

Abstract
Using a Ca2+ imaging system and fura-2 AM (5 μm) we showed that exposure of polarised monolayers of human bronchial epithelial cells (16HBE14o- cell line) to aldosterone produced a fast intracellular [Ca2+] ([Ca2+]i) decrease, in 70 % of cells. Exposure to aldosterone (1 nm) reduced the [Ca2+]i by 39 ± 9 nm (n = 282, P < 0.0001) within 10 min, from a basal [Ca2+]i of 131 ± 19 nm (n = 282).
The effect of aldosterone on [Ca2+]i was not affected by inhibitors of the classical genomic pathway, cycloheximide (1 μm) or spironolactone (10 μm). The aldosterone-induced [Ca2+]i decrease was inhibited by thapsigargin (1 μm), pertussis toxin (24 h at 200 ng ml−1), the adenylate cyclase inhibitors 2′,3′-dideoxyadenosine (200 μm) and MDL-12,330A hydrochloride (500 μm), and the protein kinase A inhibitor RP-adenosine 3′,5′-cyclic monophosphorothioate (200 μm). In addition, treatment of 16HBE14o- monolayers with aldosterone (1 nm) inhibited by ∼30 % the large and transient [Ca2+]i increase induced by apical exposure to uridine triphosphate (UTP, 0.1 mm), a known secretagogue in airway epithelia.
Our results demonstrate for the first time that in human bronchial epithelial cells, aldosterone decreases [Ca2+]i levels via a non-genomic mechanism. The hormone-induced changes to [Ca2+]i involve stimulation of thapsigargin-sensitive Ca2+-ATPase, via G-protein-, adenylate cyclase- and protein kinase A-coupled signalling pathways.
Aldosterone is a mineralocorticoid hormone which plays a central role in the water and electrolyte balance of the body, in particular by stimulating Na+ absorption across epithelia. The major ionic transporters involved in Na+ absorption are the amiloride-sensitive epithelial Na+ channel (ENaC; Voilley et al. 1994), the basolateral Na+-K+-ATPase (Jorgensen, 1969) and a basolateral K+ channel (Tsuchiya et al. 1992; Urbach et al. 1994). These ion channels and pump are known to be sensitive to aldosterone in different types of epithelia (Urbach et al. 1996). In airway epithelia, contradictory aldosterone effects on Na+ absorption have been described. Aldosterone has been reported to enhance, within a few hours to a few days, the amiloride-sensitive transepithelial Na+ absorption in canine tracheal epithelium (Cullen & Welsh, 1987), in frog lung epithelium (Fisher & Clauss, 1990) and in human airway epithelial cells (Kunzelmann et al. 1996). However, more recently it has been demonstrated that, in murine airway epithelia, aldosterone does not have a significant effect on amiloride-sensitive Na+ absorption (Grubb & Boucher, 1998). Aldosterone has also been described to attenuate Cl− conductance (Kunzelmann et al. 1996). The aldosterone effect is classically described as a genomic mechanism involving binding to a cytosolic receptor, translocation to the nucleus and protein synthesis. Corticoid receptors are present in the lung (Ballard et al. 1974; Krozowski & Funder, 1981) and aldosterone stimulates ENaC expression in rat lung primary cultures (Champigny et al. 1994).
Besides the genomic effect of aldosterone, which affects ionic transport only after a latency of a few hours, there is growing evidence for rapid, non-genomic effects of steroid hormones. In epithelial cells such as distal colon and frog skin, aldosterone regulates K+ channel activity within less than 10 min (Urbach et al. 1996; Maguire et al. 1999). Aldosterone stimulates an increase in intracellular Ca2+ concentration ([Ca2+]i), pHi and protein kinase activity in human distal colon and in mouse cortical collecting duct (Doolan & Harvey, 1996a, b; Winter et al. 1999; Harvey & Higgins, 2000). The present paper is the first report of a rapid and non-genomic effect of aldosterone in lung epithelium. We show that aldosterone, at physiological concentrations (0.1 nm to 1 μm) reduces the level of basal [Ca2+]i and partially inhibits the [Ca2+]i increase induced by secretagogues in human bronchial epithelial cells, via a non-genomic mechanism, involving activation of thapsigargin-sensitive Ca2+-ATPase and a protein kinase A (PKA) signalling pathway.
METHODS
Cell culture
In this study we used the human bronchial epithelial 16HBE14o- cell line, which is a post-crisis SV40-transformed cell line, derived from the surface epithelium of mainstream second-generation bronchi (Cozens et al. 1994). Ion transport studies indicate that it retains transport properties typical of freshly isolated surface airway epithelial cells and is morphologically similar with tight junctions and cilia (Cozens et al. 1994). The epithelial cells were grown at 37 °C in Eagle's minimal essential medium (EMEM, Biowhittaker) supplemented with 10 % fetal bovine serum, 1 %l-glutamine, and 1 % penicillin-streptomycin (Cozens et al. 1994). The tissue culture flasks were coated using a solution of fibronectin (Becton Dickinson, Bedford, MA, USA), collagen (Vitrogen 100, Celtrix, Palo Alto, CA, USA) and bovine serum albumin (BSA; Sigma). Confluent monolayers were grown from cells isolated using trypsin (0.025 % trypsin, 1 % polyvinylpyrolidone, 0.02 % EGTA in a Hepes-buffered saline solution).
