Ca2+ entry into neurones: Ca2+ entry through VGCCs, by reverse mode Na+–Ca2+ exchange, or by Ca2+ entry through voltage-dependent Na+ channels.
Classification of VGCCs involved in action potential-evoked axonal Ca2+ transients
The axonal Ca2+ transient was reduced by about 80 % by the non-selective VGCC blocker Cd2+ making it likely that most of the Ca2+ enters through VGCCs. Ca2+ entry into axons of rat cerebellar Purkinje neurones (Callewaert et al. 1996) is abolished by ω-agatoxin IVA indicating Ca2+ entry through P-type VGCCs. In our experiments, Ca2+ transients were insensitive to P/Q-type Ca2+ channel blockers. In neonatal rat optic nerve axons, 58 % of the Ca2+ influx was through N-type Ca2+ channels (Sun & Chiu, 1999) but in adult rat optic nerve axons, only L-type Ca2+ channels were found to be present (Brown et al. 2001). Our studies suggest that 40 % of the Ca2+ enters through N-type VGCCs with no entry through L-type channels, similar to that in neonatal rat optic nerve axons. These findings suggest that the types of VGCCs expressed in neurones can be very different in both central and peripheral axons and may well be neurone specific and dependent on its stage of development. It seems likely that the type and relative distribution of VGCCs will have important functional consequences.
The relative contributions of N- and P/Q-type VGCCs to transmitter release differ according to the stimulation frequency used to activate the sympathetic nerves innervating the mouse vas deferens (Waterman, 1997). In the present study the effects of N- and P/Q-type VGCC blockers on the action potential-evoked axonal Ca2+ transient were independent of stimulation frequency.
Cd2+-resistant Ca2+ entry
It is possible that the residual 20 % Cd2+-resistant component of the Ca2+ transient results from Ca2+ entry through ‘conventional’ Na+ channels. Baker et al. (1971) previously reported that depolarization of the squid axon led to a leakage of Ca2+ into the axon; a small increase (∼1 %) was detected and it was concluded that Ca2+ entered through voltage-dependent Na+ channels. It is also possible that Ca2+ enters neurones through Na+ channels operating in a ‘slip-mode’ conductance state (Santana et al. 1998). An alternative, plausible explanation is that Ca2+ enters through non-selective cation channels, which are difficult to investigate pharmacologically.
Ca2+ influx in rat CNS white matter appears to proceed via reverse mode Na+–Ca2+ exchange during anoxia (Stys et al. 1992). However, in our experiments bepridil, which blocks Na+–Ca2+ exchange, had no effect on the Ca2+ transients. In addition, the lack of effect of bepridil on the action potential-evoked Ca2+ transient indicates that the Na+–Ca2+ exchanger is not responsible for Ca2+ extrusion from postganglionic sympathetic axons. An alternative likely candidate is the plasma membrane Ca2+-ATPase.
Intracellular stores
Ca2+ entry into some neurones can be amplified by CICR from ryanodine-sensitive intraneuronal stores. This mechanism has been shown to operate in sympathetic ganglia (Lipscombe et al. 1988; Hua et al. 1993) and in sympathetic nerve terminals (Smith & Cunnane, 1996). However, our data suggest that CICR does not occur in sympathetic axons. Thus, there seem to be clear regional differences in the expression of CICR mechanisms in neurones. Interestingly, Sun & Chiu (1999) also reported that pharmacological manipulations of internal Ca2+ stores with ryanodine and thapsigargin had no significant effect on the axonal Ca2+ transients in the rat optic nerve.
Similarly, on the basis of the experiments with CCCP, we could find no evidence for the role of mitochondria in mediating the axonal Ca2+ transients even with high frequency stimulation.
Which K+ channel(s) is involved in membrane repolarization during an action potential?
