

Abstract
Neurosteroid modulation of GABAA receptors present on dentate granule cells (DGCs) acutely isolated from epileptic (epileptic DGCs) or control rats (control DGCs) was studied by application of GABA with or without the modulators and by measuring the amplitude of peak whole-cell currents.
In epileptic DGCs, GABA efficacy (1394 ± 277 pA) was greater than in control DGCs (765 ± 38 pA).
Allopregnanolone enhanced GABA-evoked currents less potently in epileptic DGCs (EC50= 92.7 ± 13.4 nm) than in control DGCs (EC50= 12.9 ± 2.3 nm).
Pregnenolone sulfate inhibited GABA-evoked currents with similar potency and efficacy in control and epileptic DGCs.
Diazepam enhanced GABA-evoked currents less potently in epileptic (EC50= 69 ± 14 nm) compared to the control DGCs (EC50= 29.9 ± 5.7 nm).
There were two different patterns of zolpidem modulation of GABAA receptor currents in the epileptic DGCs. In one group, zolpidem enhanced GABAA receptor currents but with reduced potency compared to the control DGCs (EC50= 134 ± 20 nmvs. EC50= 52 ± 13 nm). In the second group of epileptic DGCs zolpidem inhibited GABAA receptor currents, an effect not observed in control DGCs.
Epileptic DGCs were more sensitive to Zn2+ inhibition of GABAA receptor currents (IC50= 19 ± 6 μm) compared to control (IC50= 94.7 ± 7.9 μm).
This study demonstrates significant differences between epileptic and control DGCs. We conclude that (1) diminished sensitivity of GABAA receptors of epileptic DGCs to allopregnanolone can increase susceptibility to seizures; (2) reduced sensitivity to diazepam and zolpidem, and increased sensitivity to Zn2+ indicate that loss of allopregnanolone sensitivity is likely to be due to altered subunit expression of postsynaptic GABAA receptors present on epileptic DGCs; and (3) an inverse effect of zolpidem in some epileptic DGCs demonstrates the heterogeneity of GABAA receptors present on epileptic DGCs.
Temporal lobe epilepsy (TLE) is a common form of drug-refractory epilepsy in which resection of the hippocampus can render a majority of patients seizure-free (Spencer & Spencer, 1994). TLE is frequently accompanied by hippocampal sclerosis, which refers to neuronal loss and gliosis in CA1, CA3 and the hilus, while granule cells in the dentate gyrus are relatively preserved (Sloviter, 1994). It is believed that the principal cells of the dentate gyrus, the dentate granule cells (DGCs), gate propagation of paroxysmal activity through a tri-synaptic circuit in the hippocampus: (1) enthorinal cortex to dentate gyrus, (2) dentate gyrus to CA3 pyramidal cells via mossy fibres, and (3) CA3 to CA1 via Schafer collaterals (Walther et al. 1986; Lambert & Jones, 1989; Jones & Lambert, 1990; Lothman et al. 1992). However, the gating function of dentate gyrus can collapse (Behr et al. 1998) and it can generate robust paroxysmal discharges (Lothman et al. 1991; Lothman et al. 1992; Stringer & Lothman, 1989).
The epileptic dentate gyrus undergoes multiple changes in excitatory and inhibitory neurotransmission that can allow the breakdown of its function. For example, in the synaptic reorganization of the dentate gyrus, axons of DGCs sprout collaterals to the inner molecular layer of the dentate gyrus (Tauck & Nadler, 1985; Sutula et al. 1989; Houser, 1992), and excitatory synapses form between DGCs and sprouted mossy fibres resulting in recurrent excitatory circuits (Wuarin & Dudek, 1996). The potency of NMDA is increased, as are the amplitude and duration of NMDA receptor-mediated EPSPs (Mody & Heinemann, 1987; Mody et al. 1988; Kohr & Mody, 1994).
Plasticity of γ-aminobutyric acid (GABA)-mediated inhibition may also contribute to the altered function of DGCs in TLE. Early studies on animal models of TLE demonstrated increased benzodiazepine binding in the dentate gyrus of kindled rats (Fanelli & McNamara, 1983; Shin et al. 1985) and increased Cl− influx in synaptosomes isolated from dentate gyri of kindled rats (Titulaer et al. 1995a). In electrophysiological studies, increased inhibition in the dentate gyrus was demonstrated by increased paired pulse depression in kindling (de Jonge & Racine, 1987), which persisted for periods of up to 8 weeks following the last seizure (Oliver & Miller, 1985). More recent studies showed that the amplitude of GABAA receptor-mediated miniature inhibitory postsynaptic currents (mIPSCs) is increased in DGCs after kindling (Otis et al. 1994) and was associated with an increased number of GABAA receptors in DGCs in kindled rats (Nusser et al. 1998a). An increased density of GABAA receptors was found in DGCs in the pilocarpine model of TLE in rats (Gibbs et al. 1997), and in neurons isolated from hippocampi of patients with intractable chronic TLE (Brooks-Kayal et al. 1998). Despite enhancement of GABAA receptor currents in DGCs, the gating function of these neurons breaks down during seizures. A proposed mechanism for this breakdown is abnormal modulation of GABAA receptors by zinc (Zn2+). Zn2+ decreased rise time, peak amplitude, and the decay time constant of mIPSCs in DGCs of kindled rats (Buhl et al. 1996). Similarly, in rats with lithium/pilocarpine-induced TLE, Zn2+ inhibits whole-cell GABAA receptor currents in DGCs (Gibbs et al. 1997; Brooks-Kayal et al. 1998).
In addition to Zn2+, the GABAA receptor is modulated by neuroactive steroids, such as allopregnanolone and pregnenolone sulfate (PS) (Macdonald & Olsen, 1994). Neuroactive steroids are a group of compounds synthesized in the brain by glial cells from circulating steroid hormones (progesterone, oestrogen, testosterone, and cortisone) or de novo from cholesterol (Compagnone & Mellon, 2000). One of the most potent endogenous modulators of the GABAA receptor function is the tertahydro-derivative of progesterone, 3α-hydroxy-5α-pregnane-20-one (3α,5α-THP, or allopregnanolone) (Lambert et al. 1996). Physiological, nanomolar concentrations of allopregnanolone have an anticonvulsant effect (Belelli et al. 1989; Kokate et al. 1999a). PS is a natural sulfated derivative of pregnenolone. In contrast to allopregnanolone, PS potently inhibits GABAA receptors (Majewska et al. 1988). The potential physiological significance of PS was demonstrated in studies that assessed the spatial memory performance of cognitively impaired, aged rats. In these studies, cognitive impairment was correlated by bilateral intra-hippocampal injection of PS (Vallee et al. 1997). The neurosteroid modulation of GABAA receptors on dentate granule cells of naive and epileptic rats has not yet been studied and is the focus of the current study.
METHODS
Induction of TLE
All experimental procedures on rats were performed according to the protocol approved by the University of Virginia Animal Use and Care Committee. The method for inducing TLE in rats has been described in detail in the past (Lothman et al. 1989). Briefly, rats were anaesthetized with ketamine (50 mg kg−1) and xylazine (40 mg kg−1) and implanted with a pair of bipolar stimulating electrodes in the left posterior ventral hippocampus (AP 3.6, ML 4.0, DV 5.0 from dura; incisor bar at +5.0). After 1 week of recovery, the left hippocampus was stimulated with 10 s trains of 50 Hz, 1 ms, 400 mA biphasic square wave current pulses delivered every 13 s for 90 min to induce status epilepticus (SE) (Lothman et al. 1989). Approximately 4-6 weeks after the stimulation, the rats developed spontaneous limbic seizures with the motor component. For this study, seizures were documented either by continuous EEG recording or by direct observation of the spontaneous seizure (Bertram et al. 1997). The epileptic animals were killed at least 24 h after the last seizure, and DGCs were isolated from these rats. The responses from stimulated but non-epileptic rats were no different from age-matched naive rats so the data from these animals were pooled. All animals were housed individually under conditions of a 12 h:12 h light-dark cycle and had free access to food and water.
Acute isolation of DGCs
DGCs were isolated according to the protocol of Kay & Wong (1986), later modified (Kapur & Coulter, 1995). Rats were decapitated under halothane anaesthesia, and the brains were dissected free and chilled to 4 °C for 1 min in a piperazine-N,N‘-bis(2-ethanesulphonic acid) (Pipes) buffer solution composed of (mm): NaCl 120, KCL 5, CaCl2 1.5, MgCl2 1, d-glucose 25, Pipes 20 (pH 7.0) (all chemicals from Sigma, St Louis, MO, USA, unless noted otherwise). The region containing the hippocampus was blocked. The brain was blot-dried, mounted on a vibratome (Camden Instruments, UK) stage with the caudal part facing up, and 400-500 μm coronal slices containing hippocampus were cut. The slices were allowed to recover for at least 1 h in Pipes solution before being transferred to an enzyme solution. Slices were incubated in continuously oxygenated Pipes solution, containing 3 mg ml−1 type XXIII protease enzyme from Aspergillus oryzae, at 32 °C for 45 min.. The enzyme incubation was terminated by transferring the slice into enzyme-free Pipes solution, and after a minimum of 30 min of recovery, DGCs were isolated from caudal parts of both left and right ventral hippocampi. The dentate gyrus was dissected from the rest of the slice under a microscope and cut into 0.5 mm3 chunks. These chunks were triturated through 0.5 mm and 0.3 mm diameter fire-polished glass pipettes and the isolated neurons were plated on 35 mm plastic Petri dishes. DGCs were identified by their typical oval shape and single process. DGCs acutely isolated from epileptic rats were referred to as ‘epileptic DGCs’ and those isolated from control animals were referred to as ‘control DGCs’.