Calcium spectrofluorescence
[Ca2+]i was determined in confluent 16HBE14o- cell monolayers grown on fibronectin-collagen-BSA-treated glass coverslips. The cells were loaded with 5 μm of the Ca2+-sensitive fluorescent probe fura-2 acetoxymethyl ester (fura-2 AM) for 30 min, in the dark, at room temperature (22 °C). The cells were washed twice in Hepes-buffered Krebs-Heinsleit solution (NaCl 140 mm, KCl 5 mm, CaCl2 2 mm, MgCl2 1 mm, Hepes 10 mm, Tris-HCl 10 mm, glucose 10 mm, pH 7.4, 280–290 mosmol l−1). The coverslips were mounted on the stage of an inverted microscope equipped for epi-fluorescence (Diaphot 200, Nikon, The Netherlands). The light from a xenon lamp (Osram, Germany) was filtered through alternating 340 nm and 380 nm filters (Nikon), mounted on a motorized chopper under computer control (Starwise Fluo system, Imstar, France). The emission fluorescence produced after fura-2 excitation was filtered at 510 nm. The transmitted light image was detected using an intensified CCD video camera (Darkstar, Photonics Sciences, UK) coupled to the microscope. The fluorescence obtained at each excitation wavelength (F340 and F380) depends upon the level of Ca2+ binding to fura-2, according to an in vivo calibration performed using a range of EGTA-buffered Ca2+ solutions of the fura-2 free acid. The [Ca2+]i was calculated automatically by a computer program (Starwise, Imstar) using the Grynkiewicz equation (Grynkiewicz et al. 1985):
where K′ is the product of the dissociation constant of Ca2+ binding with fura-2, R is the experimental ratio of F340 and F380 and Rmax and Rmin are the values of R under saturating and Ca2+-free conditions, respectively.
The cells were exposed to various aldosterone concentrations (10−11 to 10−6m) made up from a 10−2m stock solution of hormone dissolved in methanol. In a control experiment we tested the effect of 0.01 % methanol, corresponding to the maximum amount of solvent to which the cells were exposed.
Not all cells responded to hormonal treatment. The Kruskal-Wallis test (non-parametric ANOVA) was applied to the [Ca2+]i changes detected before and after hormonal exposure. When P < 0.05, the cell was considered to be responsive.
The mean [Ca2+]i variations (Δ[Ca2+]i) given in the Results were determined in responsive cells only, using the following equation: Δ[Ca2+]i = [Ca2+]-5to0 - [Ca2+]20to25, where [Ca2+]-5to0 is the mean [Ca2+]i measured during the 5 min prior to hormone treatment and [Ca2+]20to25 is the [Ca2+]i measured between 20 and 25 min after hormone treatment. The average basal [Ca2+]i values given in the text were calculated from [Ca2+]-5to0 of responsive cells only.
All results are presented as mean values ± s.e.m. (n = number of cells). Most of the data were analysed by ANOVA. Student's t test was used to compare unpaired data. P < 0.05 was taken as statistically significant.
RESULTS
Rapid effect of aldosterone on [Ca2+]i
Exposure of 16HBE14o- monolayers to physiological concentrations (0.1 nm to 1 μm) of aldosterone produced a rapid and significant decrease of [Ca2+]i (Fig. 1 and Fig. 2). The [Ca2+]i decrease was observed in 70 % of the 16HBE14o- cells, and in the remaining cells tested no change in [Ca2+]i was detected. The [Ca2+]i started to decrease within 30 s after exposure to aldosterone and reached a plateau value at 10–15 min (Fig. 1). In responding cells, after 20–25 min exposure to 1 nm aldosterone, the [Ca2+]i was reduced by 39 ± 9 nm (n = 282, P < 0.0001) from a basal value of 131 ± 19 nm (n = 282). Ninety-minute-long records following the beginning of aldosterone exposure indicated that the [Ca2+]i reached a new sustained lower steady-state value. The magnitude of the [Ca2+]i decrease was dependent on the aldosterone concentration. As shown in Fig. 2, the most potent effect on [Ca2+]i was reached at 1 nm aldosterone. At very low hormone concentration (0.01 nm) no significant change in [Ca2+]i was detected. In order to verify that spontaneous [Ca2+]i variations in themselves were not responsible for the [Ca2+]i decrease observed with aldosterone, we used the same experimental protocol of light exposure and image acquisition on non-aldosterone-treated cell preparations. Resting [Ca2+]i levels were not significantly changed 20 min after the beginning of the recording in non-treated cells (Δ[Ca2+]i = 0.9 ± 3 nm, n = 123 cells, P > 0.1). We also verified that the methanol used as the solvent for the hormone was not responsible for the [Ca2+]i decrease observed with aldosterone. The [Ca2+]i levels were not significantly changed 15 min after exposure to 0.01 % methanol (Δ[Ca2+]i = 8 ± 7 nm, n = 20 cells, P > 0.1; Fig. 2).
Figure 1. Aldosterone effect on [Ca2+]i in 16HBE14o- monolayers.
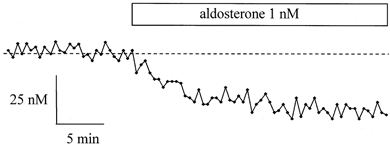
Typical record of [Ca2+]i variations upon exposure of a 16HBE14o- monolayer to 1 nm aldosterone. The [Ca2+]i was measured in a single cell. The dashed line indicates the average [Ca2+]i measured in the cell before aldosterone treatment.
Figure 2. Dose-response relationship of the aldosterone-induced [Ca2+]i decrease.
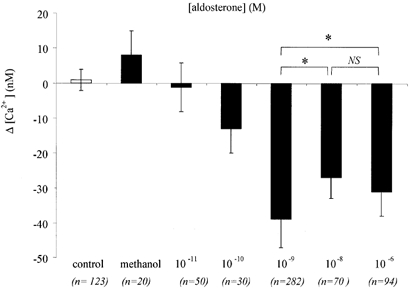
Variations in [Ca2+]i as a function of aldosterone concentration. Significant changes to [Ca2+]i were obtained upon addition of aldosterone over the range 0.1 nm to 1 μm (overall P < 0.0001, ANOVA). The solvent methanol (0.01 %) alone did not produce any significant [Ca2+]i change. *P < 0.05 from t test.
