

Abstract
The present study was undertaken to determine whether calponin (CaP) participates in the regulation of vascular smooth muscle contraction and, if so, to investigate the mechanism.
By PCR homology cloning, the cDNA sequence of ferret basic (h1) CaP was determined and phosphorothioate antisense and random oligonucleotides were synthesized and introduced into strips of ferret aorta by a chemical loading procedure.
Treatment of ferret aorta with CaP antisense oligonucleotides resulted in a decrease in protein levels of CaP to 54 % of that in random sequence-loaded muscles, but no change in the protein levels of caldesmon (CaD), actin, desmin or extracellular regulated protein kinase (ERK).
Contraction in response to phenylephrine or a phorbol ester was significantly decreased in antisense-treated muscles compared to random sequence-loaded controls. Neither basal intrinsic tone nor the contraction in response to 51 mm KCl was significantly affected by antisense treatment.
During phenylephrine contractions, phospho-ERK levels increased, as did myosin light chain (LC20) phosphorylation. Phenylephrine-induced ERK phosphorylation and CaD phosphorylation at an ERK site were significantly decreased by CaP antisense. Increases in myosin light chain phosphorylation were unaffected.
The data indicate that CaP plays a significant role in the regulation of contraction and suggest that in a tonically active smooth muscle CaP may function as a signalling protein to facilitate ERK-dependent signalling, but not as a direct regulator of actomyosin interactions at the myofilament level.
Calponin (CaP) is a relatively recently discovered 32-36 kDa smooth muscle-specific protein whose function is controversial (Takahashi et al. 1988; Takahashi & Nadal-Ginard, 1991; Horowitz et al. 1996). There have been two mechanisms proposed by which CaP might regulate smooth muscle contractility. One suggestion is that CaP, an actin-binding protein with some homology to troponin, directly inhibits actin-activated Mg2+-ATPase activity of myosin. CaP has been shown to bind actin and inhibit the actin-activated Mg2+-ATPase activity of myosin in vitro (Takahashi et al. 1986; Mezgueldi et al. 1995). When CaP is phosphorylated in vitro, its actin-binding and myosin inhibitory properties are diminished (Takahashi et al. 1986; Winder & Walsh, 1990, 1993; Horiuchi & Chacko, 1991; Makuch et al. 1991; Winder et al. 1991). However, it is not clear whether CaP is phosphorylated in vivo during smooth muscle contraction (Bárány et al. 1991; Gimona et al. 1992; Bárány & Bárány, 1993; Winder et al. 1993; Winder & Walsh, 1993; Nagumo et al. 1994; Adam et al. 1995; Rokolya et al. 1996; Pohl et al. 1997). Furthermore, it has been questioned whether CaP plays a significant role in regulating smooth muscle actomyosin in vivo since its location does not seem to be compatible with a physiological role in directly regulating myosin ATPase activity (Marston, 1991; North et al. 1994; Parker et al. 1994, 1998; Mabuchi et al. 1996; Menice et al. 1997). A CaP knockout mouse lacking h1, a basic CaP, has recently been reported (Yoshikawa et al. 1998; Matthew et al. 2000; Takahashi et al. 2000). In phasically active smooth muscle from this mouse an increase in shortening velocity was observed, consistent with a role for CaP in directly interfering with actomyosin activity. However, these authors also reported changes in tropomyosin and actin levels as well as in caldesmon (CaD) mobility in these animals. It is possible that the change in actin levels caused the observed change in shortening velocity. Furthermore, agonist activation of tonic smooth muscles was not investigated in these studies.
In contrast, it has also been suggested that CaP may facilitate agonist-dependent signal transduction. CaP, unlike other actin-binding proteins which inhibit actomyosin ATPase activity (such as CaD and troponin), has been reported to undergo an apparent agonist-induced translocation in ferret vascular smooth muscle cells (Parker et al. 1994, 1998; Menice et al. 1997). Others have also reported difficulty in isolating CaP from thin filament preparations unless special procedures were used (Lehman, 1989, 1991). Recently, this laboratory has reported that CaP (a) co-immunoprecipitates with extracellular regulated protein kinase (ERK) and with the Ca2+-independent isoform of protein kinase C (PKCε) in ferret aorta homogenates, (b) co-localizes with ERK and PKCε in cells and (c) directly binds ERK and PKCεin vitro (Menice et al. 1997; Leinweber et al. 1999, 2000). The N-terminal ‘CH domain’ of CaP binds ERK (Leinweber et al. 1999), while the C-terminal half of CaP binds the regulatory domain of PKC and facilitates the activation of PKC in vitro (Leinweber et al. 2000). Based on these results, we have speculated that CaP may function as an adaptor protein connecting the PKC cascade to the ERK cascade.
In the present study, we used an antisense approach to acutely down-regulate the CaP content of smooth muscle cells of the ferret aorta. The results obtained indicate that CaP plays a significant role in the regulation of smooth muscle contraction and suggest that CaP functions as a signalling protein to facilitate ERK-dependent signalling and CaD phosphorylation at an ERK site during agonist-induced contractions of tonic smooth muscle.
METHODS
Partial cloning of CaP by RT-PCR
The amino acid sequence of basic CaP from different vertebrates was compared and two oligonucleotides from homologous regions were designed as primers. Antisense 5′-CCC TTG TTG CTG CCC ATC TG-3′ and sense (degenerate) 5′-CAA CTT CAT GGA T/CGG CCT C-3′ sequences were synthesized. Total RNA was isolated from ferret aorta using TRIzol reagent (Gibco BRL). First strand cDNA synthesis was performed (cDNA synthesis kit from Clontech) using random hexamer at a reaction volume of 20 μl. A 2 μl volume of this reaction product was PCR amplified with these sense and antisense primers under the following conditions: 94 °C 5 min, 10 cycles (touchdown): 94 °C 45 s, 60-61 °C 30 s, 72 °C 40 s followed by 25 cycles of 94 °C 45 s, 55 °C 30 s and 72 °C 40 s. A final extension for 7 min was applied at 72 °C. One product of about 500 bp was cloned into the TOPO TA vector (Invitrogen) and sequenced.
cDNA library and cloning
Total RNA was isolated from ferret aorta as described above. HybriZAP 2.1 cDNA library was made from the custom library production facility of Stratagene. Screening of the library was performed according to instructions from the supplier. The partial clone DNA fragment was used as a probe in screening the library. As many as 14 different primary clones were sequenced and found to be the same sequence. The nucleotide sequence and the deduced amino acid sequence are presented in Fig. 1.
Figure 1. Nucleotide and deduced amino acid sequence of ferret basic CaP as determined with a full-length cDNA clone isolated from a ferret aorta cDNA library.

