

Abstract
Arachidonic acid (AA) exerts multiple physiological and pathophysiological effects in the brain. By continuously measuring the intracellular pH (pHi) and Ca2+ levels ([Ca2+]i) in primary cultured rat cerebellar granule cells, we have found, for the first time, that 20 min treatment with 10 μm AA resulted in marked increases in Ca2+ and H+ levels in both the cytosol and nucleus.
A much higher concentration (40 mm) of another weak acid, propionic acid, was needed to induce a similar change in pHi. The [Ca2+]i increase was probably caused by AA-induced activation of Ni2+-sensitive cationic channels, but did not involve NMDA channels or the Na+-Ca2+ exchanger.
AA-induced acidosis occurs by a different mechanism involving predominantly the passive diffusion of the un-ionized form of AA, rather than a protein carrier, as proposed by Kamp & Hamilton for fatty acids (FAs) in artificial phospholipid bilayers (the ‘flip-flop’ model). The following results, which are similar to those observed in lipid bilayers, support this conclusion: (1) FAs containing a -COOH group (AA, linoleic acid, α-linolenic acid, and docosahexaenoic acid) induced intracellular acidosis, whereas a FA with a -COOCH3 group (AA methyl ester) had little effect on pHi, (2) a FA amine, tetradecylamine, induced intracellular alkalosis, and (3) the AA-/FA-induced pHi changes were reversed by bovine serum albumin.
Further evidence in support of a passive diffusion model, rather than a membrane protein carrier, is that: (1) there was a linear relationship between the initial rate of acid flux and the concentration of AA (2-100 μm), (2) acidosis was not inhibited by 4,4′-diisothiocyanatostilbene-2,2′-disulphonic acid, a potent inhibitor of the plasma membrane FA carrier protein, and (3) the involvement of most known H+-related membrane carriers and H+ conductance has been ruled out.
Since AA can be released under both physiological and pathophysiological conditions, the possible significance of the AA-evoked increases in H+ and Ca2+ in both the cytoplasm and nucleoplasm is discussed.
Arachidonic acid (AA), a non-esterified fatty acid (FA), and its metabolites have been suggested to exert many biological effects in neurones (Katsuki & Okuda, 1995). For example, when N-methyl-d-aspartate (NMDA) channels are opened by glutamate, AA is released by calcium-dependent activation of phospholipase A2 (PLA2; Dumuis et al. 1988); this potentiates the NMDA current (Miller et al. 1992) and acts as a retrograde messenger to maintain long-term potentiation (LTP; Schaechter & Benowitz, 1993). In addition to this important physiological role, it has been suggested that AA accumulation resulting from extensive activation of NMDA channels is relevant to glutamate-induced neurotoxicity (Rothman et al. 1993). The massive amount of AA and other FAs released during brain ischaemia, seizures and traumatic stimuli (Umemura et al. 1992) has also been suggested to cause neuronal damage (Farooqui et al. 1997).
There have only been a few reports of FAs (including AA) causing marked intracellular acidosis in living cells, the cell types involved being pancreatic β-cells, adipocytes and cardiac cells (Hamilton et al. 1994; Civelek et al. 1996; Wu et al. 2000), other studies having been performed on artificial phospholipid vesicles (Kamp & Hamilton, 1992, 1993; Kamp et al. 1995). The mechanism proposed by Kamp & Hamilton (1992) for FA (oleic acid)-induced acidosis, the ‘flip-flop’ model, involves simple diffusion of free FAs across the lipid bilayer, rather than carrier-mediated transport. In contrast, in many other cell types, including platelets, macrophages and neutrophils, the addition of ≈5-10 μm AA induces a marked alkalosis (i.e. pHi increase; Kapus et al. 1994; Nanda et al. 1994; Henderson et al. 1995). This is explained by the AA-induced activation of NADPH oxidase and the generation of O2-., resulting in depolarization of the membrane potential (Vm) and activation of an outward H+ current (as the depolarized Vm is less negative than the H+ equilibrium potential, EH), as confirmed by patch-clamp experiments (Kapus et al. 1994; Nanda et al. 1994; Henderson et al. 1995).
A reduction in pHi and an increase in cytosolic and nuclear Ca2+ levels have been shown to greatly influence the gene expression or phosphorylation of nuclear proteins (see Finkbeiner & Greenberg, 1998 for review) and neuronal injury (Choi, 1988; Dubinsky, 1995). In the present paper, we show for the first time that in neurones, an intriguing and profound cytoplasmic acidosis (≈0.32 pH units), rather than alkalosis, is induced by the addition of AA/FAs at a concentration as low as 10 μm. This is much lower than the concentrations of weak acids (e.g. 40 mm propionic acid) required to induce an intracellular acidosis of similar magnitude. Moreover, an increase in the intracellular Ca2+ concentration ([Ca2+]i) was also observed on the addition of AA. As far as we are aware, this is also the first study in any cell type to show that AA can cause an increase in both H+ and Ca2+ levels in the nucleus. Since AA has significant physiological and pathophysiological effects in neurones, and since it is one of the major FAs that accumulate in the synapse during LTP or brain ischaemia, the possible mechanism involved in these intriguing AA-induced intracellular increases in H+ and Ca2+ has been investigated. In addition, the possible physiological or pathophysiological role of the AA-induced cytoplasmic and nuclear H+ and Ca2+ changes is discussed.