Glucocorticoid effect on [Ca2+]i
The effects on [Ca2+]i of glucocorticoid hormones such as hydrocortisone and triamcilone acetonide were also tested in 16HBE14o- cells. The same protocol as in aldosterone experiments was used and the mean [Ca2+]i changes were determined at 20–25 min after hormone exposure. The results given in Fig. 3 indicate that hydrocortisone or triamcilone acetonide did not produce any significant change in [Ca2+]i when tested at 1 nm (P > 0.1), the concentration at which aldosterone produced the maximum decrease in [Ca2+]i. However, at a higher concentration (1 μm), both glucocorticoids produced an [Ca2+]i response (P < 0.001). Taken together, these results indicate that mineralocorticoids are more potent than glucocorticoids in producing an [Ca2+]i decrease in 16HBE14o- cells.
Figure 3. Glucocorticoid effect on [Ca2+]i.
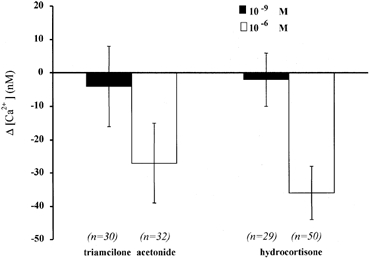
Variations in [Ca2+]i measured 15–20 min after exposure of 16HBE14o- monolayers to the glucocorticoids hydrocortisone and triamcilone acetonide, at two different concentrations (1 nm and 1 mm).
Effects of external Na+ replacement, orthovanadate and thapsigargin
In order to identify the mechanism by which aldosterone produced a decrease of [Ca2+]i in 16HBE14o- monolayers, we tested a possible involvement of the Na+–Ca2+ exchanger and Ca2+ pumps.
The effect of aldosterone on the Na+–Ca2+ exchange activity was investigated by replacement of all external Na+ by a non-permeant cation, N-methyl d-glucamine (NMDG). As shown in Fig. 4A and B, replacing Na+ resulted in an [Ca2+]i increase of 45 ± 4 nm (n = 29). This is the expected result if Na+–Ca2+ exchange contributes to the maintenance of basal Ca2+ levels. However, the absence of external Na+ did not affect the aldosterone-induced reduction of [Ca2+]i. In a Na+-free bathing solution, the [Ca2+]i was decreased by 32 ± 3 nm (n = 29) upon exposure of the cells to aldosterone (1 nm) (Fig. 4A), which was not significantly different from that obtained in Na+-containing solutions (P > 0.1). This result indicates that Na+–Ca2+ exchange activity is not involved in the aldosterone-induced [Ca2+]i variations.
Figure 4. Effects of NMDG, orthovanadate and thapsigargin.
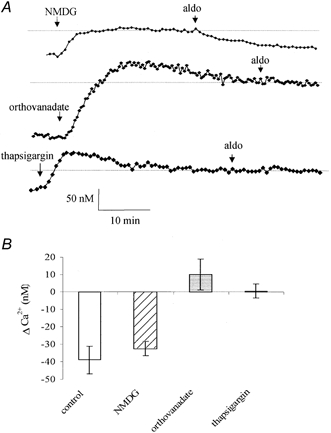
A, representative [Ca2+]i change upon replacement of Na+ by NMDG, or treatment of the monolayers to orthovanadate (1 mm) or thapsigargin (1 μm) followed by exposure to aldosterone (aldo, 1 nm). B, average [Ca2+]i variations induced by aldosterone in control conditions, and between 15 and 20 min after Na+ replacement (NMDG), or orthovanadate or thapsigargin treatment.
Intracellular Ca2+ may be reduced by extrusion via the Ca2+-ATPase pump. Therefore, we tested the effect of sodium orthovanadate, a P-type Ca2+-ATPase inhibitor, on the aldosterone-induced decrease of [Ca2+]i. As shown in Fig. 4A, orthovanadate produced an [Ca2+]i increase followed by a slow decline to basal values. Treatment of the 16HBE14o- monolayers with orthovanadate (1 mm) produced a large [Ca2+]i increase of 198 ± 23 nm (n = 51). After orthovanadate (1 mm) treatment, the aldosterone effect on [Ca2+]i was abolished (Δ[Ca2+]i = 10 ± 9 nm, n = 51, P > 0.1). A lower orthovanadate concentration (10 μm) affected neither the basal [Ca2+]i nor the [Ca2+]i decrease induced by aldosterone (1 nm). This result indicated that a Ca2+-ATPase contributes to the basal Ca2+ concentration and is also involved in the aldosterone-induced decrease of [Ca2+]i. Orthovanadate is a non-specific ATPase inhibitor and its effects at high concentration could implicate either plasma membrane or intracellular organelle Ca2+-ATPases.
In order to further investigate the nature of the Ca2+-ATPase involved in the aldosterone-induced decrease of [Ca2+]i, we tested the effect of thapsigargin, a more specific antagonist of the endoplasmic reticulum (ER) Ca2+-ATPase. Exposure of the 16HBE14o- monolayers to thapsigargin (1 μm) resulted in an increase of [Ca2+]i by 115 ± 11 nm (n = 70) followed by a slow decline to a plateau value (Fig. 4A), indicating the contribution of the thapsigargin-sensitive Ca2+-ATPase to the maintenance of the basal levels of [Ca2+]i. In the presence of thapsigargin, the aldosterone-induced [Ca2+]i decrease was completely abolished. Aldosterone (1 nm), added 15 min after the beginning of the thapsigargin exposure did not produced any significant change in [Ca2+]i (Δ[Ca2+]i = −0.5 ± 4 nm, n = 70, P > 0.2; Fig. 4B). This latter result suggests that the [Ca2+]i decrease induced by aldosterone occurred via stimulation of a thapsigargin-sensitive Ca2+ pump and uptake of Ca2+ into the ER.