Whole-cell patch clamp recordings of GABAA receptor currents
Thin-walled borosilicate patch electrodes of 1.5 mm external diameter (Sutter Instruments, Novato, CA, USA) were pulled on a horizontal Flaming Brown P-97 model puller (Sutter Instruments) using a 2-stage pull to a final resistance of 6-8 MΩ. Patch electrodes were filled with recording solution containing (mm): Trizma phosphate (dibasic) 110, Trizma base 28, EGTA 11, MgCl2 2, CaCl2 0.5; pH 7.40; the osmolarity was 270-275 mosmol l−1. ATP disodium salt, in a final concentration of 3 mm, was included in the intracellular solution before recording. The extracellular medium contained (mm): NaCl 155, KCl 3, MgCl2 1, CaCl2 3 and Hepes-Na+ 10; pH 7.4. The osmolarity was 314-322 mosmol l−1. Whole-cell currents were recorded at room temperature with an Axopatch 200A amplifier (Axon Instruments) and low-pass filtered at 2 kHz with an 8 pole Bessel filter prior to digitization, storage and display using standard patch clamp techniques (Hamill et al. 1981). Acutely isolated neurons were studied on the stage of an inverted Nikon microscope. Cell capacitance was compensated and the capacitance value was obtained from the potentiometer on the amplifier. Currents were displayed on-line on a Gould Viper TA11 digital chart recorder. Additionally, currents were recorded and stored on a Pentium II personal computer using the Axoscope 7.0 program (Axon Instruments) and digitized at 400 Hz. Peak currents were measured manually from the computer recordings and confirmed by chart paper recording.
Allopregnanolone (Tocris, Ballwin, MO, USA), PS, zolpidem (Research Biochemicals, Natick, MA, USA), and diazepam were dissolved in dimethysulphoxide (DMSO) and stock solutions were stored at -20 °C. On the day of the experiment, stock solutions of drugs were diluted in extracellular solution. ZnCl2 (Fluka, Buchs, Switzerland) was first dissolved in distilled water at a concentration of 50 mm and then diluted in the extracellular solution to a final concentration ranging from 1 μm to 1 mm. GABA was dissolved in the extracellular solution.
Drugs were applied to the DGCs with a modified U-tube ‘multipuffer’ application system (Bormann, 1992; Greenfield & Macdonald, 1996) with the tip of the application pipette placed 100-200 μm from the neuron. Data from each DGC were fitted to a four-parameter logistic equation (equation for a sigmoid curve):
where I is the peak GABAR current at a given GABA concentration. The Hill slope, EC50, and Imax (maximal current) were derived from the equation that best fitted the observed data by the least square fit method using a curve fitting program (Graphpad Prism 3.0) on an IBM PC-compatible computer. All values are reported as means ±s.e.m.
RESULTS
Increased efficacy of GABA in the epileptic DGCs
Whole-cell GABA-evoked currents were recorded from control and epileptic DGCs. GABA at 10 μm was applied to the DGC repeatedly, 3-5 times, and an increase in peak amplitude (run-up) occurred during the first several minutes of recordings, possibly due to the adjustment of the cell to the ATP load. To avoid the confounding effect of run-up, data collection was started after stable responses to GABA were achieved. Control and epileptic DGCs had similar capacitances (12-15 pF). GABA at 30 μm elicited significantly larger currents in the epileptic DGCs (838 ± 179 pA, n = 5,) than in control DGCs (443 ± 27 pA, n = 6, P = 0.015, Student's unpaired t test). In order to understand the mechanism of larger currents in epileptic DGCs, the GABA concentration response relationship was studied by applying multiple concentrations of GABA (1 μm to 1 mm) to epileptic and control DGCs. The amplitudes of peak currents recorded in response to each concentration of GABA were recorded, averaged and fitted to an equation for sigmoidal function (see Methods). The potency (EC50) and efficacy (maximum current elicited) were derived from the equation with the best fit to the sigmoidal function in control and epileptic DGCs. EC50 of GABA in the epileptic DGCs was 32.6 ± 6 μm (n = 5), similar to that in the control DGCs -29.2 ± 4 μm, (n = 6, P = 0.72, unpaired t test; Fig. 1 and Table 1). However, the maximal GABAA receptor current recorded in epileptic DGCs, 1394 ± 277 pA, was larger than in control DGCs, 765 ± 38 pA (Fig. 1 and Table 1). In epileptic DGCs, the Hill coefficient was 0.8 ± 0.57 (n = 5), and in control DGCs it was 1.05 ± 0.17 (n = 6). The difference was not statistically significant (P = 0.04, unpaired t test; Table 2). This increased GABA efficacy in epileptic DGCs is most likely to reflect an increased density of GABAA receptors on epileptic DGCs, since the capacitance (and hence cell surface area) of control and epileptic DGCs was similar (12-15 pF).
Figure 1. GABA-elicited currents in control and epileptic DGCs.
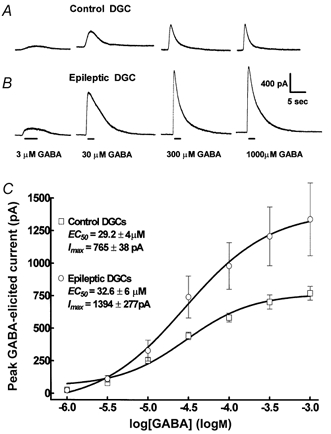
A, in a control DGC, increasing concentrations of GABA (1 μm to 1 mm) elicited currents with increasing amplitude of peak current. B, the same concentrations of GABA evoked larger currents in an epileptic DGC then in control. In this and all subsequent figures concentration of the drug is indicated at the bottom of the traces. Duration of the drug application (bars) is indicated below the traces. C, concentration of GABA (log[GABA]) and GABA-elicited peak current (pA) relationships were plotted for control (□, n = 6) and epileptic (○, n = 5) DGCs. In this and all subsequent figures, each point represents the mean of the amplitude of peak current for a given concentration of GABA, and error bars show s.e.m. The data were fitted to the equation of a sigmoid curve (see Methods) and the lines indicate the best fit to the data. EC50 and the Hill coefficients were derived from the equation of the sigmoidal function that best fitted the data.
Table 1.
Potency of allosteric modulators of GABAA receptor in control and epileptic DGCs
| Drug | Epileptic DGCs | Control DGCs |
|---|---|---|
| GABA | 32.6 ± 6.1 μM | 29.2 ± 4.3 μM |
| Allopregnanolone | 92.7 ± 13.4 nM* | 12.9 ± 2.3 nM |
| PS | 17.4 ± 2 μM | 13.1 ± 1.6 μM |
| Diazepam | 69 ± 14 nM* | 29.9 ± 5.7 nM |
| Zolpidem | 93.7 ± 6.5 nM* | 44.2 ± 8.6 nM |
| Zn2+ | 19 ± 6 μM* | 94.7 ± 7.9 μM |
Values are means ±s.e.m.
P < 0.05.
Table 2.
Hill coefficients derived from the equation that best fitted the data
| Drug | Epileptic DGCs | Control DGCs |
|---|---|---|
| GABA | 0.8 ± 0.57 | 1.05 ± 0.17 |
| Allopregnanolone | 1.39 ± 0.24 | 1.33 ± 0.35 |
| PS | 0.64 ± 0.07 | 0.93 ± 0.23 |
| Diazepam | 1.2 ± 0.2 | 1.4 ± 0.24 |
| Zolpidem | 0.99 ± 0.55 | 1.4 ± 0.6 |
| Zn2+ | −0.9 ± 0.35 | −0.7 ± 0.22 |
Values are means ±s.e.m. The Hill slopes of the fits for GABA and various allosteric modulators of GABAA receptor were not significantly different.
Allopregnanolone enhancement of GABAA receptor currents
Allopregnanolone enhancement of GABAA receptor currents in control and epileptic DGCs was studied. In studies of augmentation of GABA responses by positive allosteric modulators such as allopregnanolone, 10 μm GABA was used to elicit control responses, since this concentration was 0.5 log units less than the EC50 in control and epileptic DGCs, and allowed discrimination of peak current enhancement. Allopregnanolone (10 nm) enhancement of 10 μm GABA-evoked currents in epileptic DGCs was significantly diminished (12.3 ± 3 %, n = 5), compared to that in control DGCs, (52.3 ± 6 %, n = 6, P = 0.001, unpaired t test; Fig. 2A and B, and Table 1).