Amino acid residues are given in one-letter code above the respective codons. The numbers on the right represent the nucleotide position. Antisense oligonucleotide used in the study is from the region underlined. The arrow indicates the position of degenerate oligonucleotides used in RT-PCR for partial cloning of the gene. The nucleotide sequence has been submitted to GenBank. The accession number of this sequence is AF323674.
Tissue preparation
Ferrets (Marshal Farms, North Rose, NY, USA) were killed with an overdose of chloroform in a ventilation hood, in agreement with procedures approved by the Institutional Animal Care and Use Committee. The thoracic aorta was quickly removed and immersed in oxygenated (95 % O2-5 % CO2) physiological saline solution (PSS) composed of (mm): 120 NaCl, 5.9 KCl, 25 NaHCO3, 11.5 dextrose, 2.5 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4 (pH 7.4). The aorta was cleaned of all adherent connective tissue, and the endothelium was removed by gentle abrasion with a cell scraper.
Contraction measurements
Circular strips (3 mm wide) were prepared as previously described (Jiang & Morgan, 1989) and attached to a force transducer. The strips were allowed to equilibrate at 37 °C for at least 1 h and challenged with a depolarizing solution containing 51 mm KCl (PSS in which 45.1 mm NaCl has been stoichiometrically replaced by KCl). Muscle strips were then washed and allowed to equilibrate for 1 h before beginning the experiment. Throughout the study, forces were expressed as percentages of forces obtained on day 1 of the experiment. The means ±s.e.m. of forces on day 1 for 51 mm KCl, 10−5m phenylephrine and intrinsic tone were: 1.63 ± 0.09 g, 1.57 ± 0.10 g and 0.87 ± 0.03 g, respectively, for antisense-treated muscles (n = 27), 1.51 ± 0.08 g, 1.56 ± 0.10 g and 0.88 ± 0.04 g for random-treated muscles (n = 21) and 1.54 ± 0.07 g, 1.63 ± 0.12 g and 0.91 ± 0.03 g for sham-loaded muscles (n = 24).
Oligodeoxynucleotide loading and organ culture
The antisense oligonucleotide was generated from a sense 21-mer sequence of ferret CaP (5′-TT GGC ACC AGC TGG AAA ACA T-3′, see Fig. 1). All oligodeoxynucleotides were synthesized as fluorescein isothiocyanate-conjugated (FITC)-tagged phosphorothioates, using a Millipore Expedite nucleic acid synthesis system, cleaved from the reaction column by 30 % ammonium hydroxide and purified using a Poly-Pak cartridge (Glen Research, Sterling, VA, USA). For most experiments, the effects of antisense (5′-AT GTT TTC CAG CTG- GTG CCA A-3′) were compared to those of a random sequence (5′-CG TGG TAT AAA ACC GAT CAC G-3′) as well as a sham-loaded and a time control.
A method originally developed to load aequorin into smooth muscle and referred to as a ‘chemical loading procedure’ (Morgan & Morgan, 1984) was used to introduce the oligonucleotides into the cells of the vascular strip. This method has been used to load diverse substances such as peptides, heparin and DNA into smooth muscle tissue (Kobayashi et al. 1989; Lesh et al. 1995; Earley et al. 1998; Kim et al. 2000; Hulvershorn et al. 2001) and is sometimes referred to as a ‘reversible permeabilization method’ (Lesh et al. 1995; Johnson et al. 1996) even though the mechanism of the loading procedure is unknown and evidence is lacking that the cell membrane is permeabilized. The lack of change in any measurable cell function and the lack of loss of small molecules argue that the mechanism does not involve permeabilization (DeFeo & Morgan, 1985; Johnson et al. 1985; Rembold & Murphy, 1986; Morgan & Jiang, 1987). Briefly, muscles were soaked for 30-120 min each in a series of four solutions at 2 °C. The compositions of the solutions were as follows. Solution I: EGTA 10 mm, Na2ATP 5 mm, KCl 120 mm, MgCl2 2 mm, Tes 20 mm; solution II: EGTA 0.1 mm, Na2ATP 5 mm, KCl 120 mm, MgCl2 2 mm, Tes 20 mm, oligodeoxynucleotide 25 μm; solution III: EGTA 0.1 mm, Na2ATP 5 mm, KCl 120 mm, MgCl2 10 mm, Tes 20 mm; solution IV: NaCl 120 mm, KCl 5.8 mm, dextrose 11 mm, NaHCO3 25 mm, MgCl2 10 mm, NaH2PO4 1.4 mm. The pH of all solutions was titrated to 7.0. After adding solution IV the [Ca2+] was raised to the normal value of 2.5 mm in gradual steps in order to avoid damage to the preparation from the ‘Ca2+ paradox’ (Zimmerman & Hülsmann, 1966). Tissues were kept overnight at room temperature in a mixture of PSS and Dulbecco's modified Eagle's medium (1:1) in the presence of penicillin (25 units ml−1), streptomycin (25 mg ml−1) and nystatin (50 units ml−1). This procedure was repeated daily for 3 days in order to obtain a sufficient loading of oligodeoxynucleotides to decrease the level of the target protein significantly. The viability of the preparation and contractile function were tested daily by measuring the response to KCl PSS.
To confirm the loading of FITC-tagged oligonucleotides, fluorescence was quantified using a ×40 objective (NA 0.85) on a Nikon Diaphot 300 microscope attached to a Photometrics CH250 cooled CCD camera. An aperture on the excitation side of the objective lens was closed until vignetting was observed to reduce the scatter of light and to minimize the contribution of out of focus focal planes. Images were acquired from at least 10 optical sections for each muscle. For each section the average fluorescence intensity from each of five 100 × 100 pixel boxes was determined using PMIS Image Processing Software (EHD imaging GmbH, Damme, Germany). The average of the five values for each section was then plotted against position (mm) into the tissue.
Measurements of LC20 phosphorylation
LC20 phosphorylation was measured by a previously published method (Kim et al. 2000). Muscle strips were quick frozen by immersion in a dry ice-acetone slurry containing 10 % trichloroacetic acid (TCA) and 10 mm dithiothreitol (DTT). Muscles were stored at -80 °C until used. Tissues were brought to room temperature in a dry ice-acetone-TCA-DTT mixture, then ground with glass pestles and washed three times with ether to remove the TCA. Tissues were extracted in 100 μl of sample buffer containing 20 mm Tris base and 23 mm glycine (pH 8.6), 8.0 m urea, 10 mm DTT, 10 % glycerol and 0.04 % bromophenol blue. Samples (20 μl) were electrophoresed at 400 V for 2.5 h after a 30 min pre-run in 1.0 mm mini-polyacrylamide gels containing 10 % acrylamide-0.27 % bisacrylamide, 40 % glycerol, 20 mm Tris base and 23 mm glycine, pH 8.6. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes and subjected to immunoblot with a specific LC20 antibody (1:1500, Sigma). Anti-mouse IgG (goat) conjugated with horseradish peroxidase was used as a secondary antibody (1:2000, Calbiochem). Bands were detected with enhanced chemiluminescence (ECL) (Super Signal kit, Pierce Chemical Co., Rockford, IL, USA) visualized on films and analysed by NIH Image (US National Institutes of Health Research Services Branch). Care was taken to ensure that saturation of the signal did not occur at any step in the processing. Moles phosphate per mole light chain was calculated by dividing the density of the phosphorylated band by the sum of the densities of the phosphorylated plus the unphosphorylated bands.