Cycloheximide and spironolactone effects
The rapidity of the [Ca2+]i decrease in 16HBE14o- cell monolayers exposed to aldosterone suggests that this regulation does not occur through the classical genomic pathway. In order to verify this hypothesis, we tested the effect of pre-treatment of the monolayer with cycloheximide, a protein synthesis inhibitor, and spironolactone, a classical antagonist of the type I mineralocorticoid receptor, on the [Ca2+]i decrease induced by aldosterone (Fig. 5).
Figure 5. Effects of cycloheximide and spironolactone.
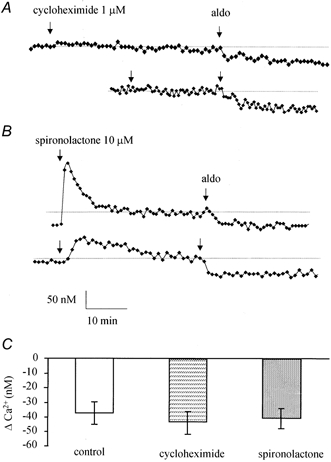
Representative [Ca2+]i change in 16HBE14o- monolayers pre-treated for 15 min with cycloheximide (1 μm, A) or spironolactone (10 μm, B) and exposed to aldosterone (1 nm). C, average [Ca2+]i variations induced by aldosterone in control conditions and after cycloheximide or spironolactone treatment.
Cycloheximide (1 μm) alone did not produce a significant change in basal [Ca2+]i (Δ[Ca2+]i = 18 ± 15 nm, n = 28, P > 0.05). After cycloheximide treatment, aldosterone (1 nm) produced an [Ca2+]i decrease of 48 ± 8 nm (n = 28), which was not significantly different from the aldosterone-induced [Ca2+]i decrease observed with aldosterone alone (P > 0.1). In contrast, spironolactone (10 μm) alone produced a transient [Ca2+]i increase of 113 ± 31 nm (n = 25). However, spironolactone treatment did not affect the aldosterone-induced decrease of [Ca2+]i. In the presence of spironolactone, aldosterone (1 nm) induced a decrease in [Ca2+]i by 41 ± 7 nm (n = 25), similar to the control aldosterone response (P > 0.1).
Role of G-proteins, adenylate cyclase and PKA in the aldosterone response
The role of G-proteins, adenylate cyclase and PKA signalling was also investigated in the aldosterone-induced [Ca2+]i decrease.
The role of G-proteins was tested by incubation of 16HBE14o- monolayers with pertussis toxin (200 ng ml−1) for 24 h. Pertussis toxin treatment did not significantly affect the basal [Ca2+]i (Δ[Ca2+]i = 24 ± 19 nm, n = 60, P > 0.1) but completely abolished the [Ca2+]i decrease induced by 1 nm aldosterone (Δ[Ca2+]i = 7 ± 4 nm, n = 60, P > 0.1; Fig. 6).
Figure 6. Effects of G-protein, adenylate cyclase and PKA inhibition.
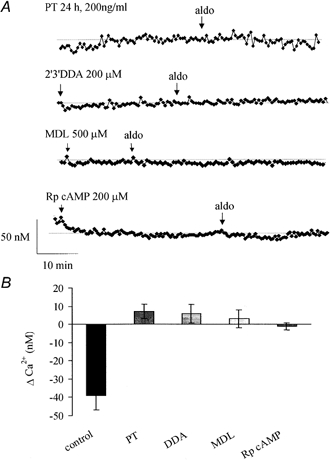
A, representative [Ca2+]i recording during treatment with pertussis toxin (PT), DDA, MDL or Rp-cAMP followed by exposure to aldosterone (1 nm). B, average [Ca2+]i variations induced by aldosterone in control conditions or after treatment with inhibitor.
The role of adenylate cyclase was also explored using two different inhibitors: 2′,3′-dideoxyadenosine (DDA, 200 μm) and MDL-12,330A hydrochloride (MDL, 500 μm). Neither of these two inhibitors significantly affected the basal [Ca2+]i (DDA pre-treatment Δ[Ca2+]i = 30 ± 33 nm, n = 78, P > 0.1, and MDL Δ[Ca2+]i = 3 ± 7 nm, n = 80, P > 0.1). However, both inhibitors completely abolished the [Ca2+]i decrease normally induced by aldosterone (1 nm). In cells pre-treated with DDA or MDL, aldosterone exposure did not produce a significant change in [Ca2+]i (Δ[Ca2+]i = 7 ± 4 nm, n = 88, P > 0.1 and Δ[Ca2+]i = 3 ± 5 nm, n = 90, P > 0.1, respectively; Fig. 6).
The effect of RP-adenosine 3′,5′-cyclic monophosphorothioate triethylammonium salt (Rp-cAMP, 200 μm), a PKA inhibitor, was also tested. Rp-cAMP did not significantly affect the basal [Ca2+]i (Δ[Ca2+]i = −20 ± 22 nm, n = 80, P > 0.1) but completely abolished the [Ca2+]i decrease induced by aldosterone (Δ[Ca2+]i = −1 ± 2 nm, n = 80, P > 0.1; Fig. 6).
Effect of aldosterone on the UTP-stimulated [Ca2+]i response
Intracellular Ca2+ is a known secretagogue signal and a reduction in cell Ca2+ levels may antagonize the secretory event. In order to investigate a possible anti-secretory role of aldosterone, the effect of the hormone was tested on the [Ca2+]i response to external UTP, a known secretagogue in airway epithelial cells. Apical exposure of 16HBE14o- monolayers to UTP alone produced a large and transient increase in [Ca2+]i (Fig. 7A), which was observed in 100 % of the cells tested. Upon apical exposure of the monolayers to UTP (0.1 mm) the [Ca2+]i was increased from 128 ± 18 to 534 ± 36 nm (n = 90) and rapidly returned to a plateau value of 180 ± 24 nm (n = 90), measured 5 min after UTP addition (Fig. 7A). Pre-exposure of the monolayer to aldosterone for 20 min caused a reduction in the [Ca2+]i response to apical UTP. As shown in Fig. 7B, aldosterone (0.1 nm) treatment reduced the amplitude of the peak Ca2+ increase. After aldosterone, apical UTP (0.1 mm) exposure produced an increase in [Ca2+]i from 100 ± 13 nm to 290 ± 27 nm (n = 150). This latter response (Δ[Ca2+]i with aldosterone = 200 ± 35 nm, n = 150 cells) was significantly reduced compared to the UTP response produced in non-aldosterone-treated monolayers (Δ[Ca2+]i without aldosterone = 386 ± 34 nm, n = 62, P < 0.02).