METHODS
Chemicals and solutions
All test solutions were prepared in Hepes-buffered modified Tyrode solution, containing (mm): NaCl 118, KCl 4.5, MgCl2 1.0, CaCl2 2.0, glucose 11, Hepes 10; the pH being adjusted to 7.4 with NaOH at 37 °C, unless otherwise specified. A 10 mm stock solution of AA in ethanol was prepared and stored under N2. When chemicals were added at concentrations greater than 5 mm, the fraction of NaCl was reduced accordingly to keep the osmolarity constant. Unless otherwise specified, all chemicals were purchased from Sigma (St Louis, MO, USA). U78517F, an O2-./OH. scavenger, was a generous gift from Dr E. J. Jacobsen (Medicinal Chemistry Research Unit, Pharmacia & Upjohn Co., Kalamazoo, MI, US).
Primary culture of cerebellar granule cells
All procedures described in this study were approved by the Animal Care and Use Committee of the College of Medicine, National Taiwan University.
Rat cerebellar granule cells were prepared and cultured essentially as described previously (Chen et al. 1999). In brief, 7-day-old Wistar rats (of either sex) were killed by cervical dislocation, then decapitated. The cerebella were removed, minced and dissociated with 0.025 % trypsin for 15 min at 37 °C. The dissociated cells were suspended in basal modified Eagle's medium containing 10 % fetal calf serum, 25 mm KCl, 2 mm glutamine and 50 μg ml−1 of gentamycin, then plated on poly-l-lysine-coated, 24 mm coverslips, and maintained in a humidified 5 % CO2 incubator. Cytosine arabinoside (10 μm) was added 24 h after plating, to kill and arrest the replication of non-neuronal cells, especially astrocytes. After 6-7 days in culture, the purity of the granule cells was generally greater than 90 %.
Measurement of pHi
Cells were loaded for 10-15 min at room temperature with 5 μm 2′,7′-bis(carboxyethyl)-5,6-carboxyfluorescein acetoxymethyl ester (BCECF AM; Molecular Probes). A small group of cells (approximately five cells for each experiment) were alternately excited with 490 and 440 nm wavelength light, using a filter wheel (Cairn Research, Faversham, UK), rotating at 32 Hz. The BCECF fluorescence was measured using an inverted microscope (Nikon Diaphot) converted for epifluorescence. The ratio of the 510 nm emission at 490 nm excitation over that at 440 nm excitation was calculated and converted to a linear pH scale (see below) by in situ calibration at pH 5.5 and 9.5, using the nigericin technique (Rink et al. 1982; Wu et al. 1999) when required. All data were corrected for background fluorescence (cell-free area). The following equation was used to convert the fluorescence ratio into pHi:
where R is the ratio of the 510 nm fluorescence at 490 nm excitation over that at 440 nm excitation, Rmax and Rmin are the maximum and minimum ratio values from the data curve, respectively, and the pKa (-log of dissociation constant) is 7.16. F440,min/F440,max is the ratio of the fluorescence measured at 440 nm of Rmin and Rmax.
Measurement of [Ca2+]i
The method for measuring [Ca2+]i (Wu et al. 1997; Chen et. al. 1999) was similar to that used for pH measurement. In brief, cells were loaded for 60 min at room temperature with 5 μm fura-2 AM (Molecular Probes). The ratio of the emission at 510 nm at the excitation wavelengths of 340 and 380 nm was calculated and converted to [Ca2+]i using the following equation (Grynkiewicz et al. 1985):
where R is the ratio of the 510 nm fluorescence at 340 nm excitation over that at 380 nm excitation. All data were corrected for background fluorescence (cell-free area). Calibration constants were obtained by adding 5 μm ionomycin to a solution containing 10 mm Ca2+ (Rmax) or to a calcium-free solution containing 10 mm EGTA (Rmin). A Kd of 224 nm was used (Grynkiewicz et al. 1985). Sf2/Sb2 is the ratio of the 510 nm emissions at 380 nm excitation determined at Rmin and Rmax. We tested whether the Kd for fura-2 was altered when the pHi changed, using an in vitro test (fura-2 free acid with different pH values, from pH 6.0 to 8.0), and found that in the pH range of 6.3-8.0, the 340/380 ratio showed little change.
Electrophysiology
The whole-cell configuration of the patch-clamp technique was used to record ionic currents (Su et al. 1997) in granule cells. During measurement of the proton current, possible contamination by K+, Na+, Ca2+, or Cl− currents was prevented by bathing the cell in calcium-free, Mg2+/caesium aspartate-containing, Hepes (100 mm)-buffered solution (pHo 7.4) and dialysing internally with a caesium aspartate, Hepes (100 mm)-buffered solution (pHi 7.1). Membrane currents were determined by 3 s depolarizing or hyperpolarizing steps from the holding potential of -40 mV to potential levels between +60 and -120 mV (see Fig. 5).