Western blot analysis
Tissues were quick frozen at the end of the final day of the experiment, day 4, as for LC20 phosphorylation, above. Samples were homogenized in a buffer containing 20 mm Mops, 4 % SDS, 10 % glycerol, 10 mm DTT, 20 mmβ-glycerophosphate, 5.5 μm leupeptin, 5.5 μm pepstatin, 20 KIU aprotinin, 2 mm Na3VO4, 1 mm NaF, 100 μm ZnCl2, 20 μm 4-(2-aminoethyl)benzenesulphonyl fluoride (AEBSF) and 5 mm EGTA. Protein-matched samples (modified Lowry protein assay, DC Protein Assay Kit, Bio-Rad) were electrophoresed on SDS-PAGE (Protogel, National Diagnostics), transferred to PVDF membranes and subjected to immunostaining and densitometry, as above, using the appropriate antibody. The success of protein matching was confirmed by Naphthol Blue Black staining of the membrane and densitometry of the actin band. Any mismatch of lane loading was corrected by normalization to actin staining. Each set of samples (antisense, random, sham) from an individual experiment was run on the same gel and densitometry was performed on the same film.
Antibodies
The mouse monoclonal CaP antibody (1:500 000) and rabbit polyclonal desmin antibody (1:1000) were obtained from Sigma (St Louis, MO, USA). Rabbit polyclonal CaD antibody (1:10 000) was a generous gift from K. Mabuchi (BBRI). The rabbit polyclonal p44/p42 MAPK antibody (1:1000) and rabbit polyclonal phospho-p44/p42 (Thr-202/Tyr-204) MAPK antibody (1:1000) were obtained from New England Biolabs (Beverly, MA, USA). The phospho-CaD Ser-789 antibody was produced by L. Adam (Bristol Myers Squibb Co., Princeton, NJ, USA) and has been previously characterized (D'Angelo et al. 1999).
Statistics
Each set of data was expressed as a mean ±s.e.m. Student's unpaired t test was used to determine the statistical significance of the means between groups of two with P < 0.05 taken as significant.
RESULTS
Isolation and sequencing of cDNA encoding ferret basic CaP
RT-PCR was performed with total RNA of ferret aorta using two oligonucleotides (see arrows, Fig. 1). The amplified DNA obtained corresponded to the size of the expected product. This was cloned into a TOPO TA vector, sequenced and found to be 508 bp in length. The deduced amino acid sequence of this amplified DNA showed considerable homology with that of basic CaP of other vertebrates. This fragment was used in screening a HybriZAP 2.1 custom cDNA library (Stratagene). Fourteen different primary clones were isolated and sequenced; 11 of these were complete cDNA clones of 1129 nucleotides in length (excluding poly(A) tail) with an open reading frame of 297 amino acid residues (Fig. 1).
The amino acid sequence of the ferret basic CaP gene was compared with those from rat, pig, mouse, human and chicken and a high degree of homology was found between all sequences. In fact, 97-98 % identity exists between all sequences except for the chicken where the identity with ferret CaP is 83 %.
Antisense oligonucleotides to CaP load into cells throughout vascular rings
As described in Methods, aortic strips were loaded with CaP antisense or random sequence oligonucleotides, or sham loaded. To confirm adequate loading, all oligonucleotides were FITC labelled and the fluorescence of the preparation was quantified at the end of the experiment (Fig. 2A and B). Statistical analysis of the increase in fluorescence obtained after loading with FITC-tagged antisense or random oligonucleotides showed this to be highly significant in comparison to the sham-loaded preparations. There was no significant difference between the amounts of the antisense and random oligonucleotides that were taken up by the tissue (Fig. 2A). It was also noted that uniform fluorescence was observed throughout the thickness of the muscle in all cases (Fig. 2B).
Figure 2. Effect of CaP antisense or random sequence loading on the level of fluorescence and on CaD, desmin and CaP protein levels.
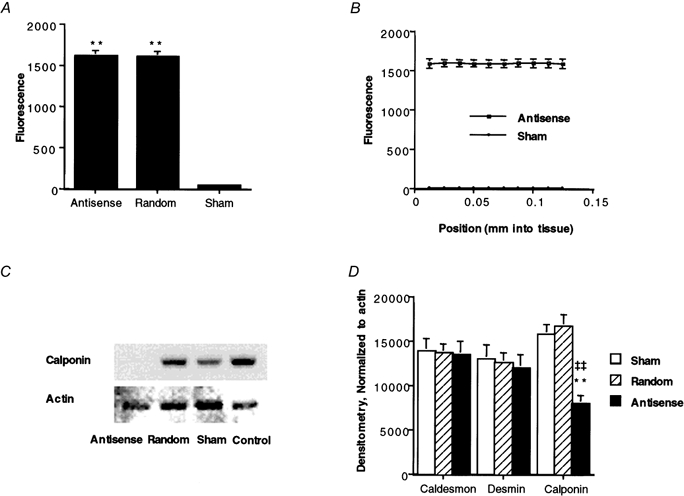
A, quantification of fluorescence (in arbitrary units) of FITC-oligonucleotide-loaded muscles versus sham-loaded muscles. **P < 0.01 compared to sham-loaded muscles. B, uniform fluorescence was observed throughout the thickness of the muscle. C, Western blot for CaP (upper panel) in comparison to actin levels (lower panel) from the same homogenates, determined by Naphthol Blue Black staining. D, the average levels of CaD, desmin and CaP in antisense-loaded (▪), random sequence-loaded ( ) or sham-loaded (□) muscles (n = 13-18). **P < 0.01 for antisense- compared to random sequence-loaded muscles. ‡‡P < 0.01 for antisense- compared to sham-loaded muscles. All samples were protein matched.
) or sham-loaded (□) muscles (n = 13-18). **P < 0.01 for antisense- compared to random sequence-loaded muscles. ‡‡P < 0.01 for antisense- compared to sham-loaded muscles. All samples were protein matched.