Figure 2. Allopregnanolone enhancement of GABA-elicited currents.
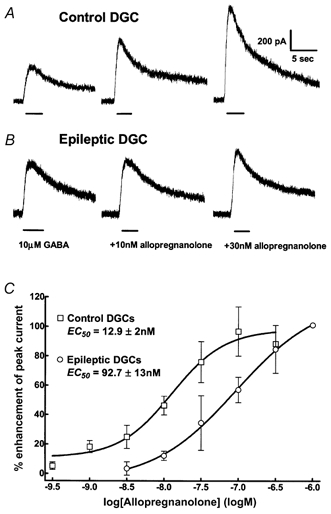
A, in a control DGC, current elicited by 10 μm GABA compared to that elicited by co-application of 10 and 30 nm allopregnanolone with GABA. The peak current elicited by GABA was robustly enhanced by 10 and 30 nm allopregnanolone. B, in contrast, the same concentrations of allopregnanolone poorly enhanced GABAA receptor currents in an epileptic DGC. C, allopregnanolone concentration-response curves were obtained for control (□, n = 6) and epileptic DGCs (○, n = 5). Note that in epileptic DGCs the curve shifted to the right.
A detailed characterization of allopregnanolone enhancement of GABAA receptor-mediated currents was studied in control and epileptic DGCs by co-applying multiple concentrations (300 pm to 1 μm) of allopregnanolone with 10 μm GABA. The peak amplitude of currents elicited by application of 10 μm GABA alone was the control response and augmentation of current was measured as the percentage increase above control response. In order to avoid the effects of possible run-down on the actual modulation of GABA currents, GABA was applied before and after the application of each concentration of allopregnanolone. The amplitude of the current elicited by co-application of a given concentration of allopregnenolone and 10 μm GABA was compared to the mean current amplitude of those two GABA-elicited currents. When the amplitude of a current elicited by the second application of GABA was smaller than 90 % of that elicited by the first application, the recording was excluded from further analysis.
In epileptic DGCs, there was a significant right shift of the allopregnanolone concentration-response curve. The potency (EC50) of allopregnanolone enhancement of GABA-evoked currents was markedly diminished in epileptic DGCs (92.7 ± 13 nm, n = 5) compared to that in control DGCs (12.9 ± 2 nm, n = 6, P = 0.002, t test; Fig. 2 and Table 1). In the epileptic DGCs, the Hill coefficient was 1.39 ± 0.24, similar to that in control DGCs, 1.33 ± 0.35 (Table 2). Allopregnanolone efficacy was similar in control and epileptic DGCs; the maximal augmentation of GABA current by allopregnanolone was 96 ± 13.4 % in epileptic DGCs and 106.5 ± 10.8 % in control DGCs (n = 5. P = 0.56, unpaired t test).
PS inhibition of GABAA receptor currents
PS modulation of GABAA receptor currents was compared in epileptic and control DGCs. In epileptic DGCs, PS (30 μm) inhibited 30 μm GABA-evoked currents to 24.4 ± 5 % of that evoked by 30 μm GABA alone (n = 10). In control DGCs, peak current amplitude evoked by co-application of 30 μm PS and 30 μm GABA was 27.4 ± 4 % (n = 9) of that evoked by 30 μm GABA alone. There was no difference in inhibition of GABA current by 30 μm PS in epileptic and control DGCs (P = 0.64, unpaired t test). The PS concentration (300 nm to 300 μm)-GABAA receptor current inhibition relationship was studied further in epileptic and control DGCs. The potency and efficacy of PS inhibition of GABAA receptor currents were also similar in epileptic and control DGCs. In epileptic DGCs, the IC50 for PS was 17.4 ± 2 μm (n = 10) and in control DGCs it was 13.1 ± 1 μm (n = 14; Fig. 3 and Table 1). The maximal inhibition of GABA currents by PS was similar in epileptic DGCs (7.2 ± 0.7 %, n = 7) and control DGCs (7.6 ± 1.9 %, n = 6, P = 0.82, unpaired t test; Fig. 3). The Hill coefficient of the fit was 0.93 ± 0.23 in control DGCs, and 0.64 ± 0.07 (n = 10, P = 0.72; Table 2) in epileptic DGCs. Therefore, neither the potency nor the efficacy of PS inhibition of GABAA receptor-mediated currents is altered in DGCs in chronic temporal lobe epilepsy.
Figure 3. Inhibition of GABA-elicited currents by PS.
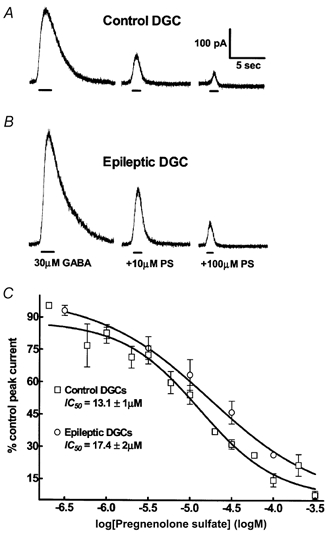
A, in a control DGC, current elicited by 30 μm GABA compared to that elicited by co-application of 10 and 100 μm pregnenolone sulfate (PS) and GABA. The peak current elicited by GABA was strongly inhibited by 30 and 100 μm PS. B, in an epileptic DGC, inhibition of GABA currents by the same concentrations of PS was not different from the control cell. C, PS concentration-DGC GABAA receptor current relationships. PS concentration- response curves were obtained for control (□, n = 14) and epileptic DGCs (○, n = 10).
Diazepam enhancement of GABAA receptor currents
One possible mechanism of diminished allopregnanolone sensitivity of GABAA receptors in epileptic DGCs could be the altered subunit composition of the receptor. Allosteric modulation of GABA receptor function by allopregnanolone is markedly decreased when the α4 (Smith et al. 1998a) or δ subunits are expressed (Zhu et al. 1996). Both α4 and δ subunits are known to reduce diazepam sensitivity of GABAA receptors (Saxena & Macdonald, 1994). Diazepam (30 nm) enhanced 10 μm GABA-evoked currents by 19.9 ± 3.9 % (n = 8) in epileptic DGCs (Fig. 4) and by 32 ± 2.4 % (n = 8, P = 0.014, t test) in control DGCs. A detailed diazepam concentration (3 nm to 1 μm)-response relationship was studied to further characterize this difference in diazepam effect in control and epileptic DGCs. The EC50 for diazepam enhancement of GABA-evoked currents in epileptic DGCs was 69 ± 14 nm (n = 9) and 29.9 ± 5.7 nm in control DGCs (n = 8, P = 0.04, t test; Fig. 4 and Table 1). The Hill coefficient remained unchanged, 1.4 ± 0.24 versus 1.2 ± 0.2 (Table 2). The maximal diazepam-induced enhancement of 10 μm GABA-elicited currents was similar in epileptic (61.2 ± 7 %, n = 9) and control DGCs (63.6 ± 3 %, n = 8, P = 0.77, paired t test). Therefore, the diazepam concentration response-curve is shifted to the right in epileptic DGCs compared to controls.
Figure 4. Diazepam enhancement of 10 μm GABA-elicited currents.
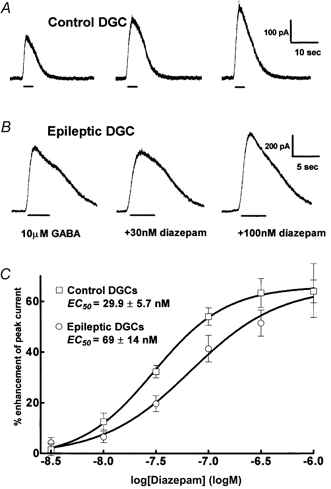
A, in a control DGC co-application of 30 and 100 nm diazepam with 10 μm GABA strongly enhanced 10 μm GABA-elicited currents. B, in an epileptic DGC, 30 nm diazepam did not enhance GABA-elicited current, and 100 nm diazepam was less effective than in the control cell. C, diazepam concentration-DGC GABAA receptor current relationships. Diazepam concentration- response curves were obtained for control (□, n = 8) and epileptic DGCs (○, n = 9). The maximal enhancement of GABAA receptor currents remained similar for control and epileptic cells.
Zolpidem modulation of GABAA receptor function in the epileptic DGCs
Zolpidem is a subunit-selective benzodiazepine type I receptor agonist with high affinity for α1-subunit-containing GABAA receptors. The α1 subunit-containing GABAA receptors are robustly enhanced by allopregnanolone (Shingai et al. 1991), but suppression of the subunit diminishes allopregnanolone sensitivity (Brussaard et al. 1997). In this study, we identified two different patterns of zolpidem modulation of GABAA receptor function in epileptic DGCs. In one group of epileptic DGCs, zolpidem (100 nm) enhanced 10 μm GABA-evoked currents by 16.2 ± 10 % (n = 5), while in control DGCs, the enhancement was 54.1 ± 9.8 %, (n = 5, P = 0.02, t test). In the second group of epileptic DGCs (n = 3), 100 nm zolpidem inhibited 10 μm GABA-evoked currents by 8.1 ± 3.3 %. It is important to note that epileptic DGCs, in which zolpidem inhibited GABAA receptor currents, were isolated from the same animals in which zolpidem enhanced GABA-evoked currents. Zolpidem did not inhibit GABA-evoked currents in any control DGCs tested. Each group of epileptic DGCs was evaluated in detail.