Figure 7. Effect of UTP on [Ca2+]i in 16HBE14o- monolayers.
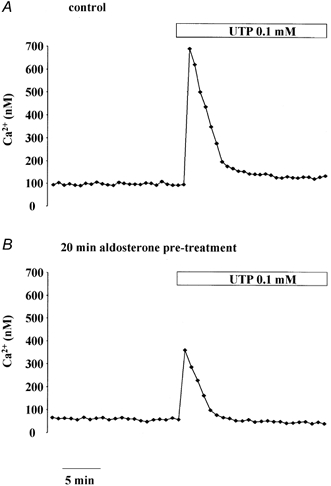
Representative [Ca2+]i recording before and after apical exposure to UTP (0.1 mm) in 16HBE14o- monolayers either not treated (A) or pre-treated for 30 min with aldosterone (0.1 nm, B).
DISCUSSION
In this paper, we report for the first time a rapid and non-genomic effect of aldosterone in human bronchial epithelial cells. Aldosterone induced a reduction in [Ca2+]i at physiological plasma concentrations. The normal human plasma aldosterone level is reported to be 0.2 nm (Al-Dujaili & Edwards, 1978; Vetter et al. 1993) and can increase to 1–5 nm with a low Na+ diet or in pathological cases such as primary hyper-aldosteronism (Vetter et al. 1993; Koren et al. 1997). We verified that the methanol used to dissolve the corticosteroids and the light exposure of the fluorescent dye (photobleaching) were not responsible for the observed [Ca2+]i decrease in this study.
The comparison of aldosterone with glucocorticoid hormone potency indicated a mineralocorticoid specificity for the [Ca2+]i decrease. The role of the type I mineralocorticoid receptor was investigated using spironolactone. The stimulation of a transient [Ca2+]i increase by spironolactone alone was unexpected, but the mechanism involved was not investigated further in this study. However, the insensitivity of the aldosterone-induced [Ca2+]i response to spironolactone and to cycloheximide and the rapidity of the [Ca2+]i response (seconds) strongly indicate that aldosterone does not act via binding to the type I mineralocorticoid receptor, nor does the response involve protein synthesis. The hormone-induced [Ca2+]i regulation probably occurred through a non-genomic mechanism.
A non-genomic effect of aldosterone has already been described in other epithelia such as distal colon (Doolan et al. 1996a; Maguire et al. 1999; Winter et al. 1999), T84 cells (Doolan et al. 1996b), frog skin principal cells (Urbach et al. 1996) and MDCK cells (Gekle et al. 1996). The present paper is the first report of a non-genomic effect of aldosterone in an airway epithelium.
The [Ca2+]i-reducing effect of aldosterone was observed in intact polarized monolayers grown on fibronectin-coated coverslips. Seventy per cent of the cells tested showed an [Ca2+]i response to aldosterone; the remaining cells showed no response. In several non-epithelial tissues, steroids have been reported to also decrease [Ca2+]i. For example, aldosterone has been shown to inhibit the Ca2+ influx in skeletal muscle (Passaquin et al. 1998). 17 β-Oestradiol has been reported to rapidly produce an [Ca2+]i decrease in cardiac myocytes (Jiang et al. 1992) and in coronary arterial smooth muscle (Prakash et al. 1999; Murphy & Khalil, 1999). Corticosteroids cause a decrease in [Ca2+]i in human B lymphoblasts (Gardner & Zhang, 1999), in leukocytes and in airway smooth muscle (Chabra et al. 1999). However, in isolated vascular smooth muscle cells aldosterone has been reported to stimulate a non-genomic increase of [Ca2+]i (Wehling et al. 1994). In epithelial cells such as isolated rat colonic epithelial cells, T84 cells and murine cortical collecting duct cells aldosterone has been described to rapidly increase [Ca2+]i (Doolan et al. 1996a, b; Maguire et al. 1999; Harvey & Higgins, 2000). These heterogeneous responses suggest the involvement of different receptors and signalling pathways in the rapid non-genomic response to aldosterone and other steroid hormones.
In our study, the [Ca2+]i increases observed after replacement of Na+, and sodium orthovanadate and thapsigargin treatment indicate that the activity of a Na+–Ca2+ exchanger and a Ca2+-ATPase contribute to the maintenance of the basal [Ca2+]i concentration. Inhibition of the fast aldosterone effect by thapsigargin strongly suggests that the [Ca2+]i decrease is due to the stimulation of the Ca2+-ATPase pump of the ER. There is no previous report of such an effect of aldosterone on the thapsigargin-sensitive Ca2+-ATPase. However, it has been observed that glucocorticoid treatment stimulates Ca2+-ATPase activity in human smooth muscle and in leukocytes of asthmatic patients where a decrease in the activity of the Ca2+-ATPase was reported (Chabra et al. 1999). The increase of Ca2+-ATPase activity by other steroids has also been described in sarcoplasmic reticulum membrane preparations (Whiting et al. 2000).