Antisense oligonucleotides to CaP, but not random sequence oligonucleotides, decrease CaP protein levels
Western blots of day 4 muscles were obtained to monitor the protein levels of CaP. Figure 2C shows a Western blot of a particularly successful experiment in which CaP protein levels from the antisense-treated muscle were decreased essentially 100 % compared to those from a random sequence-loaded muscle, a sham-loaded muscle and a time control. On average, CaP protein levels were decreased by antisense loading to 54 ± 5.1 % (n = 20) of those in paired, random sequence-loaded muscles (Fig. 2D). For comparison, CaP levels in sham-loaded muscles were 97 ± 7.6 % of those in random sequence-loaded muscles. Actin levels, determined by Naphthol Blue Black staining of the immunoblot membrane (Fig. 2C), were monitored to confirm equal protein loading of lanes and to show specificity of the antisense treatment. On average, actin protein levels were not significantly changed by antisense loading. By densitometry, actin levels in antisense-loaded muscles averaged 108 ± 5.7 % of those in paired, random sequence-loaded muscles.
The levels of CaD, desmin and CaP were determined by Western blot analysis (Fig. 2D). In individual experiments, any inequality in lane loading was corrected by normalization to the actin level. As can be seen in Fig. 2D, there were no significant changes in CaD or desmin levels in CaP antisense-treated muscles compared to random sequence- or sham-loaded muscles.
Contractility is maintained in organ culture
Contractility was assessed daily by exposure of the muscles to 51 mm KCl. The force generated in response to 51 mm KCl was normalized to the response of each muscle on day 1 (Fig. 3A). In serum-free organ culture, there was no significant decrease in the amplitude of the KCl-induced contraction with time in any group. Furthermore, there was no significant difference in the amplitude of the KCl contraction between the antisense-loaded muscles and either the sham-loaded or random sequence-loaded muscles on any day. However, a tendency was noted for all muscles to show a slight increase in contractile amplitude on days 2 and 3, perhaps representing a recovery from the trauma of dissection on day 1 (Fig. 3A). We also measured the rate of force development (as the time to development of 50 % peak force) in these muscles. These values did not change significantly either, and on day 4 they were: 167 ± 8.9 s (n = 18), 169 ± 15 s (n = 14) and 148 ± 10 s (n = 10) for antisense-, random- and sham-treated muscles, respectively.
Figure 3. Effect of CaP antisense loading on KCl- or PE-induced contraction, intrinsic tone and Ca2+-independent contractions.
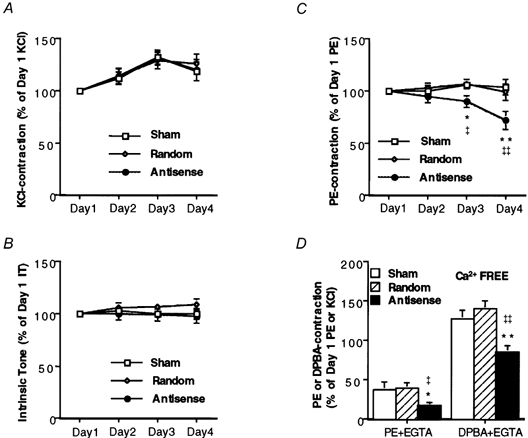
A, magnitude of steady-state (10-15 min) contractile response to 51 mm KCl on the indicated number of days after loading with oligonucleotides. Forces are normalized to the amplitude of contraction of each muscle in response to 51 mm KCl on day 1 (n = 10-12). B, magnitude of intrinsic tone, normalized to the amplitude of the intrinsic tone on day 1 (n = 10). Intrinsic tone is defined as the change in active basal tone at constant length when the temperature is changed between 37 °C and 2 °C. C, magnitude of steady-state (10-15 min) contractile response to 10 μm PE on the indicated number of days after loading with oligonucleotides. Forces are normalized to the amplitude of the contraction of each muscle to PE on day 1 (n = 10-12). D, effect of CaP antisense loading on Ca2+-independent contractions (PE- and DPBA-induced contraction in the presence of EGTA) (n = 8). *P < 0.05, **P < 0.01 compared to random sequence-loaded muscles. ‡P < 0.05, ‡‡P < 0.01 compared to sham-loaded muscles. DPBA contractions are normalized to KCl contractions on day 1 and PE contractions are normalized to PE contractions on day 1.
A basal active intrinsic tone has previously been demonstrated to be present in ferret aorta (Pawlowski & Morgan, 1992). The mechanism of this tone is unknown but it is eliminated by cooling and presumably requires crossbridge cycling. Antisense loading had no effect on the magnitude of intrinsic tone in these preparations (Fig. 3B).
CaP antisense decreases agonist-induced contractility
Contractility was assessed daily by exposure of the muscles to 10 μm phenylephrine (PE). The muscles loaded with the antisense sequence showed statistically significant decreases in the amplitude of the PE-induced contraction on day 3 (8.3 ± 4.9 % decrease) and on day 4 (26 ± 7.2 % decrease) (Fig. 3C) compared to day 1. In contrast, there was no statistically significant change in contractility in preparations loaded with the random oligonucleotide sequence or in sham-loaded preparations. The rate of contraction due to exposure to PE was measured as the time to the development of half the peak force and did not change with antisense treatment. The time to development of 50 % peak force after the addition of PE was: 87 ± 9.4 s (n = 18), 80 ± 10 s (n = 14) and 74 ± 6.1 s (n = 10) in antisense-, random- and sham-treated muscles, respectively.
In day 4 tissues, after the PE-induced contraction reached a steady state, 3 mm EGTA was added. The steady-state portion of the contraction that persists after the addition of EGTA corresponds to the previously described Ca2+-independent contractile tone (Collins et al. 1992) and has been suggested to involve thin filament regulation (Menice et al. 1997; Dessy et al. 1998). Muscles loaded with antisense produced a contraction that was 46 % of that in random sequence-loaded muscles and 44 % of that in sham-loaded muscles in response to PE in a Ca2+-free solution (Fig. 3D).
The phorbol ester 12-deoxyphorbol 13-isobutyrate 20-acetate (DPBA) has previously been reported to contract ferret aorta in the absence of any measurable increase in LC20 phosphorylation (Jiang & Morgan, 1989) and is thus likely to involve thin filament regulation. The response to 3 μm DPBA was determined in day 4 tissues in Ca2+-free PSS, after the determination of the amplitude of the Ca2+-independent phenylephrine contraction. The muscles loaded with antisense sequence showed a 39 % decrease in the amplitude of the DPBA-induced contraction compared to the random sequence-loaded muscle (Fig. 3D). In contrast, there was no statistically significant difference in the response to DPBA between preparations loaded with the random sequence or sham loaded.
Antisense oligonucleotides to CaP do not decrease LC20 phosphorylation
It is generally accepted that calcium-calmodulin (Ca2+-CaM)-dependent activation of myosin light chain kinase (MLCK) and the subsequent phosphorylation of the 20 kDa myosin light chains are a major regulatory mechanism for smooth muscle contractility. In an effort to determine the mechanism of the antisense-induced changes in contractility, we measured LC20 phosphorylation levels in antisense-treated muscles quick frozen after 5 min exposure to 10 μm PE. In all muscles, PE treatment resulted in a significant increase in LC20 phosphorylation levels (Fig. 4) but there was no significant difference between antisense-treated muscles and random sequence- or sham-loaded muscles in PE-induced LC20 phosphorylation.