Figure 5. I–V curves obtained from the averaged data for 5-6 cells.
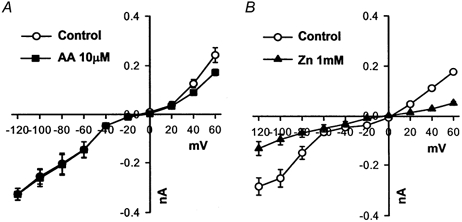
The experimental protocol was similar to that in Fig. 4. Current amplitudes obtained at 3 s of depolarizing and hyperpolarizing pulses were plotted against the depolarizing and hyperpolarizing potentials.
Confocal imaging measurement of cytosolic and nuclear Ca2+ and pH
Optical sectioning allows independent nuclear and cytoplasmic fluorescent signals to be obtained. Fluo-3 AM (5 μm, loaded at room temperature for 40 min) or BCECF AM (5 μm, loaded for 10 min at room temperature) was used to measure the Ca2+ or pH signal in the cytosolic and nuclear compartments, respectively, in a single neurone using a Zeiss LSM 510 confocal laser scanning imaging system equipped with an inverted Zeiss microscope and a × 100, numerical aperture 1.3, oil-immersion objective. At the end of each experiment, the nucleus was identified by labelling with the specific nuclear marker propidium iodide (PI). To reduce photobleaching and damage to cells by laser illumination, the excitation intensity was reduced to 0.3-0.5 % with a tuneable filter. For Ca2+ and pH measurements, the cell was excited every 15 s at a wavelength of 488 nm by a 25 mW argon ion laser. The X–Y plane images (512 pixels × 512 pixels) of the entire selected cell were generated from fluorescence emission images collected with a 505-550 nm band-pass filter. Because fluo-3 is a single-wavelength dye, we used an arbitrary relative fluorescence ratio value, R = F/Fo, where Fo is the fluo-3 fluorescence intensity at the resting [Ca2+], rather than an absolute [Ca2+], as an indicator of the relative [Ca2+]. Since the confocal system was not equipped with a 440 nm laser (i.e. BCECF excitation was at 488 nm), R = F/Fo was also used as a measure of relative pH changes. The nuclear contour was confirmed by specific staining with the PI at the end of each experiment. PI excitation was achieved with the 543 nm laser line and images were collected using a 585 nm long-pass filter.
Data analysis and processing were performed as described previously (Lipp et al. 1997). In brief, all data were corrected for background (cell-free area) and plotted for two different regions of interest (ROI), the soma and the nucleus. The boundaries of the cell were not included in the ROI (Lipp et al. 1997).
Statistics
All results are expressed as the mean ±s.e.m. for a given number of experiments (n). Statistical differences were compared using Student's paired or non-paired t test, and a P value of < 0.05 was considered significant.
RESULTS
The resting level of pHi in granule cells in the nominally bicarbonate-free, Hepes-buffered medium was 7.11 ± 0.01 (n = 32). Continuous perfusion with 10 μm AA induced a profound intracellular acidosis (0.32 ± 0.01 pH units, n = 60, Fig. 1A). When increasing concentrations of AA (2-100 μm, Fig. 1B) were used, the amplitude of the AA-induced acidosis (ΔpHi) plateaued at a concentration of 10 μm (Fig. 1C).
Figure 1. Arachidonic acid (AA)-induced intracellular acidosis in rat cerebellar granule cells.
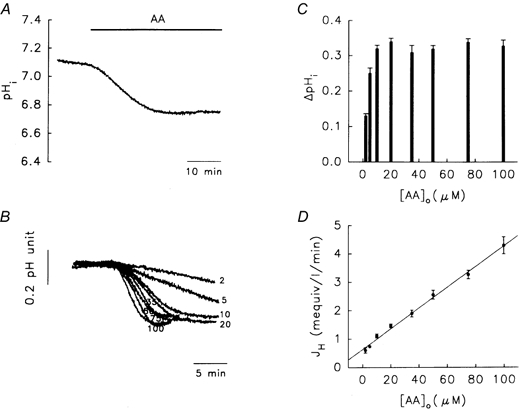
A, intracellular acidosis induced by continuous perfusion with 10 μm AA for 15-20 min. B and C, acidosis produced by different concentrations of [AA]o, i.e. 2, 5, 10, 20, 35, 50, 75 and 100 μm. ΔpHi is the amplitude of the intracellular acidosis produced (C). D, initial rate of pHi change (JH) versus [AA]o (data from B; the correlation coefficient for line fitting was 0.98). In C and D, the data are the means of at least five experiments. All experiments were performed in Hepes-buffered solutions (pHo 7.4) at 37 °C.
To avoid masking effects due to the activation of acid extrusion/buffering mechanisms during AA treatment, the initial rate of acid flux (JH=βi×ΔpHi min−1, where intrinsic buffering power βi= 33.3 ± 0.3 mm; n = 9 at pHi 7.1; Vaughan-Jones & Wu, 1990; Wu et al. 1994) was calculated and found to increase linearly with increasing AA concentration over the range 2-100 μm (Fig. 1D). In subsequent experiments, we used 10 μm AA to investigate the mechanism(s) involved, since this concentration gave an optimal response, and many physiological and/or pathophysiological effects of AA can be seen at this concentration (Miller et al. 1992; Katsuki & Okuda, 1995; Farooqui et al. 1997).