Modulation of protein kinase activities has been reported as a major signalling pathway in the non-genomic response to steroids in epithelia cells, but different mechanisms and different types of protein kinases are involved. In distal colon, the non-genomic stimulation of a Na+-H+ exchanger by aldosterone is dependent on protein kinase C (PKC) activity, but not cAMP-dependent protein kinase (PKA) activity (Doolan & Harvey, 1996a; Maguire et al. 1999). However, 17 β-oestradiol stimulates both PKC and PKA activities in rat colonic cells (Doolan et al. 2000) and in pulmonary vascular smooth muscle (Farhat et al. 1996). In 16HBE14o- cells, we have shown that the aldosterone-induced [Ca2+]i decrease is dependent on PKA activity. This observation is consistent with the inhibiton of the aldosterone-induced [Ca2+]i decrease by adenylate cyclase antagonists. In addition, stimulation of cAMP and PKA activity has been reported to reduce [Ca2+]i in pancreatic β cells, where this effect was also sensitive to thapsigargin (Yaekura & Yada, 1998).
The pertussis toxin sensitivity of the aldosterone effect on [Ca2+]i in 16HBE14o- cells is consistent with the stimulation of G-proteins by aldosterone described in other epithelial cells (Ling et al. 1990; Sariban-Sohraby et al. 1995; Gekle et al. 1998). Furthermore, in rat distal colonic crypt, the non-genomic effect of aldosterone on Na+-H+ exchange activity was demonstrated to be pertussis toxin dependent (Winter et al. 1999).
Taken together, our results suggest that a coupling between pertussis toxin-sensitive G-proteins, adenylate cyclase activation and cAMP production, PKA activation and finally ER Ca2+-ATPase stimulation is the signalling pathway involved in the rapid decrease in [Ca2+]i stimulated by aldosterone in human bronchial epithelium.
The physiological role of steroid hormone action in airway epithelial cells is controversial. In canine tracheal epithelium (Cullen & Welsh, 1987) and in frog airway epithelia (Illek et al. 1990), aldosterone has been reported to stimulate amiloride-sensitive Na+ current and also to stimulate the expression of ENaC in rat lung epithelium (Champigny et al. 1994). However, Kunzelmann et al. (1996) reported no correlation between the level of ENaC mRNA and Na+ transport rate in human airway epithelia. More recently, Grubb & Boucher (1998) reported that aldosterone failed to significantly affect the rate of amiloride-sensitive Na+ absorption in murine airway epithelia. They reported that aldosterone failed to significantly affect the rate of amiloride-sensitive Na+ absorption in murine airway epithelia. In addition, it has been recently reported that aldosterone stimulates a serum- and glucocorticoid-dependent kinase (sgk) expression and that co-expression of the epithelial Na+ channel with this kinase increases the amiloride-sensitive current (Chen et al. 1999). However, the rapid stimulation (30 min) of sgk mRNA has so far been detected in rat kidney and colon but not in the lung (Shigaev et al. 2000). These data and our results presented here may indicate that the role of aldosterone in the airway is not primarily to enhance Na+ absorption but rather to limit Cl− secretion and fluid loss. In human airway epithelia, the inhibition of Cl− secretion by aldosterone has been reported (Kunzelmann et al. 1996) and an anti-secretory effect of aldosterone has also been described in other epithelia. In human distal colon and frog skin epithelium, aldosterone stimulates the activity of ATP-sensitive K+ (KATP) channels involved in Na+ absorption (Urbach et al. 1996; Maguire et al. 1999), and in human colon aldosterone additionally inhibits the activity of Ca2+-activated K+ (KCa) channels involved in Cl− secretion (Maguire et al. 1999). In airway epithelia, nucleotides stimulate Cl− secretion via an intracellular Ca2+ increase and activation of a Ca2+-sensitive Cl− channel (Mason et al. 1991). Here, we found that aldosterone inhibited the Cai2+-mobilizing effect of UTP. Because of its potential therapeutic effect as a secretagogue in cystic fibrosis, the effects of UTP on [Ca2+]i and Cl− secretion have been largely studied in human airway epithelial cells (Mason et al. 1991; Stutts et al. 1994). Purino-receptors of type P2Y2 and P2Y6 have been identified in the apical membrane of human airway epithelial cells. The UTP-induced [Ca2+]i responses that we observed here are in agreement with other reports (Parr et al. 1994; Walsh et al. 2000). The antagonistic effect of aldosterone on a Cai2+-mobilizing secretagogue has implications for its use in cystic fibrosis, where steroids are used as an adjunct therapy.
In conclusion, our results provide the first evidence for a rapid, non-genomic reduction of [Ca2+]i induced by aldosterone, in airway epithelia. We postulate that the [Ca2+]i decrease results from stimulation of thapsigargin-sensitive Ca2+ pumps via a G-protein-coupled PKA signalling pathway.
Acknowledgments
We wish to thank N. Helix for her expert technical assistance. This work was funded by the Wellcome Trust (055695/Z/98/Z/ CH/TG/JF). The 16HBE14o- cell line was obtained as a gift from Dr D. C. Gruenert (Human Molecular Genetics, Department of Medicine, University of Vermont, Burlington, VT, USA).