Figure 4. Effect of CaP antisense loading on LC20 and CaD phosphorylation.
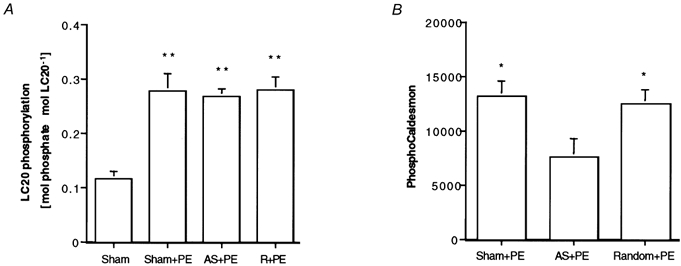
A, comparison of LC20 phosphorylation levels as determined by glycerol urea gels. **P < 0.01 compared to resting muscles. No differences occurred between PE-stimulated tissues loaded with antisense (AS) or random (R) sequences or sham loaded (n = 6). B, comparison of CaD phosphorylation levels as determined by an antibody specific for phosphorylation of Ser-789, between antisense-, random- or sham-loaded muscles. Values are protein matched and normalized to actin levels as determined by Naphthol Blue Black staining to correct for any differences in lane loading. *P < 0.05 compared to antisense-treated muscles (n = 4).
Antisense oligonucleotides to CaP decrease agonist-induced changes in CaD phosphorylation
CaD is an actin binding protein that has been suggested to be involved in thin filament regulation, possibly as the downstream target of a PKC- and ERK-dependent pathway. We used a phospho-CaD antibody to monitor phosphorylation of CaD at Ser-789, an ERK site, in PE-treated muscles. As is shown in Fig. 4B, there was a significant decrease in PE-induced CaD phosphorylation in antisense-treated muscles compared to random- or sham-loaded muscles.
Antisense oligonucleotides to CaP decrease agonist-induced increases in ERK phosphorylation
To confirm a role for CaP in an ERK-dependent pathway, we measured the levels of ERK and phospho-ERK in antisense-treated muscles quick frozen after 5 min exposure to 10 μm PE. As can be seen in Fig. 5A, there was no significant decrease in the level of ERK in antisense-treated muscles compared to random- or sham-loaded muscles. As previously described (Dessy et al. 1998), PE treatment resulted in a severalfold increase in phospho-ERK levels in control muscles. However, there was a significant decrease in the PE-induced ERK phosphorylation in antisense-treated muscles compared to random- or sham-loaded muscles (Fig. 5B).
Figure 5. Effect of CaP antisense loading on ERK and phospho-ERK protein levels.
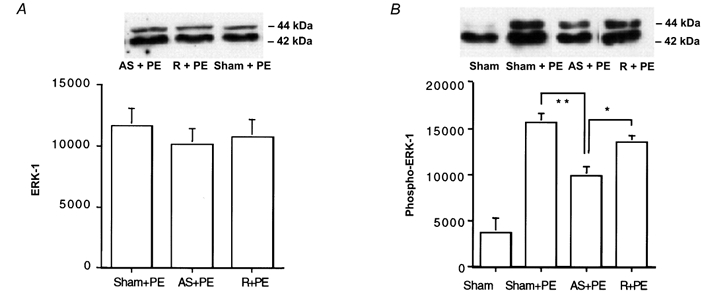
Upper panel, typical blot; lower panel, average densitometry results. A, the level of ERK (n = 8) in unstimulated muscles. B, the level of the PE-induced increase in phospho-ERK (n = 6). *P < 0.05, **P < 0.01 for antisense-loaded muscles compared to random sequence-loaded muscles and sham-loaded muscles. Values are protein matched and normalized to actin levels as determined by Naphthol Blue Black staining to correct for any differences in lane loading.
DISCUSSION
The main finding of the present study is that down-regulation of CaP protein levels in differentiated smooth muscle resulted in a significant inhibition of agonist-induced contractility. Thus, these results indicate that endogenous CaP is a physiologically important regulator of vascular tone. However, as mentioned above, in vitro studies have demonstrated that CaP can inhibit actin-activated myosin ATPase activity. Furthermore, in a study on toad stomach cells (Malmqvist et al. 1997), the chemical extraction of CaP resulted in a sustained increase in basal tone. It is to be noted, however, that the signal transduction in this muscle appears to be quite different from that in vascular smooth muscle, both in the unusual agonist-induced translocation of PKC away from the cell membrane as well as in the fact that the cells do not appear to contract to phorbol esters (Meininger et al. 1999). If CaP had a physiologically important role in inhibiting myosin ATPase activity in the intact ferret aorta smooth muscle cell, then the down-regulation of CaP should lead to an increase in basal tone, i.e. a generalized increase in contractility. Such a result has been reported when CaD was down-regulated using an antisense approach (Earley et al. 1998) or by a peptide antagonist (Katsuyama et al. 1992). Alternatively, Haeberle (1994) has reported that, in an in vitro motility assay, the addition of CaP caused a decreased sliding velocity, as expected, but also caused an increase in the force exerted on stationary actin filaments. He hypothesized that the increased force was due to a decreased rate of dissociation of high-affinity actomyosin complexes. Presumably, in the intact fibre this effect would lead to a generalized increase in basal tone under control conditions and a decrease in basal tone in response to CaP down-regulation. In contrast, in the present study, there were no changes in the level of basal, intrinsic tone when endogenous CaP was down-regulated, only a decrease in the amplitude of agonist-induced contractions. Thus, the results of the present study are not consistent with a role for CaP in directly regulating actomyosin interactions at the myofilament level in ferret aorta smooth muscle.
We have previously suggested an alternative role for CaP as an adaptor molecule that facilitates PKC and ERK-dependent signalling, leading to contraction of smooth muscle (Menice et al. 1997; Leinweber et al. 1999, 2000). This suggestion was based on in vitro protein chemistry, immunoprecipitation and cell imaging studies. The in vivo results presented here for CaP antisense-treated muscles are consistent with this suggestion, in that acute CaP down-regulation caused a decrease in agonist-induced contractility. In this regard, it is also of interest that CaP antisense inhibited both phenylephrine-induced contractions and DPBA-induced contractions but not KCl-induced contractions. DPBA is a phorbol ester known to activate PKC. Phenylephrine has been shown to cause the translocation and, presumably, activation of the Ca2+-independent isoform of PKC, PKCε, in ferret aorta cells (Khalil et al. 1992) as well as the activation of ERK (Dessy et al. 1998) in these same cells. The phorbol ester-induced contraction of ferret aorta, as well as the part of the phenylephrine contraction that persists in the absence of extracellular calcium, occurs without a detectable change in LC20 phosphorylation levels (Jiang & Morgan, 1989; Menice et al. 1997). Thus, the fact that the antisense-induced decrease in the amplitude of the phenylephrine contraction in the presence of extracellular calcium occurred without a detectable change in the levels of LC20 phosphorylation, but with a significant decrease in ERK phosphorylation levels, is also consistent with a role for CaP as a signalling molecule that facilitates ERK-dependent signalling.