In a group of eight epileptic DGCs, the relationship between zolpidem concentration (3 nm to 1 μm) and the enhancement of GABAA receptor currents was evaluated. In these epileptic DGCs, zolpidem enhanced GABA currents with lower potency (EC50= 134 ± 20 nm) than in control DGCs (EC50= 52.2 ± 12.6 nm, n = 6, P = 0.007, t test; Fig. 5 and Table 1). The Hill coefficient was 0.99 ± 0.55 in epileptic DGCs and it was 1.4 ± 0.6 (n = 8) in control DGCs (Table 2). In these epileptic DGCs, maximal enhancement of 10 μm GABA-evoked currents by zolpidem was 61.3 ± 12.4 %, similar to that in the control DGCs (66.9 ± 9 %, n = 6, P = 0.64, t test). In this group of epileptic DGCs, the zolpidem concentration- response curve was shifted to the right compared to control DGCs.
Figure 5. Heterogeneity of zolpidem responses in epileptic DGCs.
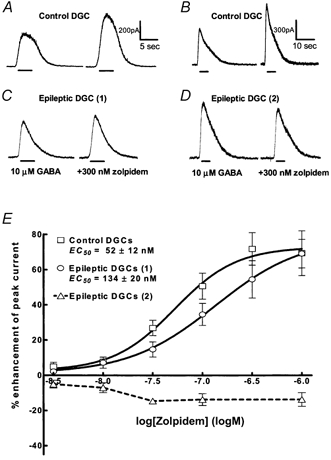
A and B, in the two control DGCs co-application of 10 μm GABA with 300 nm zolpidem robustly enhanced 10 μm GABA-elicited current. C, in one group of epileptic DGCs 300 nm zolpidem enhancement of 10 μm GABA-elicited current was reduced compared to control DGCs. D, in a group of epileptic DGCs 300 nm zolpidem inhibited 10 μm GABA-elicited current, as demonstrated by the current traces. E, zolpidem concentration-DGC GABAA receptor current relationships. Zolpidem concentration-response curves were obtained for control (□, n = 7) and epileptic DGCs. Note that in one population of epileptic DGCs (○, n = 7) zolpidem enhanced GABAA receptor currents, whereas in the other population (▵, dashed line, n = 8) zolpidem inhibited GABA currents.
Zolpidem inhibition of GABA currents in the second group of epileptic DGCs was also characterized further (n = 8). Zolpidem inhibition occurred at 10 nm (9.4 ± 1.7 %) and increased insignificantly at 1 μm (14.6 ± 2.1 %, P = 0.69). These results indicated that zolpidem inhibited GABAA receptor currents in the second group of epileptic DGCs. This pattern of zolpidem modulation is qualitatively different from that found in control and other epileptic DGCs since zolpidem consistently enhanced GABAA receptor currents in these neurons.
Zn2+ modulation of GABAA receptor currents in epileptic DGCs
Increased Zn2+ inhibition of GABA-evoked currents in DGCs was reported in the kindling model (Buhl et al. 1996), in the lithium/pilocarpine model of epilepsy (Gibbs et al. 1997) and in DGCs isolated from hippocampi of patients with intractable TLE (Brooks-Kayal et al. 1998; Shumate et al. 1998). Zn2+ (30 μm) inhibited peak amplitudes of 30 μm GABA-elicited currents more profoundly in epileptic DGCs (44.2 ± 3 %, n = 6) compared to control DGCs (32.2 ± 3.3 %, n = 7, P = 0.024, t test; Fig. 6). The Zn2+ concentration (1 μm to 1 mm)-GABAA receptor current inhibition relationship was studied in epileptic and control DGCs. In control, the IC50 was 94.7 ± 7.9 μm, and the Hill coefficient was -0.7 ± 0.2 (n = 11; Table 2); in epileptic DGCs, the IC50 was 19 ± 6 μm, and the Hill coefficient was -0.9 ± 0.35 (n = 9, P = 0.02, t test). These results indicated that the potency of Zn2+ in inhibiting GABAA receptor currents was increased in epileptic DGCs. Maximal inhibition of GABAA receptor currents by ZnCl2 tended to be higher in epileptic DGCs compared to control DGCs. However, the difference did not reach a statistically significant value. In control DGCs, 300 μm ZnCl2 inhibited GABA currents by 67.7 ± 2.6 % (n = 5). In epileptic DGCs, the same concentration of ZnCl2 inhibited GABA currents by 77.5 ± 2.6 % (n = 5, P = 0.08, t test).
Figure 6. Zn2+ inhibition of GABA-elicited currents.
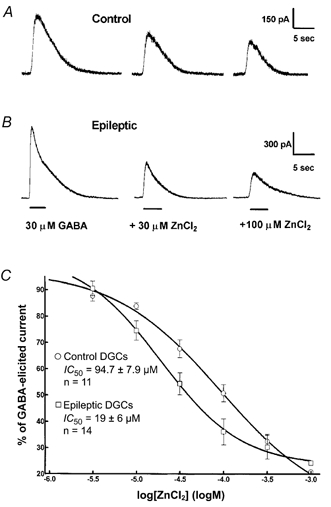
A, co-application of 30 and 100 μm Zn2+ with 30 μm GABA caused inhibition of 30 μm GABA-elicited currents in a control DGC, B, inhibition was more profound in an epileptic DGC. C, Zn2+ concentration- DGC GABAA receptor current relationships. Concentration-response curves were obtained for control (○, n = 11) and epileptic DGCs (□, n = 14). Note the increased potency of Zn2+ in inhibiting of GABAA receptor currents, evoked in epileptic DGCs. The maximal enhancement of GABAA receptor currents remained similar for control and epileptic cells.
DISCUSSION
This study demonstrated that (1) the efficacy of GABA is increased in epileptic DGCs compared to the control DGCs; (2) the sensitivity of GABAA receptors to allopregnanolone enhancement is markedly reduced in epileptic DGCs, whereas PS-mediated inhibition of GABAA receptor currents remains unchanged; (3) the sensitivity to diazepam enhancement of GABAA receptor currents is diminished in epileptic DGCs compared to controls; (4) there are two groups of epileptic DGCs with respect to modulation of GABAA receptors by zolpidem: in one there was decreased augmentation of GABA currents by zolpidem compared to the control, whereas in the other zolpidem inhibited GABAA receptor currents; and (5) Zn2+ inhibition of GABAA receptors is increased in epileptic DGCs.
We studied the properties of GABAA receptors on epileptic DGCs studied in acutely isolated neurons. Acute isolation of neurons has certain advantages and limitations. A clear benefit is that detailed concentration- response relationships and the potency and efficacy of various drugs can be studied in individual neurons. A drawback is that enzymatic treatment followed by mechanical isolation of differentiated neurons may damage synaptic elements, which are better preserved in slice preparations. However, some key findings made in slice preparations, such as increased GABAA receptor currents (Nusser et al. 1998a) and increased Zn2+ sensitivity of GABAA receptor-mediated currents in the dentate gyrus in kindling (Buhl et al. 1996) have been replicated in acutely isolated neurons (Gibbs et al. 1997; Shumate et al. 1998) It is therefore likely that the findings regarding neurosteroids would be valid in a more intact slice preparation.
Acute isolation of neurons may shift the ratio of extrasynaptic to synaptic receptors present on an acutely isolated cell towards more extrasynaptic receptors. In cerebellar granule cells, extrasynaptic receptors, which mainly mediate tonic inhibition, contain the δ subunit. (Nusser et al. 1998b). Since the δ subunit may alter sensitivity of GABAA receptors to neuroactive steroids (Zhu et al. 1996), it is possible that the diminished allopregnanolone sensitivity observed in the current study is due to a shift in the ratio of extrasynaptic to synaptic receptors. However, since the protocol for the cell isolation was identical for both control and epileptic dentate gyri, it appears unlikely that the procedure of isolation would have resulted in a group-specific altered proportion of GABAA receptors. Clearly, it is important to localize specific receptor subunits in epileptic DGCs and that is the subject of a separate ongoing study.
The augmentation and inhibition of GABAA receptor currents were studied by co-application of drugs with 10 and 30 μm GABA, respectively, because we intended to detect differences in the affinity and potency of the drug between control and epileptic cells. These concentrations are useful for studying the pharmacological modulation of GABAA receptors but do not mimic the GABA concentration in the synaptic cleft.