References
- Al-Dujaili EA, Edwards CR. The development and application of a direct radioimmunoassay for plasma aldosterone using 125I-labeled ligand-comparison of three methods. Journal of Clinical Endocrinology Metabolism. 1978;46:105–113. doi: 10.1210/jcem-46-1-105. [DOI] [PubMed] [Google Scholar]
- Ballard PL, Baxter JD, Higgins SJ, Rousseau GG, Tomkins GM. General presence of glucocorticoid receptors in mammalian tissues. Endocrinology. 1974;94:998–1002. doi: 10.1210/endo-94-4-998. [DOI] [PubMed] [Google Scholar]
- Champigny G, Voilley N, Lingueglia E, Friend V, Brabry P, Lazdunski M. Regulation of expression of the lung amiloride sensitive Na+ channel by steroid hormones. EMBO Journal. 1994;13:2177–2181. doi: 10.1002/j.1460-2075.1994.tb06494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SY, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, Firestone GL, Verrey F, Pearce D. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proceedings of the National Academy of Sciences of the USA. 1999;96:2514–2519. doi: 10.1073/pnas.96.5.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabra SK, Khanduja A, Jain D. Decreasing sodium-potassium and calcium adenosine triphosphatase activity in asthma: modulation by inhaled and oral corticosteroids. Indian Journal of Chest Disease and Allied Sciences. 1999;41:15–26. [PubMed] [Google Scholar]
- Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, Finkbeiner WE, Widdicombe JH, Gruenert DC. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. American Journal of Respiratory Cell and Molecular Biology. 1994;10:38–47. doi: 10.1165/ajrcmb.10.1.7507342. [DOI] [PubMed] [Google Scholar]
- Cullen JJ, Welsh MJ. Regulation of sodium absorption by canine tracheal epithelium. Journal of Clinical Investigation. 1987;79:73–79. doi: 10.1172/JCI112811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolan CM, Harvey BJ. Modulation of cytosolic protein kinase C and calcium ion activity by steroid hormones in rat distal colon. Journal of Biological Chemistry. 1996a;271:8763–8767. doi: 10.1074/jbc.271.15.8763. [DOI] [PubMed] [Google Scholar]
- Doolan CM, Harvey BJ. Rapid effects of steroid hormones on free intracellular calcium in T84 colonic epithelial cells. American Journal of Physiology. 1996b;271:C1935–1941. doi: 10.1152/ajpcell.1996.271.6.C1935. [DOI] [PubMed] [Google Scholar]
- Doolan CM, Condliffe SB, Harvey BJ. Rapid non-genomic activation of cytosolic cyclic AMP-dependent protein kinase activity and [Ca2+]i by 17beta-oestradiol in female rat distal colon. British Journal of Pharmacology. 2000;129:1375–1386. doi: 10.1038/sj.bjp.0703193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhat MY, Abi-Younes S, Dingaan B, Vargas R, Ramwell PW. Estradiol increases cyclic adenosine monophosphate in rat pulmonary vascular smooth muscle cells by a nongenomic mechanism. Journal of Pharmacology and Experimental Therapeutics. 1996;276:652–657. [PubMed] [Google Scholar]
- Fisher H, Clauss W. Regulation of Na+ channels in frog lung epithelium: a target tissue for aldosterone action. Pflügers Archiv. 1990;416:62–67. doi: 10.1007/BF00370222. [DOI] [PubMed] [Google Scholar]
- Gardner JP, Zhang L. Glucocorticoid modulation of Ca2+ homeotasis in human B lymphoblasts. Journal of Physiology. 1999;514:385–396. doi: 10.1111/j.1469-7793.1999.385ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekle M, Golenhofen N, Oberleithner H, Silbernag S. Rapid activation of Na+/H+ exchange by aldosterone in renal epithelial cells requires Ca2+ and stimulation of a plasma membrane proton conductance. Proceedings of the National Academy of Sciences of the USA. 1996;93:10500–10504. doi: 10.1073/pnas.93.19.10500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekle M, Silbernagl S, Wunsch S. Non-genomic action of the mineralocorticoid aldosterone on cytosolic sodium in cultured kidney cells. Journal of Physiology. 1998;15:255–263. doi: 10.1111/j.1469-7793.1998.255bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb BR, Boucher RC. Effect of in vivo corticosteroids on Na+ transport across airway epithelia. American Journal of Physiology. 1998;275:C303–308. doi: 10.1152/ajpcell.1998.275.1.C303. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Harvey BJ, Higgins H. Nongenomic effects of aldosterone on Ca2+ in M-1 cortical collecting duct cells. Kidney International. 2000;57:1395–1403. doi: 10.1046/j.1523-1755.2000.00981.x. [DOI] [PubMed] [Google Scholar]
- Jiang C, Poole-Wilson PA, Sarrel PM, Mochizuki S, Collins P, Macleod KT. Effect of 17 beta-oestradiol on contraction, Ca2+ current and intracellular free Ca2+ in guinea-pig isolated cardiac myocytes. British Journal of Pharmacology. 1992;106:739–745. doi: 10.1111/j.1476-5381.1992.tb14403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen PL. Regulation of the (Na+ equals K+)-activated ATP hydrolyzing enzyme system in rat kidney. II. The effect of aldosterone on the activity in kidneys of adrenalectomized rats. Biochimica et Biophysica Acta. 1969;18:326–334. [PubMed] [Google Scholar]
- Koren W, Postnov IY, Postnov YV. Increased Na+/H+ exchange in red blood cells of patients with primary aldosteronism. Hypertension. 1997;29:587–591. doi: 10.1161/01.hyp.29.2.587. [DOI] [PubMed] [Google Scholar]
- Krozowski Z, Funder JW. Mineralocorticoid receptors in the rat lung. Endocrinology. 1981;109:1811–1813. doi: 10.1210/endo-109-6-1811. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Kathofer S, Hipper A, Gruenert DC, Greger R. Culture-dependent expression of Na+ conductances in airway epithelial cells. Pflügers Archiv. 1996;431:578–586. doi: 10.1007/BF02191906. [DOI] [PubMed] [Google Scholar]