We have previously reported that PE activation leads to both ERK activation and CaD phosphorylation at ERK sites and that both events are down-regulated by the MAPK/ERK kinase (MEK) inhibitor PD098059 (Dessy et al. 1998). The fact that CaP antisense-treated muscles had significantly lower phosphorylation of CaD at Ser-789 is further evidence linking CaP, ERK and CaD in a signalling cascade. Thus, our results and the results of others, as recently reviewed by Morgan & Gangopadhyay (2001), are consistent with a model whereby PE- or DPBA-induced stimulation leads to PKC activation. At the time of PKC activation, PKC binds CaP, which also binds ERK. The three proteins translocate to the cell membrane as a complex. At the membrane, ERK co-distributes with MEK and is phosphorylated. Phosphorylation of ERK releases ERK from the membrane and targets ERK to CaD on the actin filaments. The phosphorylation of CaD by ERK contributes to a disinhibition of the contractile proteins and an increased contractility.
It is of interest to compare our results using an antisense approach to acutely down-regulate CaP levels to those reported for the CaP knockout mouse lacking h1 basic CaP (Yoshikawa et al. 1998; Matthew et al. 2000). In this mouse, no change in tonic contractile force was detected but shortening velocity was increased during KCl contraction. Our results in the present study are in agreement with the results from the CaP knockout mouse in that we also saw no decrease in the amplitude of the KCl tonic contraction. Takahashi et al. (2000) reported that an increase in shortening velocity during KCl contraction in the aorta of the CaP knockout mouse occurred only after 5 min of stimulation but not after 1 min and suggested that this may indicate that CaP plays a role in the genesis of the latch state of smooth muscle, a phenomenon primarily associated with tonic contractions. However, these authors did not investigate agonist-induced contractions.
Matthew et al. (2000) reported that there is no detectable change in the amplitude of agonist-induced contractions in permeabilized muscles from the CaP knockout mouse. However, these authors only studied phasic muscle in the permeabilized state; also, a decrease in actin and tropomyosin levels was seen in these muscles as well as an upward shift in CaD mobility, suggesting that compensatory mechanisms may have masked some of the functional effects of CaP knockout. It is worth pointing out that in the present study we used intact, rather than permeabilized muscles, which may have preserved signalling molecules necessary for CaP's action. It has been suggested that CaP might play a more important role in the regulation of agonist-induced contractions of tonic smooth muscle (Walsh, 2000), such as the aorta used in our study. In other investigations, Nigam et al. (1998) have reported that h1 and h2 CaPs are present in greater amounts in aortic smooth muscle from adult Sprague-Dawley rats compared to Wistar or Wistar Kyoto rats. Furthermore, the Sprague-Dawley rat aortae, containing more CaP, were more sensitive in contractile response to the α-agonist noradrenaline (norepinephrine) compared to the Wistar or Wistar Kyoto rat aortae (Nigam et al. 1998). These results are consistent with the results of the present study.
Thus, in summary, when we acutely decreased CaP protein levels to about half of the endogenous level in ferret aortic smooth muscle, results were obtained which are consistent with a significant physiological role for CaP in facilitating agonist-induced contractions in tonic smooth muscle, but are not consistent with a role in directly regulating actomyosin interactions at the myofilament level in this tissue.
Acknowledgments
We would like to thank Ms Marilyn DeMont for her expert assistance in preparing the manuscript and Dr Janet Smith of BBRI for sharing her laboratory space with us in the performance of some of these experiments. We would also like to thank Dr Katsuhide Mabuchi of BBRI for his generous gift of the caldesmon antibody and Dr Len Adam (Bristol Myers Squibb Co., Princeton, NJ, USA) for generously making available to us the phospho-CaD Ser-789 antibody. Thanks are also due to Dr Uy-Dong Sohn (Seoul, Korea) for his support of Dr H.-D. Je and Dr Katsuhito Takahashi for his comments in writing this manuscript. This research was supported by NHLBI/NIH grants HL31704 and HL42293 to K.G.M.
References
- Adam LP, Haeberle JR, Hathaway DR. Calponin is not phosphorylated during contractions of porcine carotid arteries. American Journal of Physiology. 1995;268:C903–909. doi: 10.1152/ajpcell.1995.268.4.C903. [DOI] [PubMed] [Google Scholar]
- Bárány M, Bárány K. Calponin phosphorylation does not accompany contraction of various smooth muscles. Biochimica et Biophysica Acta. 1993;1179:229–233. doi: 10.1016/0167-4889(93)90146-g. [DOI] [PubMed] [Google Scholar]
- Bárány M, Rokolya A, Bárány K. Absence of calponin phosphorylation in contracting or resting arterial smooth muscle. FEBS Letters. 1991;279:65–68. doi: 10.1016/0014-5793(91)80252-x. [DOI] [PubMed] [Google Scholar]
- Collins EM, Walsh MP, Morgan KG. Contraction of single vascular smooth muscle cells by phenylephrine at constant [Ca2+]i. American Journal of Physiology. 1992;262:H754–762. doi: 10.1152/ajpheart.1992.262.3.H754. [DOI] [PubMed] [Google Scholar]
- D'Angelo G, Graceffa P, Wang, C-L A, Wrangle J, Adam LP. Mammal-specific, ERK-dependent, caldesmon phosphorylation in smooth muscle. Journal of Biological Chemistry. 1999;274:30115–30121. doi: 10.1074/jbc.274.42.30115. [DOI] [PubMed] [Google Scholar]
- DeFeo TT, Morgan KG. Calcium-force relationships as detected with aequorin in two different vascular smooth muscles of the ferret. Journal of Physiology. 1985;369:269–282. doi: 10.1113/jphysiol.1985.sp015900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessy C, Kim I, Sougnez CL, Laporte R, Morgan KG. A role for MAP kinase in differentiated smooth muscle contraction evoked by α-adrenoceptor stimulation. American Journal of Physiology. 1998;275:C1081–1086. doi: 10.1152/ajpcell.1998.275.4.C1081. [DOI] [PubMed] [Google Scholar]