AA-induced [Ca2+]i increase: possible role of the Ca2+ increase in the AA-induced acidosis
Depolarization of the membrane potential and activation of NMDA channels both caused an increase in [Ca2+]i. An increase in [Ca2+]i induces an internal acid load via activation of the Ca2+-H+-ATPase (i.e. the - exchanger, Wu et al. 1999) in granule cells. If such an effect occurred in our system, the acidosis evoked by AA could be due to changes in [Ca2+]i, and this was therefore tested.
The basal level of [Ca2+]i in granule cells was 30 ± 10 nm (n = 56). After 15 min treatment with AA (Fig. 2A), the [Ca2+]i increased to 275 ± 15 nm (n = 10), but could be reversed by calcium-free/EGTA medium, suggesting that AA caused an influx of Ca2+. When 0.1 % ethanol, the solvent for AA, was used, there was little change in either pHi or [Ca2+]i (data not shown). AA is known to open NMDA channels in granule cells (Miller et al. 1992), but this did not account for the AA-induced increase in [Ca2+]i, since 10 μm MK-801 (an NMDA channel inhibitor) had little effect on the AA-induced increase (Fig. 2B, 260 ± 10 nm, n = 4). Since AA induces a marked influx of Ca2+, which is blocked by Ni2+, a non-specific Na+-Ca2+ exchanger inhibitor (Levi et al. 1994), it was possible that the Na+-Ca2+ exchanger might be involved in the increase in Ca2+. However, this was ruled out by the use of sodium-free medium (inhibition of the Na+-Ca2+ exchanger; Fig. 2C). Moreover, the AA-induced acidosis was not blocked by either the removal of Ca2+ from the bathing solution (Fig. 2D) or the addition of 1 mm Ni2+ (Fig. 2E), further suggesting that changes in [Ca2+]i, resulting in changes in pHi, via activation of the Ca2+-ATPase, Na+-Ca2+ exchanger, or NMDA channels, play little role in AA-induced acidosis, and that the mechanisms involved in AA-induced [H+]i and [Ca2+]i increases are different.
Figure 2. Possible role of [Ca2+]i ions in AA-induced acidosis.
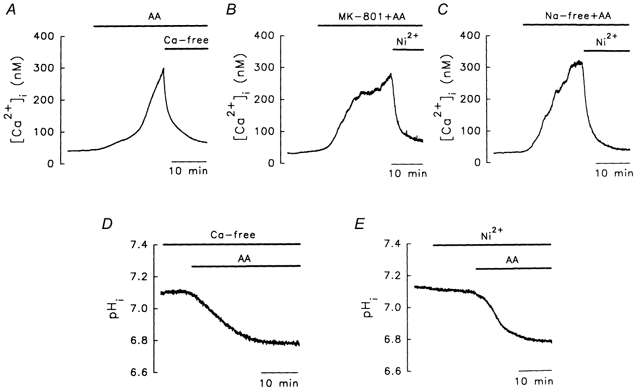
A–C, [Ca2+]i recordings. A, effect of calcium-free medium (+10 mm EGTA). B, effects of MK-801 (10 μm), an NMDA channel inhibitor, and Ni2+ (1 mm). C, lack of effect of the Na+-Ca2+ exchanger. D and E are pHi recordings. D, effect of calcium-free medium on the AA-induced acidosis. E, lack of effect of Ni2+ on the AA-induced acidosis. The concentration of AA was 10 μm.
AA-induced acidosis: lack of involvement of the Na+-H+ exchanger, Cl−-OH− exchanger, K+-H+ exchanger, free radicals, protein kinase C (PKC), or NADPH oxidase
We then investigated whether the Na+-H+ exchanger, the main acid extruder in nominally bicarbonate-free medium (Hepes-buffered solution; Yao et al. 1999), was involved. Figure 3A shows that basal pHi was reduced following the total removal of Na+ from the bathing solution (replacement by N-methyl-d-glucamine), and that an even larger AA-induced acidosis was seen under sodium-free conditions, showing that the Na+-H+ exchanger was not responsible for the acidosis, and that the real magnitude of the AA-induced acidosis was only seen when acid extrusion was blocked. The involvement of two acid loaders, the Cl−-OH− exchanger (Leem & Vaughan-Jones, 1998) and the putative - exchanger, was also tested. Figure 3B shows that resting pHi increased in the chloride-free medium, suggesting that the Cl−-OH− exchanger is possibly present in granule cells; however, the AA-induced acidosis was even greater under these conditions (Fig. 3B, Table 1, see Discussion). When 120 mm KCl (used to inhibit the K+-H+ exchanger and to depolarize the membrane potential to ≈-4 mV) in calcium-free medium (to inhibit the depolarization-induced [Ca2+]i/[H+]i increases) was used, the acidosis was still seen (Fig. 3C), again suggesting that these two acid loaders were not involved.