Loss of allopregnanolone sensitivity may increase susceptibility to seizures
This study demonstrated that GABAA receptor currents are less sensitive to allopregnanolone enhancement in epileptic DGCs compared to control DGCs. Allopregnanolone is an endogenous anticonvulsant (Belelli et al. 1989; Kokate et al. 1999a) that potentiates the antiseizure effect of flurazepam in mice (Deutsch et al. 1996) and its intraperitoneal injection evoked sleep in a manner similar to that of benzodiazepines (Lancel & Faulhaber, 1996). Intraperitoneal injections of allopregnanolone significantly increased the dose of NMDA necessary to induce seizures in rats (Budziszewska et al. 1998) and subcutaneous injections of allopregnanolone increased the latency of kainic acid-induced seizures (Frye & Scalise, 2000). This study also found that the potency and efficacy of PS remained unchanged. PS is a physiologically active sulfated derivative of pregnenolone. Endogenous PS in the hippocampus is believed to enhance memory, and age-related decline in memory function of rats correlates well with decline in hippocampal PS levels. A number of studies have shown that the physiological action of PS is reciprocal to that of allopregnanolone. PS has convulsant properties (Kokate et al. 1999b), inhibits GABAA receptor currents (Majewska et al. 1988), enhances NMDA currents (Bowlby, 1993), and exacerbates NMDA-induced death of cultured hippocampal neurons (Weaver et al. 1998). Sustained inhibition of GABA currents by PS, in parallel with its ability to enhance NMDA-mediated currents, could shift an excitatory/ inhibitory balance toward increased excitation. When augmentation of GABAA receptor-mediated currents by allopregnanolone is diminished in epileptic DGCs, this increased excitation can trigger a seizure.
Neurosteroid modulation of GABAA receptors may be of particular relevance in the catamenial exacerbation of seizures. Catamenial exacerbation of epilepsy is characterized by recurrent increased frequency of seizures at specific times during the menstrual cycle in association with rising and falling levels of progesterone and oestrogen. Oestrogen is known to be proconvulsant; progesterone is anticonvulsant (for review, see Herzog, 1999). Progesterone exerts its anticonvulsant effects largely by its conversion to allopregnanolone. A blockade of the enzymatic conversion of progesterone to allopregnanolone by finasteride resulted in a loss of the anticonvulsant properties of progesterone against pentilentetrazolium-induced seizures in mice (Kokate et al. 1999a). It was suggested that the catamenial exacerbation of seizures was due to rapid withdrawal from allopregnanolone, which was the result of the rapid decline of serum levels of progesterone prior to menses (Reddy & Rogawski, 2000). Treatment of pseudopregnant rats with the 5α reductase inhibitor finateride, which blocks synthesis of allopregnanolone from progesterone, resulted in increased susceptibility to pentylentetrazol-induced seizures (Reddy et al. 2001). The findings of the current study suggest that, in addition to changes in sex hormone concentrations during the menstrual cycle, loss of allopregnanolone enhancement of GABAA receptor function may contribute to the catamenial exacerbation of seizures.
Diazepam modulation
The present study found that the potency of diazepam enhancement of GABAA receptor currents was diminished in epileptic DGCs compared to that in controls. The efficacy remained unchanged. Diazepam and clonazepam are ‘broad spectrum’ benzodiazepines. That is, they bind to both BZ type 1 and BZ type 2 benzodiazepine receptors. In contrast to the present study, Gibbs et al. (1997) found that 100 nm clonazepam enhanced 10 μm GABA-evoked currents more robustly in epileptic DGCs compared to control DGCs. It is difficult to compare the results of the two studies further because it is unclear whether the potency or efficacy of clonazepam is increased in the lithium/pilocarpine model. In addition to chronic TLE, there are several other conditions where the brain is more susceptible to seizures, commonly referred to as hyper-excitable states. Withdrawal from alcohol is known to increase seizure susceptibility in humans and in experimental animals. A specific diazepam-insensitive GABAA receptor is expressed in hippocampal neurons during alcohol withdrawal (Mhatre et al. 1988; Mhatre & Ticku, 1989). Increased seizure activity occurs in epileptic women during the premenstrual period presumably due to physiological withdrawal of progesterone (Smith et al. 1998a) and hippocampal GABAA receptors are rendered diazepam insensitive during progesterone withdrawal (Smith et al. 1998b). Immature brains are more susceptible to seizures than adult brains and GABAA receptors present on DGCs of neonatal rats are insensitive to diazepam (Kapur & Macdonald, 1999). Finally, during prolonged seizures, GABAA receptors expressed by DGCs rapidly rendered diazepam insensitive (Kapur & Macdonald, 1997). This benzodiazepine insensitivity may lower seizure threshold since there is some evidence for the existence of an endogenous benzodiazepine agonist in the brain (Rothstein et al. 1992a, b).
Zolpidem modulation
There were two populations of epileptic DGCs. In one, zolpidem augmented GABA-evoked currents, but less than in control DGCs, whereas in the other zolpidem inhibited GABAA receptor currents. Zolpidem never inhibited GABAA receptor currents in control DGCs. The findings in the first group of DGCs were similar to those of Gibbs et al. (1997) and Brooks-Kayal et al. (1998) in lithium/pilocarpine-induced TLE. In epileptic DGCs, augmentation of GABAA receptor currents by 100 nm zolpidem was significantly diminished compared to the controls (Gibbs et al. 1997).
Zolpidem inhibited GABAA receptor currents in one population of epileptic DGCs, whereas it uniformly augmented GABA currents in control cells. While zolpidem-insensitive receptors occur in CA1 pyramidal neurons, such receptors have not been described in DGCs in the past. This finding suggested a functional heterogeneity of GABAA receptors present on epileptic DGCs. One of the factors mediating this functional heterogeneity of GABAA receptors may be increased neurogenesis in DGCs in epilepsy. Compared with other hippocampal areas, the unique property of DGCs is that they undergo prolonged postnatal neurogenesis (Altman & Das, 1966). Seizures in the pilocarpine model of TLE, as well as in perforant path stimulation, produce a marked increase in DGC proliferation and subsequent migration (Parent et al. 1997). This phenomenon might contribute to an increased number of immature DGCs in epilepsy associated with the functional properties of GABAA receptors found in DGCs in neonatal animals. Neonatal DGCs have pharmacological properties similar to those in adult epilepsy including insensitivity to diazepam (Kapur & Macdonald, 1999).
Increased number of GABAA receptors
The finding that GABA elicited larger currents in DGCs isolated from epileptic animals, whereas its potency remained the same as in the control cells, was likely to be due to the increased number of GABAA receptors on epileptic DGCs. In the present study, it is conceivable that due to the relatively slow rate of drug application by the U tube device (10-90 rise time of 50 ms; Greenfield & Macdonald, 1996) a certain fraction of GABAA receptors was desensitized before the current peaked. Low concentrations of GABA cause pronounced desensitization of GABAA receptors (Celentano & Wong, 1994) and reduce availability of GABAA receptors (Overstreet et al. 2000) thereby reducing the total number of receptors contributing to the whole-cell response. However, since the same application system was used in control and epileptic DGCs, peak GABAA receptor currents would be underestimated with similar ratios in both preparations.
Several previous studies using other experimental models of epilepsy have demonstrated increased GABA-mediated inhibition in the dentate gyrus. Paired pulse inhibition, a measure of recurrent inhibition, increasesd in dentate gyrus in the kindling model (Tuff et al. 1983; de Jonge & Racine, 1987) and persisted for periods of up to 8 weeks following the last seizure (Oliver & Miller, 1985). Receptor binding studies demonstrated increased benzodiazepine binding to synaptosomes isolated from the dentate gyrus of kindled animals (McNamara et al. 1980; Fanelli & McNamara, 1983; Shin et al. 1985). Binding of [3H]muscimol was significantly increased in dentate gyrus, whereas it was decreased in the CA1 area of kindled rats (Titulaer et al. 1994). Muscimol induced increased uptake of labelled Cl− in synaptosomes isolated from the dentate gyri of kindled rats (Titulaer et al. 1995a). A similar increase of [3H]flunitrazepam (Valdes et al. 1982; Shin et al. 1985; Titulaer et al. 1995c) and [3H]t-butylbicyclophosphorothionate (Titulaer et al. 1995b) binding was found in DGCs of kindled rats. In Lothman's model of TLE, polysynaptic IPSPs are reduced or eliminated in the CA1 area, whereas they are unchanged in the dentate gyrus (Mangan et al. 1995) indicating that GABA-mediated inhibition is preserved in the dentate gyrus in this model of TLE.
More directly comparable to the present study are published whole-cell voltage clamp recordings from DGCs in rats made epileptic by the lithium/pilocarpine injection. GABAA receptor currents were increased in DGCs acutely isolated from rats in the lithium/ pilocarpine model of TLE (Gibbs et al. 1997) and from the dentate gyri of patients who underwent temporal lobectomy (Gibbs et al. 1996). An increased number of functionally active GABAA receptors had been proposed as a main factor contributing to the increased amplitude of mIPSCs in the dentate gyrus of kindled rats (Otis et al. 1994). An increased number of GABAA receptors on DGCs after kindling was confirmed by immunogold staining (Nusser et al. 1998a).