- Earley JJ, Su X, Moreland RS. Caldesmon inhibits active crossbridges in unstimulated vascular smooth muscle: an antisense oligodeoxynucleotide approach. Circulation Research. 1998;83:661–667. doi: 10.1161/01.res.83.6.661. [DOI] [PubMed] [Google Scholar]
- Gimona M, Sparrow MP, Strasser P, Herzog M, Small JV. Calponin and SM 22 isoforms in avian and mammalian smooth muscle. Absence of phosphorylation in vivo. European Journal of Biochemistry. 1992;205:1067–1075. doi: 10.1111/j.1432-1033.1992.tb16875.x. [DOI] [PubMed] [Google Scholar]
- Haeberle JR. Calponin decreases the rate of cross-bridge cycling and increases maximum force production by smooth muscle myosin in an in vitro motility assay. Journal of Biological Chemistry. 1994;269:12424–12431. [PubMed] [Google Scholar]
- Horiuchi KY, Chacko S. The mechanism for the inhibition of actin-activated ATPase of smooth muscle heavy meromyosin by calponin. Biochemical and Biophysical Research Communications. 1991;176:1487–1493. doi: 10.1016/0006-291x(91)90455-g. [DOI] [PubMed] [Google Scholar]
- Horowitz A, Menice CB, Laporte R, Morgan KG. Mechanisms of smooth muscle contraction. Physiological Reviews. 1996;76:967–1003. doi: 10.1152/physrev.1996.76.4.967. [DOI] [PubMed] [Google Scholar]
- Hulvershorn J, Gallant C, Wang, C-L W, Dessy C, Morgan KG. Calmodulin levels are dynamically regulated in living vascular smooth muscle cells. American Journal of Physiology – Heart Physiology. 2001;280:1422–H1426. doi: 10.1152/ajpheart.2001.280.3.H1422. [DOI] [PubMed] [Google Scholar]
- Jiang MJ, Morgan KG. Agonist-specific myosin phosphorylation and intracellular calcium during isometric contractions of arterial smooth muscle. Pflügers Archiv. 1989;413:637–643. doi: 10.1007/BF00581814. [DOI] [PubMed] [Google Scholar]
- Johnson JA, Gray MO, Karliner JS, Chen CH, Mochly-Rosen D. An improved permeabilization protocol for the introduction of peptides into cardiac myocytes. Application to protein kinase C research. Circulation Research. 1996;79:1086–1099. doi: 10.1161/01.res.79.6.1086. [DOI] [PubMed] [Google Scholar]
- Johnson PC, Ware JA, Cliveden PB, Smith M, Dvorak AM, Salzman EW. Measurement of ionized calcium in blood platelets with the photoprotein aequorin. Comparison with Quin 2. Journal of Biological Chemistry. 1985;260:2069–2076. [PubMed] [Google Scholar]
- Katsuyama H, Wang C-L A, Morgan KG. Regulation of vascular smooth muscle tone by caldesmon. Journal of Biological Chemistry. 1992;267:14555–14558. [PubMed] [Google Scholar]
- Khalil RA, Lajoie C, Resnick MS, Morgan KG. Ca2+-independent isoforms of protein kinase C differentially translocate in smooth muscle. American Journal of Physiology. 1992;263:C714–719. doi: 10.1152/ajpcell.1992.263.3.C714. [DOI] [PubMed] [Google Scholar]
- Kim I, Je H-D, Gallant C, Zhan Q, Van Riper D, Badwey JA, Singer HA, Morgan KG. Ca2+-calmodulin-dependent protein kinase II-dependent activation of contractility in ferret aorta. Journal of Physiology. 2000;526:367–374. doi: 10.1111/j.1469-7793.2000.00367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Kitazawa T, Somlyo AV, Somlyo AP. Cytosolic heparin inhibits muscarinic and alpha-adrenergic Ca2+ release in smooth muscle. Journal of Biological Chemistry. 1989;264:17997–18004. [PubMed] [Google Scholar]
- Lehman W. 35 kDa proteins are not components of vertebrate smooth muscle thin filaments. Biochimica et Biophysica Acta. 1989;996:57–61. doi: 10.1016/0167-4838(89)90094-0. [DOI] [PubMed] [Google Scholar]
- Lehman W. Calponin and the composition of smooth muscle thin filaments. Journal of Muscle Research and Cell Motility. 1991;12:221–224. doi: 10.1007/BF01745110. [DOI] [PubMed] [Google Scholar]
- Leinweber B, Parissenti AM, Gallant C, Gangopadhyay SS, Kirwan-Rhude A, Leavis PC, Morgan KG. Regulation of protein kinase C by the cytoskeletal protein calponin. Journal of Biological Chemistry. 2000;275:40329–40336. doi: 10.1074/jbc.M008257200. [DOI] [PubMed] [Google Scholar]
- Leinweber BD, Leavis PC, Grabarek Z, Wang C-L A, Morgan KG. Extracellular regulated kinase (ERK) interaction with actin and the calponin homology (CH) domain of actin-binding proteins. Biochemical Journal. 1999;344:117–123. [PMC free article] [PubMed] [Google Scholar]
- Lesh RE, Somlyo AP, Owens GK, Somlyo AV. Reversible permeabilization. A novel technique for the intracellular introduction of antisense oligonucleotides into intact smooth muscle. Circulation Research. 1995;77:220–230. doi: 10.1161/01.res.77.2.220. [DOI] [PubMed] [Google Scholar]
- Mabuchi K, Li Y, Tao T, Wang CL. Immunocytochemical localization of caldesmon and calponin in chicken gizzard smooth muscle. Journal of Muscle Research and Cell Motility. 1996;17:243–260. doi: 10.1007/BF00124246. [DOI] [PubMed] [Google Scholar]
- Makuch R, Birukov K, Shirinsky V, Dabrowska R. Functional interrelationship between calponin and caldesmon. Biochemical Journal. 1991;280:33–38. doi: 10.1042/bj2800033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmqvist U, Trybus KM, Yagi S, Carmichael J, Fay FS. Slow cycling of unphosphorylated myosin is inhibited by calponin, thus keeping smooth muscle relaxed. Proceedings of the National Academy of Sciences of the USA. 1997;94:7655–7660. doi: 10.1073/pnas.94.14.7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston SB. Properties of calponin isolated from sheep aorta thin filaments. FEBS Letters. 1991;292:179–182. doi: 10.1016/0014-5793(91)80862-w. [DOI] [PubMed] [Google Scholar]
- Matthew JD, Khromov AS, McDuffie MJ, Somlyo AV, Somlyo AP, Taniguchi S, Takahashi V. Contractile properties and proteins of smooth muscles of a calponin knockout mouse. Journal of Physiology. 2000;529:811–824. doi: 10.1111/j.1469-7793.2000.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meininger GA, Moore EDW, Schmidt DJ, Lifshitz LM, Fay FS. Distribution of active protein kinase C in smooth muscle. Biophysical Journal. 1999;77:973–984. doi: 10.1016/S0006-3495(99)76948-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menice CB, Hulvershorn J, Adam LP, Wang C-L A, Morgan KG. Calponin and mitogen-activated protein kinase signaling in differentiated vascular smooth muscle. Journal of Biological Chemistry. 1997;272:25157–25161. doi: 10.1074/jbc.272.40.25157. [DOI] [PubMed] [Google Scholar]