It has previously been shown, using focal extracellular recording in rodent vas deferens, that TEA prolongs the nerve terminal impulse, an effect believed to result from the blockade of Ca2+-dependent K+ channels (Stjärne et al. 1991; Brock & Cunnane, 1995). Similar conclusions regarding the action of Ca2+-dependent K+ channels have been reported using optical measurements from rat optic nerve axons (Lev-Ram & Grinvald, 1987). In the present study, TEA increased the amplitude of the axonal Ca2+ transient. However, it would appear that TEA was not acting on large conductance Ca2+-activated K+ channels, as the specific blocker iberiotoxin had no effect on the axonal Ca2+ transient. Apamin, which blocks small conductance Ca2+-activated K+ channels, was also ineffective. It is possible that TEA acts on 4-AP-sensitive, delayed rectifier channels (see Hille, 1992). Like TEA, 4-AP increased the amplitude of the Ca2+ transient at all stimulation frequencies employed. It follows that either delayed rectifier (KV) or A-like (KA) K+ channels (or both) are involved in regulating the configuration of the nerve action potential and hence the magnitude of the axonal Ca2+ transient.
KV and KA channels are normally isolated and studied electrophysiologically. However, in the mouse vas deferens, the axon bundles are too small to permit patch-clamp studies. As a result, the contribution of KV and KA channels to repolarization following an action potential was investigated pharmacologically by using the purified toxins pandinustoxin-K (α) and tityustoxin-K (α). Pandinustoxin-K (α) preferentially blocks the rapidly inactivating KA channels (Rogowski et al. 1996) while tityustoxin-K (α) is selective for slowly inactivating delayed rectifier type channels (Werkman et al. 1993). The current study shows that KA and KV channels contribute to axonal membrane repolarization as both toxins increased the action potential-evoked axonal Ca2+ transient, an action that was independent of stimulation frequency. However, it must be noted that tityustoxin-K (α) has an IC50 of 210 pm for blockade of KV 1.2 K+ channels (Werkman et al. 1993), and similarly pandinustoxin-K (α) has been reported to block KV 1.2 K+ channels, but with an IC50 of 32 pm (Rogowski et al. 1996) raising the possibility that they may both inhibit KV 1.2 in sympathetic axons.
Possible functions for action potential-evoked axonal Ca2+ transients
It is possible that the Ca2+ channels are simply ‘in transit’ from the cell body to the terminal. However, there is no evidence in postganglionic neurones that Ca2+ channels migrate in this manner. It is likely that Ca2+ channels are packaged in the cell body and transported to different regions of the nerve where they are functionally expressed. Based on our data, a number of points can be addressed. First, if the types and numbers of Ca2+ channels differ significantly between the terminal varicosities and main axons, that would indicate that the axonal channels are not simply ‘in transit’. In axons and varicosities about ∼50 % of the action potential-evoked Ca2+ transient is due to N-type Ca2+ channels, i.e. sensitive to ω-conotoxin GVIA and insensitive to blockers of P/Q-type Ca2+ channels. However, it is interesting to note that there are Cd2+-resistant Ca2+ channels in axons but not in varicosities (Brain & Bennett, 1997; Jackson, 2000). Second, the axonal Ca2+ channels are tightly regulated by K+ channels that are sensitive to 4-AP indicating that they may play an important physiological role.
In axons, the physiological role of Ca2+ influx following an action potential is unclear. A possible role would be to activate K+ channels, which in turn would contribute to repolarization of the action potential. TEA, which is often used as a blocker of large conductance Ca2+-activated K+ channels, greatly potentiated the action potential-evoked Ca2+ transient. In contrast, the specific blocker of large conductance Ca2+-activated K+ channels iberiotoxin had no effect. Inhibition of the small conductance Ca2+-activated K+ channels with apamin also failed to affect the Ca2+ transient. It is noteworthy that blockade of Ca2+-activated Cl− channels with niflumic acid had no effect. The results indicate that the influx of Ca2+ following an action potential is not modulated by Ca2+-dependent ion channel activity in the axon membrane.