Increased zinc sensitivity
The present study confirms the previous findings of Buhl et al. (1996) and Gibbs et al. (1997) that GABAA receptors present on epileptic DGCs have enhanced susceptibility to Zn2+ inhibition. It has been demonstrated that 200 μm Zn2+ diminished the frequency and enhanced the decay of mIPSCs recorded from DGCs of kindled rats whereas it does not alter the decay kinetics of mIPSCs in control slices. In addition, Zn2+ inhibition of GABAA receptor currents is markedly increased in DGCs acutely isolated from rats with pilocarpine-induced TLE compared to controls (Gibbs et al. 1997). These seminal studies have led to the formulation of the zinc hypothesis (Buhl et al. 1996; Coulter, 1999), which proposes a mechanism to explain the collapse of inhibition in epileptic DGCs. Furthermore, Zn2+-sensitive GABAA receptors are found on DGCs isolated from hippocampi of humans with temporal lobe epilepsy (Gibbs et al. 1996). In animal models with TLE, zinc-containing mossy fibres sprout new recurrent connections onto granule cells. Based on the proximity of zinc-containing mossy fibres and zinc-sensitive postsynaptic GABAA receptors mediating IPSCs, Buhl et al. (1996) proposed that during intense activity, zinc released from sprouted mossy fibres spills into inhibitory synapses and inhibits action of GABA on its receptor, leading to a collapse of inhibition in the epileptic dentate gyrus. The present study confirms that in another model of temporal lobe epilepsy, GABAA receptors on DGCs have increased sensitivity to Zn2+.
Mechanisms of altered GABAA receptor properties
Studies on recombinant as well as native GABAA receptors have shown that expression of α subunits is necessary to maintain allopregnanolone sensitivity which varies depending on the type of expressed α subunit. In recombinant GABAA receptors, allopregnanolone has maximal effect in the presence of an α1 subunit; and the presence of the γ2 subunit increases allopregnanolone potency (Shingai et al. 1991) A dramatic decline of GABAA receptor sensitivity to allopregnanolone occurred in hippocampal CA1 pyramidal neurons when an α4 subunit was over-expressed; normal sensitivity is restored after intrahippocampal infusion of an α4 subunit antisense deoxynucleotide (Smith et al. 1998a). β subunits do not alter the sensitivity of recombinant GABAA receptors to allopregnanolone (Maitra & Reynolds, 1998). The effect of the δ subunit expression on neurosteroid sensitivity remains uncertain. Expression of the δ subunit in recombinant GABAA receptors results in the loss of sensitivity to the analogue of allopregnanolone - 3α,21-dihydroxy-5α-pregnan-20-1 (THDOC; Zhu et al. 1996). In contrast, in mice lacking the δ subunit, there is a marked reduction of sensitivity to the sedative and anxiolytic effects of neurosteroids (Mihalek et al. 1999). This effect was the opposite of that predicted by recombinant receptor studies.
The altered expression of α1, α4 or δ subunits might explain the reduced sensitivity of GABAA receptors to allopregnanolone on epileptic DGCs. These subunits are also known to alter the diazepam, zolpidem and Zn2+ sensitivity of GABAA receptors (Macdonald & Olsen, 1994) and changes in the combination of the subunits may explain concurrent changes in the sensitivity to these drugs in epileptic DGCs. In naive rats, DGCs expressed α1, α2, α4, and δ subunit mRNA (Wisden et al. 1992; Sperk et al. 1997) and immunoreactivity for these subunits has been documented in the rat dentate gyrus (Sperk et al. 1997). Diminished α1 subunit mRNA expression and increased α4 and δ subunit mRNA expression in DGCs of epileptic rats has been reported in the lithium/pilocarpine model (Brooks-Kayal et al. 1998).
In conclusion, diminished sensitivity of DGCs to allopregnanolone and sustained sensitivity to PS in TLE, in combination with other well-described pathological changes, are likely to increase susceptibility to seizures. A detailed analysis of the functional properties of GABAA receptors on epileptic DGCs revealed a complex picture. Past studies have emphasized two properties of these receptors, namely, increased GABA efficacy and enhanced zinc sensitivity. In addition to these changes, the present study demonstrated diminished sensitivity of epileptic DGC GABAA receptors to allopregnanolone and diazepam and demonstrated heterogeneity of GABAA receptor sensitivity to zolpidem in epileptic DGCs.
Acknowledgments
This project was sponsored by the American Epilepsy Society with support from the Milken Family Foundation to Z.M. and NINDS grants KO2-NS 02081 and RO1 NS40337 to J.K. We thank Dr Kevin Kelly and Dr Patrick Mangan for editing the manuscript and John Williamson for producing and monitoring epileptic rats.
References
- Altman J, Das GD. Autoradiographic and histological studies of postnatal neurogenesis. I. A longitudinal investigation of the kinetics, migration and transformation of cells incorporating tritiated thymidine in neonate rats, with special reference to postnatal neurogenesis in some brain regions. Journal of Comparative Neurology. 1966;126:337–389. doi: 10.1002/cne.901260302. [DOI] [PubMed] [Google Scholar]
- Behr J, Lyson KJ, Mody I. Enhanced propagation of epileptiform activity through the kindled dentate gyrus. Journal of Neurophysiology. 1998;79:1726–1732. doi: 10.1152/jn.1998.79.4.1726. [DOI] [PubMed] [Google Scholar]
- Belelli D, Bolger MB, Gee KW. Anticonvulsant profile of the progesterone metabolite 5 alpha-pregnan-3 alpha-ol-20-one. European Journal of Pharmacoogy. 1989;166:325–329. doi: 10.1016/0014-2999(89)90077-0. [DOI] [PubMed] [Google Scholar]
- Bertram EH, Williamson JM, Cornett JF, Spradlin S, Chen ZF. Design and construction of a long-term continuous video-EEG monitoring unit for simultaneous recording of multiple small animals. Brain Research. 1997;2:85–97. doi: 10.1016/s1385-299x(97)00033-0. [DOI] [PubMed] [Google Scholar]
- Bormann J. U-tube drug application. In: Kettenmann H, Grantyn R, editors. Practical Electrophysiological Methods. New York: Wiley-Liss; 1992. pp. 136–140. [Google Scholar]
- Bowlby MR. Pregnenolone sulfate potentiation of N-methyl-D-aspartate receptor channels in hippocampal neurons. Molecular Pharmacology. 1993;43:813–899. [PubMed] [Google Scholar]
- Brooks-Kayal AR, Shumate MD, Lin H, Rikhter TY, Coulter DA. Selective changes in single cell GABAA receptor subunit expression and function in temporal lobe epilepsy. Nature Medicine. 1998;4:1166–1172. doi: 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- Brussaard AB, Kits KS, Baker RE, Willems WP, Leyting-Vermeulen JW, Voorn P, Smit AB, Bicknell RJ, Herbison AE. Plasticity in fast synaptic inhibition of adult oxytocin neurons caused by switch in GABAA receptor subunit expression. Neuron. 1997;19:1103–1114. doi: 10.1016/s0896-6273(00)80401-8. [DOI] [PubMed] [Google Scholar]
- Budziszewska B, Siwanowicz J, Leskiewicz M, Jaworska-Feil L, Lason W. Protective effects of neurosteroids against NMDA-induced seizures and lethality in mice. European Journal of Neuropsychopharmacology. 1998;8:7–12. doi: 10.1016/s0924-977x(97)00037-0. [DOI] [PubMed] [Google Scholar]
- Buhl EH, Otis TS, Mody I. Zinc-induced collapse of augmented inhibition by GABA in a temporal lobe epilepsy model. Science. 1996;271:369–373. doi: 10.1126/science.271.5247.369. [DOI] [PubMed] [Google Scholar]
- Celentano JJ, Wong RKS. Multiphasic desensitization of the GABAA receptor in outside-out patches. Biophysical Journal. 1994;66:1039–1050. doi: 10.1016/S0006-3495(94)80885-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compagnone NA, Mellon SH. Neurosteroids: biosynthesis and function of these novel neuromodulators. Frontiers of Neuroendocrinology. 2000;211:1–56. doi: 10.1006/frne.1999.0188. [DOI] [PubMed] [Google Scholar]
- Coulter DA. Chronic epileptogenic cellular alterations in the limbic system after status epilepticus. Epilepsia. :S401–1. doi: 10.1111/j.1528-1157.1999.tb00875.x. S23–S33. [DOI] [PubMed] [Google Scholar]
- De Jonge M, Racine RJ. The development and decay of kindling-induced increases in paired-pulse depression in the dentate gyrus. Brain Research. 1987;412:318–328. doi: 10.1016/0006-8993(87)91139-5. [DOI] [PubMed] [Google Scholar]