- Illek B, Fischer H, Clauss W. Aldosterone regulation of basolateral potassium channels in alveolar epithelium. American Journal of Physiology. 1990;259:L230–237. doi: 10.1152/ajplung.1990.259.4.L230. [DOI] [PubMed] [Google Scholar]
- Ling BN, Kemendy AE, Kokko KE, Hinton CF, Marunaka Y, Eaton DC. Regulation of the amiloride-blockable sodium channel from epithelial tissue. Molecular and Cellular Biochemistry. 1990;99:141–150. doi: 10.1007/BF00230344. [DOI] [PubMed] [Google Scholar]
- Maguire D, MacNamara B, Cuffe JE, Winter D, Doolan CM, Urbach V, O'Sullivan GC, Harvey BJ. Rapid responses to aldosterone in human distal colon. Steroids. 1999;64:51–63. doi: 10.1016/s0039-128x(98)00096-8. [DOI] [PubMed] [Google Scholar]
- Mason SJ, Paradiso AM, Boucher RC. Regulation of transepithelial ion transport and intracellular calcium by extracellular ATP in human normal and cystic fibrosis airway epithelium. British Journal of Pharmacology. 1991;103:1649–1656. doi: 10.1111/j.1476-5381.1991.tb09842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JG, Khalil RA. Decreased [Ca2+]i during inhibition of coronary smooth muscle contraction by 17b-estradiol, progesterone and testosterone. Journal Pharmacology Experimental Theapy. 1999;291:44–52. [PubMed] [Google Scholar]
- Parr CE, Sullivan DM, Paradiso AM, Lazarowski ER, Burch LH, Olsen JC, Erb L, Weisman GA, Boucher RC, Turner JT. Cloning and expression of a human P2U nucleotide receptor, a target for cystic fibrosis pharmacotherapy. Proceedings of the National Academy of Sciences of the USA. 1994;91:3275–3279. doi: 10.1073/pnas.91.8.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passaquin AC, Lhote P, Ruegg UT. Calcium influx inhibition by steroids and analogs in C2C12 skeletal muscle cells. British Journal of Pharmacology. 1998;124:1751–1759. doi: 10.1038/sj.bjp.0702036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash YS, Togaibayeva AA, Kannan MS, Miller VM, Fitzpatrick LA, Sieck GC. Estrogen increases Ca2+ efflux from female porcine coronary arterial smooth muscle. American Journal of Physiology. 1999;276:H926–934. doi: 10.1152/ajpheart.1999.276.3.H926. [DOI] [PubMed] [Google Scholar]
- Sariban-Sohraby S, Mies F, Abramow M, Fisher RS. Aldosterone stimulation of GTP hydrolysis in membranes from renal epithelia. American Journal of Physiology. 1995;268:C557–562. doi: 10.1152/ajpcell.1995.268.3.C557. [DOI] [PubMed] [Google Scholar]
- Shigaev A, Asher C, Latter H, Garty H, Reuveny E. Regulation of sgk by aldosterone and its effects on the epithelial Na+ channel. American Journal of Physiology—Renal Physiology. 2000;278:F613–619. doi: 10.1152/ajprenal.2000.278.4.F613. [DOI] [PubMed] [Google Scholar]
- Stutts MJ, Fitz JG, Paradiso AM, Boucher RC. Multiple modes of regulation of airway epithelial chloride secretion by extracellular ATP. American Journal of Physiology. 1994;267:C1442–1451. doi: 10.1152/ajpcell.1994.267.5.C1442. [DOI] [PubMed] [Google Scholar]
- Tsuchiya K, Wang W, Giebisch G, Welling PA. ATP is a coupling modulator of parallel Na+/K+-ATPase K-channel activity in the renal proximal tubule. Proceedings of the National Academy of Sciences of the USA. 1992;89:6418–6422. doi: 10.1073/pnas.89.14.6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbach V, Van Kerkhove E, Harvey BJ. Inward-rectifier potassium channels in basolateral membranes of frog skin epithelium. Journal of General Physiology. 1994;103:583–604. doi: 10.1085/jgp.103.4.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbach V, Van Kerkhove E, Maguire D, Harvey BJ. Rapid activation of KATP channels by aldosterone in principal cells of frog skin. Journal of Physiology. 1996;491:111–120. doi: 10.1113/jphysiol.1996.sp021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter W, Vetter H, Siegenthaler W. Radioimmunoassay for aldosterone without chromatography. 2. Determination of plasma aldosterone. Acta Endocrinologica. 1993;74:558–567. doi: 10.1530/acta.0.0740558. [DOI] [PubMed] [Google Scholar]
- Voilley N, Lingueglia E, Champigny G, Mattei MG, Waldmann R, Lazdunski M, Barbry P. The lung amiloride-sensitive Na+ channel: biophysical properties, pharmacology, ontogenesis, and molecular cloning. Proceedings of the National Academy of Sciences of the USA. 1994;91:247–251. doi: 10.1073/pnas.91.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D, Harvey BJ, Urbach V. CFTR regulation of intracellular Ca in normal and cystic fibrosis human airway epithelia. Journal of Membrane Biology. 2000;177:209–219. doi: 10.1007/s002320010004. [DOI] [PubMed] [Google Scholar]
- Wehling M, Ulsenheimer A, Schneider M, Neylon C, Christ M. Rapid effects of aldosterone on free intracellular calcium in vascular smooth muscle and endothelial cells: subcellular localization of calcium elevations by single cell imaging. Biochemical and Biophysical Research Communications. 1994;204:475–481. doi: 10.1006/bbrc.1994.2484. [DOI] [PubMed] [Google Scholar]
- Whiting KP, Restall CJ, Brain PF. Steroid hormone-induced effects on membrane fluidity and their potential roles in non-genomic mechanisms. Life Sciences. 2000;67:743–757. doi: 10.1016/s0024-3205(00)00669-x. [DOI] [PubMed] [Google Scholar]
- Winter DC, Schneider MF, O'Sulivan GC, Harvey BJ, Geibel JP. Rapid effects of aldosterone on Na/H exchange in isolated colonic crypts. Journal of Membrane Biology. 1999;170:17–26. doi: 10.1007/s002329900534. [DOI] [PubMed] [Google Scholar]
- Yaekura K, Yada T. [Ca2+]i-reducing action of cAMP in rat pancreatic beta-cells: involvement of thapsigargin-sensitive stores. American Journal of Physiology. 1998;274:C513–521. doi: 10.1152/ajpcell.1998.274.2.C513. [DOI] [PubMed] [Google Scholar]