It has been suggested that intracellular Ca2+ acts to regulate the safety factor for action potential propagation in cultured rat dorsal root ganglion neurones (Lüscher et al. 1996). However, action potential propagation in postganglionic sympathetic nerves innervating rodent vas deferens does not fail during repetitive stimulation (Brock & Cunnane,1987, 1992).
The axonal Ca2+ transient may drive axonal transport as it has been demonstrated that high concentrations of Ca2+ (Chan et al. 1980) increase axonal active transport. Llinás et al. (1989) and Breuer et al. (1992) report evidence that fast axonal transport may be strictly Ca2+ dependent. If similar mechanisms operate in sympathetic axons, then Ca2+ entry into axons following action potentials may be the source of Ca2+ utilized to drive axonal transport.
It is possible that there is non-exocytotic Ca2+-dependent release of neurotransmitter from axons. For example, action potentials release NO in a Ca2+-dependent fashion from all regions (including axons) of some neurones (Wiklund et al. 1997). Therefore, it is possible that substances released from axons (Weidmann, 1994) might activate receptors on Schwann cells. ATP is a likely candidate as it produces intracellular Ca2+ transients in Schwann cells by activating purinergic (P2Y) receptors (Jahromi et al. 1992; Lyons et al. 1994; Robitaille, 1995; Mayer et al. 1997). Whether this occurs in sympathetic axons is purely speculative.
The reported findings may have greater, or at least more direct, implications for nerve cell biology than for sympathetic system physiology. In addition to the issues raised above, the presence of different types/subtypes of calcium channels in different types of axons has implications for calcium channel sorting and trafficking mechanisms in main axons and varicosities.
Conclusions
The present experiments show that action potentials evoke Ca2+ transients in postganglionic sympathetic nerve axons. Axonal Ca2+ transients are mainly generated by Ca2+ entry through both N-type VGCCs, pharmacologically uncharacterized VGCCs and an unidentified Cd2+-resistant mechanism. Intracellular Ca2+ stores were not involved. The amplitude of the axonal Ca2+ transient is strongly regulated by K+ channels and pharmacological evidence suggests that KA and KV (possibly KV 1.2) channels are involved. No convincing evidence was found to suggest that Ca2+-dependent K+ or Ca2+-dependent Cl− channel activity occurs in main axons.
Acknowledgments
We would like to thank the British Heart Foundation and The Wellcome Trust for financial support.
References
- Baker PF, Hodgkin AL, Ridgway EB. Depolarization and calcium entry in squid giant axons. Journal of Physiology. 1971;218:709–755. doi: 10.1113/jphysiol.1971.sp009641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer AC, Bond M, Atkinson MB. Fast axonal transport is modulated by altering trans-axolemmal calcium influx. Cell Calcium. 1992;13:249–262. doi: 10.1016/0143-4160(92)90013-i. [DOI] [PubMed] [Google Scholar]
- Brain KL, Bennett MR. Calcium in sympathetic varicosities of mouse vas deferens during facilitation, augmentation and autoinhibition. Journal of Physiology. 1997;502:521–536. doi: 10.1111/j.1469-7793.1997.521bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock JA, Cunnane TC. Relationship between the nerve action potential and transmitter release from sympathetic postganglionic nerve terminals. Nature. 1987;326:605–607. doi: 10.1038/326605a0. [DOI] [PubMed] [Google Scholar]