- Deutsch SI, Rosse RB, Steinberg K, Morn C, Koetzner L, Riggs R, Mastropaolo J. Evaluation of in vivo interactions in mice between flurazepam and two neuroactive steroids. Pharmacology, Biochemistry and Behavior. 1996;55:323–366. doi: 10.1016/s0091-3057(96)00100-1. [DOI] [PubMed] [Google Scholar]
- Fanelli RJ, McNamara JO. Kindled seizures result in decreased responsiveness of benzodiazepine receptors to gamma-aminobutyric acid (GABA) Journal of Pharmacology and Experimental Therapeutics. 1983;226:147–150. [PubMed] [Google Scholar]
- Frye CA, Scalise TJ. Anti-seizure effects of progesterone and 3alpha,5alpha-THP in kainic acid and perforant pathway models of epilepsy. Psychoneuroendocrinology. 2000;25:407–420. doi: 10.1016/s0306-4530(99)00068-2. [DOI] [PubMed] [Google Scholar]
- Gibbs JW, Shumate MD, Coulter DA. Differential epilepsy-associated alterations in postsynaptic GABAA receptor funcction in dentate granule and CA1 neurons. Journal of Neurophysiology. 1997;77:1924–1938. doi: 10.1152/jn.1997.77.4.1924. [DOI] [PubMed] [Google Scholar]
- Gibbs JW, Zhang YF, Kao CQ, Holloway KL, Oh KS, Coulter DA. Characterization of GABAA receptor function in human temporal cortical neurons. Journal of Neurophysiology. 1996;75:1458–1471. doi: 10.1152/jn.1996.75.4.1458. [DOI] [PubMed] [Google Scholar]
- Greenfield LJ, Macdonald RL. Whole-cell and single-channel α1β1γ2S GABAA receptor currents elicited by a “multipuffer” drug application device. Pflügers Archiv. 1996;432:1080–1090. doi: 10.1007/s004240050238. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Herzog AG. Psychoneuroendocrine aspects of temporolimbic epilepsy. Part II: Epilepsy and reproductive steroids. Psychosomatics. 1999;40:102–108. doi: 10.1016/S0033-3182(99)71255-7. [DOI] [PubMed] [Google Scholar]
- Houser CR. Morphological changes in the dentate gyrus in human temporal lobe epilepsy. Epilepsy Research Supplement. 1992;7:223–234. [PubMed] [Google Scholar]
- Jones RS, Lambert JD. The role of excitatory amino acid receptors in the propagation of epileptiform discharges from the entorhinal cortex to the dentate gyrus in vitro. Experimental Brain Research. 1990;80:310–322. doi: 10.1007/BF00228158. [DOI] [PubMed] [Google Scholar]
- Kapur J, Coulter DA. Experimental status epilepticus alters GABAA receptor function in CA1 pyramidal neurons. Annals of Neurology. 1995;38:893–900. doi: 10.1002/ana.410380609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur J, Macdonald RL. Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. Journal of Neuroscience. 1997;17:7532–7540. doi: 10.1523/JNEUROSCI.17-19-07532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur J, Macdonald RL. Postnatal development of hippocampal dentate granule cell gamma-aminobutyric acidA receptor pharmacological properties. Molecular Pharmacology. 1999;55:444–452. [PubMed] [Google Scholar]
- Kay AR, Wong RK. Isolation of neurons suitable for patch-clamping from adult mammalian central nervous systems. Journal of Neuroscience Methods. 1986;16:227–238. doi: 10.1016/0165-0270(86)90040-3. [DOI] [PubMed] [Google Scholar]
- Kohr G, Mody I. Kindling increases N-methyl-D-aspartate potency at single N-methyl-D-aspartate channels in dentate gyrus granule cells. Neuroscience. 1994;62:975–981. doi: 10.1016/0306-4522(94)90336-0. [DOI] [PubMed] [Google Scholar]
- Kokate TG, Banks MK, Magee T, Yamaguchi S, Rogawski MA. Finasteride, a 5alpha-reductase inhibitor, blocks the anticonvulsant activity of progesterone in mice. Journal of Pharmacology and Experimental Therapeutics. 1999a;288:679–684. [PubMed] [Google Scholar]
- Kokate TG, Juhng KN, Kirkby RD, Llamas J, Yamaguchi S, Rogawski MA. Convulsant actions of the neurosteroid pregnenolone sulfate in mice. Brain Research. 1999b;831:119–124. doi: 10.1016/s0006-8993(99)01287-1. [DOI] [PubMed] [Google Scholar]
- Lambert JD, Jones RS. Activation of N-methyl-D-aspartate receptors contributes to the EPSP at perforant path synapses in the rat dentate gyrus in vitro. Neuroscience Letters. 1989;97:323–328. doi: 10.1016/0304-3940(89)90618-6. [DOI] [PubMed] [Google Scholar]
- Lambert JJ, Belelli D, Hill-Venning C, Callachan H, Peters JA. Neurosteroid modulation of native and recombinant GABAA receptors. Cellular and Molecular Neurobiology. 1996;16:155–174. doi: 10.1007/BF02088174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancel M, Faulhaber J. The GABAA agonist THIP (gaboxadol) increases non-REM sleep and enhances delta activity in the rat. NeuroReport. 1996;7:2241–2255. doi: 10.1097/00001756-199609020-00036. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Bertram EH, Bekenstein JW, Perlin JB. Self-sustaining limbic status epilepticus induced by ‘continuous’ hippocampal stimulation: electrographic and behavioral characteristics. Epilepsy Research. 1989;3:107–119. doi: 10.1016/0920-1211(89)90038-7. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Bertram EH, Stringer JL. Functional anatomy of hippocampal seizures. Progress in Neurobiology. 1991;37:1–82. doi: 10.1016/0301-0082(91)90011-o. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Stringer JL, Bertram EH. The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Research Supplement. 1992;7:301–313. [PubMed] [Google Scholar]
- Macdonald RL, Olsen RW. GABAA receptor channels. Annual Reviews of Neuroscience. 1994;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- McNamara JO, Peper AM, Patrone V. Repeated seizures induce long-term increase in hippocampal benzodiazepine receptors. Proceedings of the National Academy of Sciences of the USA. 1980;77:3029–3032. doi: 10.1073/pnas.77.5.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra R, Reynolds JN. Modulation of GABAA receptor function by neuroactive steroids: evidence for heterogeneity of steroid sensitivity of recombinant GABAA receptor isoforms. Canadian Journal of Physiology and Pharmacology. 1998;76:909–920. doi: 10.1139/cjpp-76-9-909. [DOI] [PubMed] [Google Scholar]
- Majewska MD, Mienville JM, Vicini S. Neurosteroid pregnenolone sulfate antagonizes electrophysiological responses to GABA in neurons. Neuroscience Letters. 1988;90:279–844. doi: 10.1016/0304-3940(88)90202-9. [DOI] [PubMed] [Google Scholar]
- Mangan PS, Rempe DA, Lothman EW. Changes in inhibitory neurotransmission in the CA1 region and dentate gyrus in a chronic model of temporal lobe epilepsy. Journal of Neurophysiology. 1995;74:829–840. doi: 10.1152/jn.1995.74.2.829. [DOI] [PubMed] [Google Scholar]
- Mhatre M, Mehta AK, Ticku MK. Chronic ethanol administration increases the binding of the benzodiazepine inverse agonist and alcohol antagonist [3H]RO 15–4513 in rat brain. European Journal of Pharmacology. 1988;153:141–145. doi: 10.1016/0014-2999(88)90599-7. [DOI] [PubMed] [Google Scholar]
- Mhatre M, Ticku MK. Chronic ethanol treatment selectively increases the binding of inverse agonists for benzodiazepine binding sites in cultured spinal cord neurons. Journal of Pharmacology and Experimental Therapeutics. 1989;251:164–168. [PubMed] [Google Scholar]
- Mihalek RM, Banerjee PK, Korpi ER, Quinlan JJ, Firestone LL, Mi ZP, Lagenaur C, Tretter V, Sieghart W, Anagnostaras SG, Sage JR, Fanselow MS, Guidotti A, Spigelman I, Li Z, Delorey TM, Olsen RW, Homanics GE. Attenuated sensitivity to neuroactive steroids in gamma-aminobutyrate type A receptor delta subunit knockout mice. Proceedings of the National Academy of Sciences of the USA. 1999;96:12905–12910. doi: 10.1073/pnas.96.22.12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody I, Heinemann U. NMDA receptors of dentate gyrus granule cells participate in synaptic transmission following kindling. Nature. 1987;326:701–704. doi: 10.1038/326701a0. [DOI] [PubMed] [Google Scholar]
- Mody I, Stanton PK, Heinemann U. Activation of N-methyl-D-aspartate receptors parallels changes in cellular and synaptic properties of dentate gyrus granule cells after kindling. Journal of Neurophysiology. 1988;59:1033–1054. doi: 10.1152/jn.1988.59.3.1033. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Hajos N, Somogyi P, Mody I. Increased number of synaptic GABAA receptors underlies potentiation at hippocampal inhibitory synapses. Nature. 1998a;395:172–177. doi: 10.1038/25999. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Sieghart W, Somogyi P. Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. Journal of Neuroscience. 1998b;18:1693–1703. doi: 10.1523/JNEUROSCI.18-05-01693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver MW, Miller JJ. Alterations of inhibitory processes in the dentate gyrus following kindling-induced epilepsy. Experimental Brain Research. 1985;57:443–447. doi: 10.1007/BF00237830. [DOI] [PubMed] [Google Scholar]