Figure 3. Lack of involvement of the Na+-H+ exchanger, Cl−-OH− exchanger, K+-H+ exchanger, free radicals, protein kinase C (PKC) and NADPH oxidase in the AA-induced acidosis.
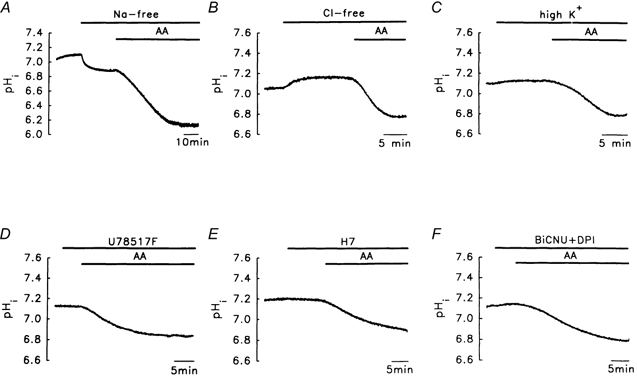
The concentration of AA used was 10 μm. A, the Na+-H+ exchanger is completely blocked by sodium-free medium; N-methyl-d-glucamine (NMDG) was isosmotically substituted for all of the (sodium-free conditions). B, the Cl−-OH− exchanger is inhibited by chloride-free solution ( was totally substituted by gluconate). C, a high-KCl medium (120 mm NaCl was replaced by 120 mm KCl), thus inhibiting the putative K+-H+ exchanger. D, free radical removal. U78517F (20 μm) is an O2-./OH. scavenger. E, PKC inhibition. H7 (200 μm) is a PKC inhibitor. F, NADPH oxidase. Diphenyleneiodonium (DPI, 10 μm) and N,N-bis(2-chloroethyl)-N-nitrosourea (BiCNU; 100 μm) are, respectively, NADPH oxidase and glutathione reductase inhibitors.
Table 1.
Intracellular acidosis induced by the addition of 10 μM arachidonic acid (AA) under various conditions
| Treatment | ΔpHi (pH units) | n |
|---|---|---|
| None (control) | 0.32 ± 0.01 | 60 |
| Calcium-free medium | 0.29 ± 0.03 | 9 |
| Ni2+ | 0.32 ± 0.01 | 4 |
| Sodium-free medium | 0.68 ± 0.04 * | 6 |
| Chloride-free medium | 0.40 ± 0.03 * | 4 |
| High K+, calcium-free medium | 0.29 ± 0.03 | 4 |
| SOD + catalase | 0.34 ± 0.04 | 4 |
| U78517F | 0.27 ± 0.01 | 4 |
| H7 | 0.33 ± 0.02 | 4 |
| TPA | 0.28 ± 0.01 | 4 |
| DPI + BiCNU | 0.27 ± 0.02 | 5 |
SOD, superoxide dismutase; TPA, 12-O-tetradecanoylphorbol 13-acetate; DPI, diphenyleneiodonium; BiCNU, N,N-bis(2-chloroethyl)-N-nitrosourea; n, number of experiments performed.
P < 0.05, non-paired t test compared with the control (0.32 ± 0.01pH units, n = 60).
AA metabolism can generate free radicals, which can induce marked intracellular acidosis in glial cells (Tsai et al. 1997). However, following pretreatment with free radical scavengers (U78517F, an O2-./OH-. scavenger, Fig. 3D) or a mixture of superoxide dismutase (an O2-. scavenger) and catalase (reduction of H2O2 into H2O), no inhibitory effect was seen (Table 1).
Unsaturated FAs, including AA, can activate PKC (Katsuki & Okuda, 1995), which regulates pHi in other cell types (Chen & Wu, 1995; Liaw et al. 1998). However, treatment with either 200 μm H7 (a PKC inhibitor, Fig. 3E) or 1 μm 12-O-tetradecanoylphorbol 13-acetate (a PKC activator, Table 1) had no effect on AA-induced acidosis.
In blood cells, AA activation of NADPH oxidase can result in the production of large amounts of intracellular protons (Kapus et al. 1994; Henderson et al. 1995). If the cells possess a putative NADPH oxidase-activated H+ channel, the flux of intracellular protons through this channel could result in alkalosis. However, this seems not to be the case in granule cells, since a mixture of diphenyleneiodonium (an NADPH oxidase inhibitor) and N,N-bis(2-chloroethyl)-N-nitrosourea (BiCNU; a glutathione reductase inhibitor) had little effect on the AA-induced acidosis (Fig. 3F), suggesting either that NADPH oxidase was not activated by AA, or that there were no NADPH oxidase-activated H+ channels. These results are summarized in Table 1.
It was also possible that the AA-induced acidosis seen in the present study was mediated by activation of an electrogenic inward H+ current, if the Vm was more negative than the EH. We used the patch-clamp technique to investigate this possibility directly.