- Mezgueldi M, Mendre C, Calas B, Kassab R, Fattoum A. Characterization of the regulatory domain of gizzard calponin – interactions of the 145–163 region with F-actin, calcium-binding proteins, and tropomyosin. Journal of Biological Chemistry. 1995;270:8867–8876. doi: 10.1074/jbc.270.15.8867. [DOI] [PubMed] [Google Scholar]
- Morgan JP, Morgan KG. Stimulus-specific patterns of intracellular calcium levels in ferret portal vein smooth muscle. Journal of Physiology. 1984;351:155–167. doi: 10.1113/jphysiol.1984.sp015239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan KG, Gangopadhyay SS. Crossbridge regulation by thin filament associated proteins. Journal of Applied Physiology. 2001;91:953–962. doi: 10.1152/jappl.2001.91.2.953. [DOI] [PubMed] [Google Scholar]
- Morgan KG, Jiang MJ. Measurement of cytoplasmic Ca2+ during tonic and phasic contractions of mammalian smooth muscle. Excerpta Medica Elsevier Science Publishers: International Congress Series. 1987;725:109–123. [Google Scholar]
- Nagumo H, Sakurada K, Seto M, Sasaki Y. Phosphorylation of calponin by PKC is blocked by F-actin in vitro. Biochemical and Biophysical Research Communications. 1994;203:1502–1507. doi: 10.1006/bbrc.1994.2355. [DOI] [PubMed] [Google Scholar]
- Nigam R, Triggle CR, Jin JP. h1- and h2-calponins are not essential for norepinephrine- or sodium fluoride-induced contraction of rat aortic smooth muscle. Journal of Muscle Research and Cell Motility. 1998;19:695–703. doi: 10.1023/a:1005389300151. [DOI] [PubMed] [Google Scholar]
- North AJ, Gimona M, Cross RA, Small JV. Calponin is localised in both the contractile apparatus and the cytoskeleton of smooth muscle cells. Journal of Cell Science. 1994;107:437–444. doi: 10.1242/jcs.107.3.437. [DOI] [PubMed] [Google Scholar]
- Parker CA, Takahashi K, Tang JX, Tao T, Morgan KG. Cytoskeletal targeting of calponin in differentiated, contractile smooth muscle cells of the ferret. Journal of Physiology. 1998;508:187–198. doi: 10.1111/j.1469-7793.1998.187br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CA, Takahashi K, Tao T, Morgan KG. Agonist-induced redistribution of calponin in contractile vascular smooth muscle cells. American Journal of Physiology. 1994;267:C1262–1270. doi: 10.1152/ajpcell.1994.267.5.C1262. [DOI] [PubMed] [Google Scholar]
- Pawlowski J, Morgan KG. Mechanism of intrinsic tone in ferret vascular smooth muscle. Journal of Physiology. 1992;448:121–132. doi: 10.1113/jphysiol.1992.sp019032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl J, Winder V, Allen BG, Walsh MP, Sellers JR, Gerthoffer WT. Phosphorylation of calponin in airway smooth muscle. American Journal of Physiology. 1997;272:L115–123. doi: 10.1152/ajplung.1997.272.1.L115. [DOI] [PubMed] [Google Scholar]
- Rembold CM, Murphy RA. Myoplasmic calcium, myosin phosphorylation, and regulation of the crossbridge cycle in swine arterial smooth muscle. Circulation Research. 1986;58:803–815. doi: 10.1161/01.res.58.6.803. [DOI] [PubMed] [Google Scholar]
- Rokolya A, Walsh MP, Moreland RS. Calcium- and phorbol ester-dependent calponin phosphorylation in homogenates of swine carotid artery. American Journal of Physiology. 1996;271:H776–783. doi: 10.1152/ajpheart.1996.271.2.H776. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Hiwada K, Kokubu T. Isolation and characterization of a 34000-dalton calmodulin- and F-actin-binding protein from chicken gizzard smooth muscle. Biochemical and Biophysical Research Communications. 1986;141:20–26. doi: 10.1016/s0006-291x(86)80328-x. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Hiwada K, Kobuku T. Vascular smooth muscle calponin: a novel troponin T-like protein. Hypertension. 1988;11:620–626. doi: 10.1161/01.hyp.11.6.620. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Nadal-Ginard B. Molecular cloning and sequence analysis of smooth muscle calponin. Journal of Biological Chemistry. 1991;266:13284–13288. [PubMed] [Google Scholar]
- Takahashi K, Yoshimoto R, Fuchibe K, Fujishige A, Mitsui-Saito M, Hori M, Ozaki H, Yamamura H, Awata N, Taniguchi S, Katsuki M, Tsuchiya T, Karaki H. Regulation of shortening velocity by calponin in intact contracting smooth muscles. Biochemical and Biophysical Research Communications. 2000;279:150–157. doi: 10.1006/bbrc.2000.3909. [DOI] [PubMed] [Google Scholar]
- Walsh MP. Calponin – knocked out but not down. Journal of Physiology. 2000;529:517. doi: 10.1111/j.1469-7793.2000.00517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder SJ, Allen BG, Fraser ED, Kang HM, Kargacin GJ, Walsh MP. Calponin phosphory lation in vitro and in intact muscle. Biochemical Journal. 1993;296:827–836. doi: 10.1042/bj2960827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder SJ, Sutherland C, Walsh MP. Biochemical and functional characterization of smooth muscle calponin. Advances in Experimental Medicine and Biology. 1991;304:37–51. doi: 10.1007/978-1-4684-6003-2_5. [DOI] [PubMed] [Google Scholar]
- Winder SJ, Walsh MP. Smooth muscle calponin: inhibition of actomyosin MgATPase and regulation by phosphorylation. Journal of Biological Chemistry. 1990;265:10148–10155. [PubMed] [Google Scholar]
- Winder SJ, Walsh MP. Calponin: thin filament-linked regulation of smooth muscle contraction. Cellular Signalling. 1993;5:677–686. doi: 10.1016/0898-6568(93)90029-l. [DOI] [PubMed] [Google Scholar]
- Yoshikawa H, Taniguchi S-I, Yamamura H, Mori S, Sugimoto M, Miyado K, Nakamura K, Nakao K, Katsuki M, Shibata N, Takahashi K. Mice lacking smooth muscle calponin display increased bone formation that is associated with enhancement of bone morphogenetic protein responses. Genes to Cells. 1998;3:685–695. doi: 10.1046/j.1365-2443.1998.00214.x. [DOI] [PubMed] [Google Scholar]
- Zimmerman AN, Hülsmann WC. Paradoxical influence of calcium ions on the permeability of the cell membranes of the isolated rat heart. Nature. 1966;211:646–647. doi: 10.1038/211646a0. [DOI] [PubMed] [Google Scholar]