- Brock JA, Cunnane TC. Electrical activity at the sympathetic neuroeffector junction in the guinea-pig vas deferens. Journal of Physiology. 1988;399:607–632. doi: 10.1113/jphysiol.1988.sp017099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock JA, Cunnane TC. Impulse conduction in sympathetic nerve terminals in the guinea-pig vas deferens and the role of the pelvic ganglia. Neuroscience. 1992;47:185–196. doi: 10.1016/0306-4522(92)90131-k. [DOI] [PubMed] [Google Scholar]
- Brock JA, Cunnane TC. Effects of Ca2+ and K+ blockers on nerve impulses recorded from guinea-pig postganglionic sympathetic nerve terminals. Journal of Physiology. 1995;489:389–402. doi: 10.1113/jphysiol.1995.sp021060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, Westenbroek RE, Catterall WA, Ransom BR. Axonal L-type Ca2+ channels and anoxic injury in rat CNS white matter. Journal of Neurophysiology. 2001;85:900–911. doi: 10.1152/jn.2001.85.2.900. [DOI] [PubMed] [Google Scholar]
- Callewaert G, Eilers J, Konnerth A. Axonal calcium entry during fast ‘sodium’ action potentials in rat cerebellar Purkinje neurones. Journal of Physiology. 1996;495:641–647. doi: 10.1113/jphysiol.1996.sp021622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SY, Ochs S, Worth RM. The requirement for calcium ions and the effect of other ions on axoplasmic transport in mammalian nerve. Journal of Physiology. 1980;301:477–504. doi: 10.1113/jphysiol.1980.sp013219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AS, Moore KA, Bangalore R, Jafri MS, Weinreich D, Kao JPY. Ca2+-induced Ca2+ release mediates Ca2+ transients evoked by single action potentials in rabbit vagal afferent neurones. Journal of Physiology. 1997;499:315–328. doi: 10.1113/jphysiol.1997.sp021929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottee LJ, Lavidis NA, Bennett MR. Spatial relationships between sympathetic varicosities and smooth muscle cells in the longitudinal layer of the mouse vas deferens. Journal of Neurocytology. 1996;25:413–422. doi: 10.1007/BF02284812. [DOI] [PubMed] [Google Scholar]
- Cunnane TC, Stjärne L. Frequency-dependent intermittency and ionic basis of impulse conduction in postganglionic sympathetic fibres of guinea-pig vas deferens. Neuroscience. 1984;11:211–229. doi: 10.1016/0306-4522(84)90225-2. [DOI] [PubMed] [Google Scholar]
- Forti L, Pouzat C, Llano I. Action potential-evoked Ca2+ signals and calcium channels in axons of developing rat cerebellar interneurones. Journal of Physiology. 2000;527:33–48. doi: 10.1111/j.1469-7793.2000.00033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. 2. Sunderland MA USA: Sinauer Associates, Inc; 1992. [Google Scholar]
- Hobai IA, Bates JA, Howarth FC, Levi AJ. Inhibition of external Cd2+ of Na/Ca exchange and L-type Ca channel in rabbit ventricular myocytes. American Journal of Physiology. 1997;41:H2164–2173. doi: 10.1152/ajpheart.1997.272.5.H2164. [DOI] [PubMed] [Google Scholar]
- Hua SY, Nohmi M, Kuba K. Characteristics of Ca2+ release induced by Ca2+ influx in cultured bullfrog sympathetic neurones. Journal of Physiology. 1993;464:245–272. doi: 10.1113/jphysiol.1993.sp019633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson VM. Oxford University; 2000. Calcium dynamics in postganglionic sympathetic nerves, Dphil Thesis. [Google Scholar]
- Jahromi BS, Robitaille R, Charlton MP. Transmitter release increases intracellular calcium in perisynaptic Schwann cells in situ. Neuron. 1992;8:1069–1077. doi: 10.1016/0896-6273(92)90128-z. [DOI] [PubMed] [Google Scholar]
- Juhng KN, Kokate TG, Yamguchi S, Kim BY, Rogowski RS, Blaustein MP, Rogawski MA. Induction of seizures by the potent K+ channel-blocking scorpion venom peptide toxins tityustoxin-K (α) and pandinustoxin (α) Epilepsy Research. 1999;34:177–186. doi: 10.1016/s0920-1211(98)00111-9. [DOI] [PubMed] [Google Scholar]