- Otis TS, De Koninck Y, Mody I. Lasting potentiation of inhibition is associated with an increased number of γ-aminobutyric acid type A receptors activated during miniature inhibitory postsynaptic currents. Proceedings of the National Academy of Sciences of the USA. 1994;91:7698–7702. doi: 10.1073/pnas.91.16.7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet LS, Jones MV, Westbrook GL. Slow desensitization regulates the availability of synaptic GABAA receptors. Journal of Neuroscience. 2000;20:7914–7921. doi: 10.1523/JNEUROSCI.20-21-07914.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. Journal of Neuroscience. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS, Kim HY, Rogawski MA. Neurosteroid withdrawal model of perimenstrual catamenial epilepsy. Epilepsia. 2001;42:328–336. doi: 10.1046/j.1528-1157.2001.10100.x. [DOI] [PubMed] [Google Scholar]
- Reddy DS, Rogawski MA. Enhanced anticonvulsant activity of ganaxolone after neurosteroid withdrawal in a rat model of catamenial epilepsy. Journal of Pharmacology and Experimental Therapeutics. 2000;294:909–915. [PubMed] [Google Scholar]
- Rothstein JD, Garland W, Puia G, Guidotti A, Weber RJ, Costa E. Purification and characterization of naturally occurring benzodiazepine receptor ligands in rat and human brain. Journal of Neurochemistry. 1992a;58:2102–2115. doi: 10.1111/j.1471-4159.1992.tb10952.x. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Guidotti A, Costa E. Release of endogenous benzodiazepine receptor ligands (endozepines) from cultured neurons. Neuroscience Letters. 1992b;143:210–214. doi: 10.1016/0304-3940(92)90267-b. [DOI] [PubMed] [Google Scholar]
- Saxena NC, Macdonald RL. Assembly of GABAA receptor subunits: role of the delta subunit. Journal of Neuroscience. 1994;14:7077–7086. doi: 10.1523/JNEUROSCI.14-11-07077.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, Pedersen HB, McNamara JO. γ-Aminobutyric acid and benzodiazepine receptors in the kindling model of epilepsy: a quantitative radiohistochemical study. Journal of Neuroscience. 1985;5:2696–2701. doi: 10.1523/JNEUROSCI.05-10-02696.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shingai R, Sutherland ML, Barnard EA. Effects of subunit types of the cloned GABAA receptor on the response to a neurosteroid. European Journal of Pharmacology. 1991;206:77–80. doi: 10.1016/0922-4106(91)90149-c. [DOI] [PubMed] [Google Scholar]
- Shumate MD, Lin DD, Gibbs JW, Holloway KL, Coulter DA. GABAA receptor function in epileptic human dentate granule cells: comparison to epileptic and control rat. Epilepsy Research. 1998;32:114–288. doi: 10.1016/s0920-1211(98)00045-x. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. On the relationship between neuropathology and pathophysiology in the epileptic hippocampus of humans and experimental animals. Hippocampus. 1994;4:250–253. doi: 10.1002/hipo.450040304. [DOI] [PubMed] [Google Scholar]
- Smith SS, Gong QH, Hsu FC, Markowitz RS, Ffrench-Mullen JM, Li X. GABAA receptor alpha4 subunit suppression prevents withdrawal properties of an endogenous steroid. Nature. 1998a;392:926–930. doi: 10.1038/31948. [DOI] [PubMed] [Google Scholar]
- Smith SS, Gong QH, Li X, Moran MH, Bitran D, Frye CA, Hsu FC. Withdrawal from 3alpha-OH-5alpha-pregnan-20-one using a pseudopregnancy model alters the kinetics of hippocampal GABAA-gated current and increases the GABAA receptor alpha4 subunit in association with increased anxiety. Journal of Neuroscience. 1998b;18:5275–5284. doi: 10.1523/JNEUROSCI.18-14-05275.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer DD, Spencer SS. Hippocampal resections and the use of human tissue in defining temporal lobe epilepsy syndromes. Hippocampus. 1994;4:243–249. doi: 10.1002/hipo.450040303. [DOI] [PubMed] [Google Scholar]
- Sperk G, Schwarzer C, Tsunashima K, Fuchs K, Sieghart W. GABAA receptor subunits in the rat hippocampus I: immunocytochemical distribution of 13 subunits. Neuroscience. 1997;80:987–1000. doi: 10.1016/s0306-4522(97)00146-2. [DOI] [PubMed] [Google Scholar]
- Stringer JL, Lothman EW. Maximal dentate gyrus activation: characteristics and alterations after repeated seizures. Journal of Neurophysiology. 1989;62:136–143. doi: 10.1152/jn.1989.62.1.136. [DOI] [PubMed] [Google Scholar]
- Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Annals of Neurology. 1989;26:321–330. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. Journal of Neuroscience. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titulaer MN, Ghijsen WE, Kamphuis W, De Rijk TC, Lopes DSF. Opposite changes in GABAA receptor function in the CA1–3 area and fascia dentata of kindled rat hippocampus. Journal of Neurochemistry. 1995a;64:2615–2621. doi: 10.1046/j.1471-4159.1995.64062615.x. [DOI] [PubMed] [Google Scholar]
- Titulaer MN, Kamphuis W, Lopes DSF. Autoradiographic analysis of [35S]t-butylbicyclophosphorothionate binding in kindled rat hippocampus shows different changes in CA1 area and fascia dentata. Neuroscience. 1995b;66:547–554. doi: 10.1016/0306-4522(94)00570-u. [DOI] [PubMed] [Google Scholar]
- Titulaer MN, Kamphuis W, Lopes DSF. Long-term and regional specific changes in [3H]flunitrazepam binding in kindled rat hippocampus. Neuroscience. 1995c;68:399–406. doi: 10.1016/0306-4522(95)00158-f. [DOI] [PubMed] [Google Scholar]
- Titulaer MN, Kamphuis W, Pool CW, Van Heerikhuize JJ, Lopes DSF. Kindling induces time-dependent and regional specific changes in the [3H]muscimol binding in the rat hippocampus: a quantitative autoradiographic study. Neuroscience. 1994;59:817–826. doi: 10.1016/0306-4522(94)90286-0. [DOI] [PubMed] [Google Scholar]
- Tuff LP, Racine RJ, Adamec R. The effects of kindling on GABA-mediated inhibition in the dentate gyrus of the rat. I. Paired-pulse depression. Brain Research. 1983;277:79–90. doi: 10.1016/0006-8993(83)90909-5. [DOI] [PubMed] [Google Scholar]
- Valdes F, Dasheiff RM, Birmingham F, Crutcher KA, McNamara JO. Benzodiazepine receptor increases after repeated seizures: evidence for localization to dentate granule cells. Proceedings of the National Academy of Sciences of the USA. 1982;79:193–197. doi: 10.1073/pnas.79.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee M, Mayo W, Darnaudery M, Corpechot C, Young J, Koehl M, Le Moal M, Baulieu EE, Robel P, Simon H. Neurosteroids: deficient cognitive performance in aged rats depends on low pregnenolone sulfate levels in the hippocampus. Proceedings of the National Academy of Sciences of the USA. 1997;94:14865–700. doi: 10.1073/pnas.94.26.14865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther H, Lambert JD, Jones RS, Heinemann U, Hamon B. Epileptiform activity in combined slices of the hippocampus, subiculum and entorhinal cortex during perfusion with low magnesium medium. [DOI] [PubMed]
- Weaver CEJ, Wu FS, Gibbs TT, Farb DH. Pregnenolone sulfate exacerbates NMDA-induced death of hippocampal neurons. Brain Research. 1998;803:129–366. doi: 10.1016/s0006-8993(98)00640-4. [DOI] [PubMed] [Google Scholar]
- Wisden W, Laurie DJ, Monyer H, Seeburg PH. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. Journal of Neuroscience. 1992;12:1040–1062. doi: 10.1523/JNEUROSCI.12-03-01040.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuarin JP, Dudek FE. Electrographic seizures and new recurrent excitatory circuits in the dentate gyrus of hippocampal slices from kainate-treated epileptic rats. Journal of Neuroscience. 1996;16:4438–4448. doi: 10.1523/JNEUROSCI.16-14-04438.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WJ, Wang JF, Krueger KE, Vicini S. δ subunit inhibits neurosteroid modulation of GABAA receptors. Journal of Neuroscience. 1996;16:6648–6656. doi: 10.1523/JNEUROSCI.16-21-06648.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]