Lack of involvement of an AA-activated H+ conductance
The following experiments were performed at near-physiological pHi and pHo (7.1 and 7.4, respectively). The poorly permeant ions caesium and aspartate were used as the main constituents of the medium to minimize contamination from other currents. Families of currents generated by applying nine successive 3 s depolarizing and hyperpolarizing pulses to potentials between +60 and -120 mV were recorded before (Fig. 4A) and after (Fig. 4B) the addition of 10 μm AA for 5 min; the I–V curve is shown in Fig. 5A. Under control conditions, the inward proton current reversed at a potential of about -9 mV and showed slight outward-rectifying properties at potentials greater than 0 mV. The average slope conductances at potentials between +20 and +60 mV and between -60 and -120 mV were 5.2 ± 0.4 and 3.0 ± 0.1 nS, respectively (open circles in Fig. 5A, n = 6). On exposure to 10 μm AA for 5 min (Fig. 4B, filled squares in Fig. 5A), there was little effect on the outward (3.5 ± 0.4 nS, n = 6) and inward (3.0 ± 0.1 nS, n = 6) slope conductances (P > 0.05). It is known that 1 mm Zn2+ blocks the basal proton conductance in neurones (Meech & Thomas, 1987). In another set of experiments, 1 mm Zn2+ significantly (P < 0.05) inhibited the control current (Figs 4C and D, and 5B). The outward and inward slope conductances were, respectively, 3.3 ± 0.1 and 3.8 ± 0.5 nS (n = 8) in the absence of Zn2+, and 0.9 ± 0.1 and 1.4 ± 0.1 nS in its presence (n = 8). These results suggest that in the resting state, the granule cell probably possesses a zinc-sensitive H+ conductance that is little affected by the addition of AA. Due to the lack of effect of AA on the putative electrogenic inward H+ current (Fig. 4B and Fig. 5A), we did not test further the properties of the basal inward H+ current in granule cells.
Figure 4. Effects of AA on the proton current.
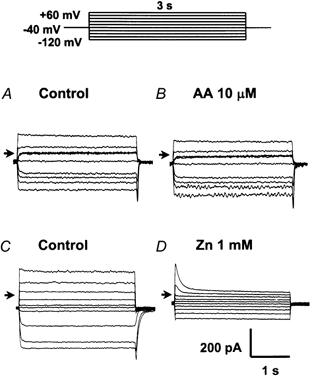
A and C, controls: the basal current. The cells were bathed in magnesium-containing, calcium-free, caesium buffer solution (pH 7.1) and dialysed with a pipette solution containing caesium (pH 7.4) for 10 min to reach equilibrium. Inward and outward proton currents under control conditions were then elicited by 3 s depolarizing and hyperpolarizing pulses to potential levels between +60 and -120 mV. B and D, typical current traces after 5 min exposure to 10 μm AA or 1 mm ZnSO4 are shown. The arrows in A–D represent 0 mV.
We next tested the possible effect of the transmembrane H+ gradient (pHo – pHi) on the 10 μm AA-induced acidosis, as shown in Table 2. AA-induced acidosis at pHo 7.4 and pHi 7.11 was ≈0.32 pH units (control). At a constant pHo 7.4, an increase (chloride-free experiment, Fig. 3B) or decrease (+ 20 mm lactate, NaCl isosmotically substituted) in the pHi resulted, respectively, in an increase (0.40 ± 0.03 pH units, n = 4) or decrease (0.15 ± 0.02 pH units, n = 4) in the amplitude of the AA-induced acidosis (ΔpHi). When pHo was increased to 7.8, pHi rose to ≈7.20 and the AA-induced acidosis was significantly reduced (0.13 ± 0.03 pH units, n = 4), but not increased (≈0.4 pH units, Table 2), as seen under chloride-free conditions. It should be noted that under all of these conditions (pHo - pHi), including the control, the H+ gradient was outwardly directed. However, when an inward proton gradient was imposed (pHo 6.8 and pHi 6.9), the AA-induced acidosis still occurred (0.22 ± 0.03 pH units, n = 4, see Table 2 and Discussion).
Table 2.
Intracellular acidosis induced by continuous perfusion with 10 μM AA under different conditions of pHo and pHi
| Treatment | pHo | pHi | (pHo–pHi) (pH units) | [AA-]i(μM) | ΔpHi (pH units) | n |
|---|---|---|---|---|---|---|
| 1 | 7.4 | 7.25 ± 0.02 | +0.15 | 1.89 | 0.40 ± 0.03 * | 4 |
| 2 | 7.4 | 7.11 ± 0.01 | +0.29 | 1.50 | 0.32 ± 0.01 | 60 |
| 3 | 7.4 | 6.90 ± 0.01 | +0.50 | 1.02 | 0.15 ± 0.02 * | 4 |
| 4 | 7.8 | 7.20 ± 0.02 | +0.60 | 1.10 | 0.13 ± 0.03 * | 4 |
| 5 | 6.8 | 6.90 ± 0.02 | −0.10 | 1.43 | 0.22 ± 0.03 † | 4 |
Treatments 1 and 2: chloride-free medium and control, respectively (data from Table 1). Treatment 3: +20 mM lactate. Treatments 4 and 5: the AA-induced acidosis at pHo 7.8 or 6.8, respectively. [AA-]i was calculated using the Henderson-Hasselbalch equation (pHo= 7.6 + log[AA-]o/[AA-H]o and pHi= 7.6 + log[AA-]i/[AA-H]i; AA was 10 μM, see text for details). n indicates the number of experiments performed.