- Kriegler S, Chiu SY. Calcium signaling of glial cells along mammalian axons. Journal of Neuroscience. 1993;13:4229–4245. doi: 10.1523/JNEUROSCI.13-10-04229.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev-Ram V, Grinvald A. Activity-dependent calcium transients in central nervous system myelinated axons revealed by the calcium indicator Fura-2. Biophysical Journal. 1987;52:571–576. doi: 10.1016/S0006-3495(87)83246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscombe D, Madison DV, Poenie M, Reuter H, Tsien RW, Tsien RY. Imaging of cytosolic Ca2+ transients arising from Ca2+ stores and Ca2+ channels in sympathetic neurons. Neuron. 1988;1:355–365. doi: 10.1016/0896-6273(88)90185-7. [DOI] [PubMed] [Google Scholar]
- Llinás R, Sugimori M, Lin JW, Leopold PL, Brady ST. ATP-dependent directional movement of rat synaptic vesicles injected into the presynaptic terminal of squid giant synapse. Proceedings of the National Academy of Sciences of the USA. 1989;86:5656–5660. doi: 10.1073/pnas.86.14.5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Lipp P, Luscher HR, Niggli E. Control of action potential propagation by intracellular Ca2+ in cultured rat dorsal root ganglion cells. Journal of Physiology. 1996;490:319–324. doi: 10.1113/jphysiol.1996.sp021146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons SA, Morell P, McCarthy KD. Schwann cells exhibit P2Y purinergic receptors that regulate intracellular calcium and are up-regulated by cyclic AMP analogues. Journal of Neurochemistry. 1994;63:552–560. doi: 10.1046/j.1471-4159.1994.63020552.x. [DOI] [PubMed] [Google Scholar]
- Mackenzie PJ, Umemiya M, Murphy TH. Ca2+ imaging of CNS axons in culture indicates reliable coupling between single action potentials and distal functional release sites. Neuron. 1996;16:783–795. doi: 10.1016/s0896-6273(00)80098-7. [DOI] [PubMed] [Google Scholar]
- McPherson PS, Kim YK, Valdivia H, Knudson CM, Takekura H, Franzini AC, Coronado R, Campbell KP. The brain ryanodine receptor: a caffeine-sensitive calcium release channel. Neuron. 1991;7:17–25. doi: 10.1016/0896-6273(91)90070-g. [DOI] [PubMed] [Google Scholar]
- Mayer C, Wächtler J, Kamleiter M, Grafe P. Intracellular calcium transients mediated by P2 receptors in the paranodal Schwann cell region of myelinated rat spinal root axons. Neuroscience Letters. 1997;224:49–52. doi: 10.1016/s0304-3940(97)13457-7. [DOI] [PubMed] [Google Scholar]
- Nagasaki K, Fleischer S. Ryanodine sensitivity of the calcium release channel of sarcoplasmic reticulum. Cell Calcium. 1988;9:1–7. doi: 10.1016/0143-4160(88)90032-2. [DOI] [PubMed] [Google Scholar]
- Richard S, Diochot S, Nargeot J, Baldy-Moulinier M, Valmier J. Inhibition of T-type calcium currents by dihydropyridines in mouse embryonic dorsal root ganglion neurons. Neuroscience Letters. 1991;132:229–234. doi: 10.1016/0304-3940(91)90308-g. [DOI] [PubMed] [Google Scholar]
- Robitaille R. Purinergic receptors and their activation by endogenous purines at perisynaptic glial cells of the frog neuromuscular junction. Journal of Neuroscience. 1995;15:7121–7131. doi: 10.1523/JNEUROSCI.15-11-07121.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogowski RS, Collins JH, O'Neill TJ, Gustafson TA, Werkman TR, Rogawski MA, Tenenholz TC, Weber DJ, Blaustein MP. Three new toxins from the scorpion Pandinus imperator selectively block certain voltage-gated K+ channels. Molecular Pharmacology. 1996;50:1167–1177. [PubMed] [Google Scholar]
- Rousseau E, Smith JS, Meissner G. Ryanodine modifies conductance and gating behavior of single Ca2+ release channel. American Journal of Physiology. 1987;253:C364–368. doi: 10.1152/ajpcell.1987.253.3.C364. [DOI] [PubMed] [Google Scholar]
- Santana LF, Gomez AM, Lederer WJ. Ca2+ flux through promiscuous cardiac Na+ channels: Slip-mode conductance. Science. 1998;279:1027–1033. doi: 10.1126/science.279.5353.1027. [DOI] [PubMed] [Google Scholar]
- Smith AB, Cunnane TC. Ryanodine-sensitive calcium stores involved in neurotransmitter release from sympathetic nerve terminals of the guinea-pig. Journal of Physiology. 1996;497:657–664. doi: 10.1113/jphysiol.1996.sp021797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AB, Cunnane TC. Multiple calcium channels control neurotransmitter release from rat postganglionic sympathetic nerve terminals. Journal of Physiology. 1997;499:341–349. doi: 10.1113/jphysiol.1997.sp021931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stjärne L, Stjärne E. Some pharmacological applications of an extracellular recording method to study secretion of a sympathetic co-transmitter, presumably ATP. Acta Physiologica Scandinavica. 1989;135:227–239. doi: 10.1111/j.1748-1716.1989.tb08572.x. [DOI] [PubMed] [Google Scholar]
- Stjärne L, Stjärne E, Msghina M, Bao JX. K+ and Ca2+ channel blockers may enhance or depress sympathetic transmitter release via a Ca2+-dependent mechanism ‘upstream’ of the release site. Neuroscience. 1991;44:673–692. doi: 10.1016/0306-4522(91)90087-5. [DOI] [PubMed] [Google Scholar]
- Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na+–Ca2+ exchanger. Journal of Neuroscience. 1992;12:430–439. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun BB, Chiu SY. N-type calcium channels and their regulation by GABAB receptors in axons of neonatal rat optic nerve. Journal of Neuroscience. 1999;19:5185–5194. doi: 10.1523/JNEUROSCI.19-13-05185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Akaike N. Calcium antagonist effect on low-threshold (T-type) calcium current in rat isolated hippocampal CA1 pyramidal neurons. Journal of Pharmacology and Experimental Therapeutics. 1991;132:229–234. [PubMed] [Google Scholar]
- Trosper TL, Philipson KD. Effects of divalent and trivalent cations on Na+–Ca2+ exchange in cardiac sarcolemmal vesicles. Biochimica et Biophysica Acta. 1983;731:63–68. doi: 10.1016/0005-2736(83)90398-x. [DOI] [PubMed] [Google Scholar]
- Wächtler J, Mayer C, Grafe P. Activity-dependent intracellular Ca2+ transients in unmyelinated nerve fibres of the isolated adult rat vagus nerve. Pflügers Archiv. 1998;435:678–686. doi: 10.1007/s004240050569. [DOI] [PubMed] [Google Scholar]
- Waterman SA. Role of N-, P- and Q-type voltage-gated calcium channels in transmitter release from sympathetic neurones in the mouse isolated vas deferens. British Journal of Pharmacology. 1997;120:393–398. doi: 10.1038/sj.bjp.0700948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidmann S. ‘Action substances’ of peripheral nerve re-visited. Experientia. 1994;50:342–345. doi: 10.1007/BF02026635. [DOI] [PubMed] [Google Scholar]
- Werkman TR, Gustafson TA, Rogowski RS, Blaustein MP, Rogawski MA. Tityustoxin-K alpha, a structurally novel and highly potent K+ channel peptide toxin, interacts with the alpha-dendrotoxin binding site on the cloned Kv1. 2 K+ channel. Molecular Pharmacology. 1993;44:430–436. [PubMed] [Google Scholar]
- Wiklund NP, Cellek S, Leone AM, Iversen HH, Gustafsson LE, Brundin L, Furst VW, Flock A, Moncada S. Visualisation of nitric oxide released by nerve stimulation. Journal of Neuroscience Research. 1997;47:224–232. doi: 10.1002/(sici)1097-4547(19970115)47:2<224::aid-jnr11>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]