P < 0.05 non-paired t test compared with the control.
P < 0.05, non-paired t test compared with treatment 1.
Other types of polyunsaturated FAs also induce intracellular acidosis, and the FA-induced pHi changes are reversed by bovine serum albumin (BSA)
Another explanation of the results was simple FA diffusion (flip-flop), mainly demonstrated to occur in lipid bilayers (Kamp & Hamilton, 1993; Kamp et al. 1995). Albumin can extract FAs from the lipid bilayer and increase the rate of efflux (Kamp & Hamilton, 1993; Kamp et al. 1995). If flip-flop were to occur in our system, an increase in the rate of efflux should be seen on the addition of FA-free BSA to granule neurones.
In the presence of AA, the addition of 0.3 % BSA resulted in a pHi recovery of 70 ± 1 % towards the resting level (Fig. 6A, n = 6). Interestingly, as shown in Fig. 6B, the initial rate of pHi recovery (R1= 0.017 ± 0.002 pH units min−1, n = 6, Fig. 6B) on switching to an AA-free solution was significantly lower than that when BSA was subsequently added (R2= 0.191 ± 0.020 pH units min−1, n = 6, P < 0.05), suggesting that BSA probably extracts AA from the lipid membrane, rather than quenching the AA remaining in the solution, since R2 was measured in an AA-free medium. This effect of BSA on pHi recovery was specific to AA-induced acidosis, since no effect was seen on acidosis (0.30 ± 0.02 pH units, n = 6, Fig. 6C) induced by another weak acid, 40 mm propionate, in the presence of a blocker of the Na+-H+ exchanger, 10 μm 5-(N-ethyl-N-isopropyl)-amiloride. Recovery from the AA-induced [Ca2+]i increase was also faster in the presence of 0.3 % BSA (Fig. 6D, R1= 15 ± 3 nm min−1, R2= 89 ± 35 nm min−1, n = 4) and, again, BSA had no effect on the [Ca2+]i increase induced by 100 mm KCl (Fig. 6E, n = 4).
Figure 6. Effects of bovine serum albumin (BSA) on the acidosis and Ca2+ changes induced by AA or other agents.
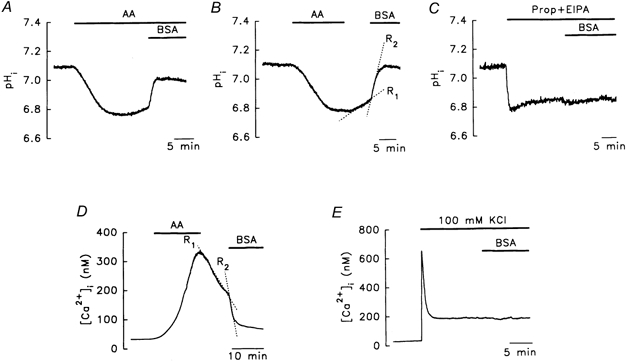
A, in the presence of AA, BSA markedly reversed AA-induced acidosis. B, the rate of pHi recovery was faster in the presence (R2) than in the absence (R1) of BSA. C, BSA did not reverse the acidosis induced by addition of 40 mm propionate in the presence of 10 μm 5-(N-ethyl-N-isopropyl)-amiloride (EIPA) buffer. D and E, the AA-induced [Ca2+]i increase was reversed by BSA, whereas the [Ca2+]i increase induced by 100 mm KCl was not. The concentrations of AA and BSA were 10 μm and 0.3 %, respectively.
We also tested other important polyunsaturated essential FA members of the Ω-6 and Ω-3 families, namely linoleic acid (the precursor of AA), α-linolenic acid, the respective parent FAs of these two families, and docosahexaenoic acid (DHA), the end metabolite of α-linolenic acid, which plays an important role in neuronal development (Simopoulos, 1996). All three FAs caused a marked intracellular acidosis (Table 3), which was reversed by 0.3 % BSA (Fig. 7A–C). In contrast, the addition of AA methyl ester had little effect on pHi (0.02 ± 0.01 pH units, n = 6, Fig. 7D). Tetradecylamine, in its neutral form, attracts intracellular protons, thus producing an intracellular alkalosis in the vesicles, and is suggested to be transported by a simple diffusion mechanism, rather than a protein carrier (Kamp & Hamilton, 1993; Kamp et al. 1995). After perfusion with 10 μm tetradecylamine, a pHi increase of 0.19 ± 0.02 pH units was seen (n = 7), which was again reversed by the addition of 0.3 % BSA (Fig. 7E).
Table 3.
Intracellular acidosis induced by the addition of different fatty acids (10 μM) under various conditions
| Treatment | ΔpHi (pH units) | n |
|---|---|---|
| Linoleic acid | 0.28 ± 0.02 | 8 |
| α-Linolenic acid | 0.31 ± 0.01 | 4 |
| DHA | 0.31 ± 0.01 | 4 |
| AA + DIDS | 0.33 ± 0.03 | 4 |
| Linoleic acid (+ SHAM) | 0.31 ± 0.01 | 4 |
| α-Linolenic acid (+ SHAM) | 0.29 ± 0.01 | 4 |
The ΔpHi under the above treatment conditions did not differ significantly (P > 0.05) from that in the control (0.32 ± 0.01pH units, n = 60). DHA, docosahexaenoic acid; DIDS, 4,4′-diiso-thiocyanatostilbene-2,2′-disulphonic acid; SHAM, salicylhydroxamic acid. n indicates the number of experiments performed.
Figure 7. Effect of other fatty acids (FAs) and FA derivatives.
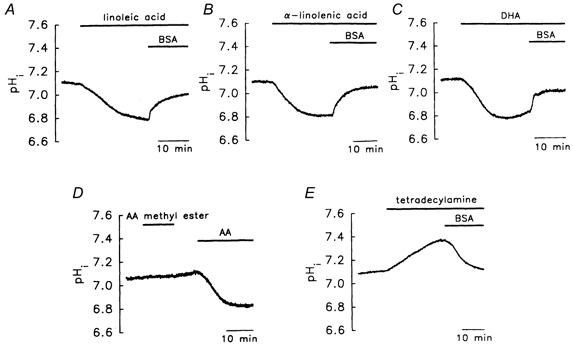
Linoleic acid (A), α-linolenic acid (B), or docosahexaenoic acid (DHA; C) caused a pHi change, while AA methyl ester did not (D). E, tetradecylamine induced an intracellular alkalosis. The concentrations of linoleic acid, α-linolenic acid, DHA, AA methyl ester and tetradecylamine were all 10 μm. The concentration of BSA was 0.3 %.
It has been suggested that FAs are transported by a transmembrane carrier protein (Abumrad et al. 1991). Figure 8A and Table 3 show that treatment with 0.2 mm 4,4′-diisothiocyanatostilbene-2,2′-disulphonic acid (DIDS), a potent inhibitor of the FA membrane transport protein (Abumrad et al. 1991), had no significant effect on the AA-evoked acidosis.
Figure 8. Lack of involvement of the transmembrane protein carrier and essential FA metabolites in AA-/FA-induced acidosis.
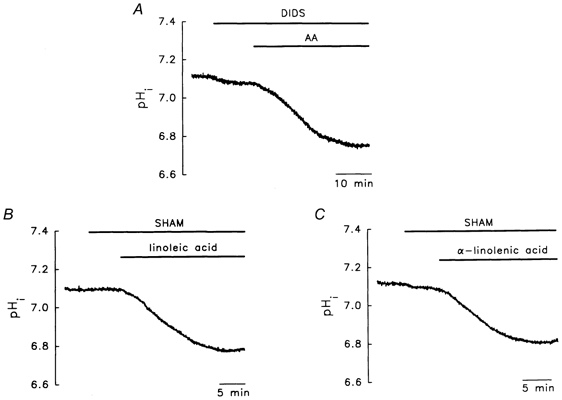
A, DIDS, an inhibitor of the protein carrier, had no effect. B and C, salicylhydroxamic acid (SHAM), an inhibitor of linoleic acid and α-linolenic acid metabolism, had no effect. The concentrations of DIDS and SHAM were 0.2 mm and 40 μm, respectively.
Most known inhibitors of the cyclooxygenase, lipoxygenase, and cytochrome P450 pathways induced marked intracellular acidosis (≈0.2-0.3 pH units). Moreover, an internal acid load induced by the addition of weak acid (20 mm lactate, ≈0.2 pH units) resulted in a smaller response to AA (≈0.15 pH units, Table 2). We therefore used the following method to test the possible involvement of AA metabolites in the acidosis.
The first enzyme involved in the metabolism of both linoleic acid (the precursor of AA) and α-linolenic acid is Δ6 desaturase (Simopoulos, 1996). Salicylhydroxamic acid (SHAM) is a potent Δ6 desaturase inhibitor (Khozin-Goldberg et al. 1999). Our results show that SHAM had little effect on the acidosis evoked by either linoleic acid (Fig. 8B) or α-linolenic acid (Fig. 8C and Table 3), suggesting that AA/FA metabolites probably do not play an important role in the production of acidosis.
AA changes cytoplasmic and nuclear H+ and Ca2+ levels
It has recently been shown that changes in pHi or nuclear [Ca2+]i play an important role in gene expression. It has been suggested that AA regulates the expression of immediate-early genes (Rao et al. 1993; Danesch et al. 1994); it was therefore of interest to know whether it had any effect on nuclear [H+] or [ Ca2+].
We used confocal microscopy to measure cytosolic and nuclear Ca2+ or pH changes (see Methods for details). Figure 9A shows that BCECF was distributed throughout the cell (green fluorescence, Fig. 9Aa), while PI was found only in the nucleus (red fluorescence, Fig. 9Ab); Fig. 9Ac shows an overlay of the two images (yellow fluorescence, ‘C’ and ‘N’ indicating the ROI in the cytosol and nucleus, respectively, to be analysed off-line).
Figure 9. Changes in H+ and Ca2+ levels in the cytosol and nucleus following addition of 10 μm AA.
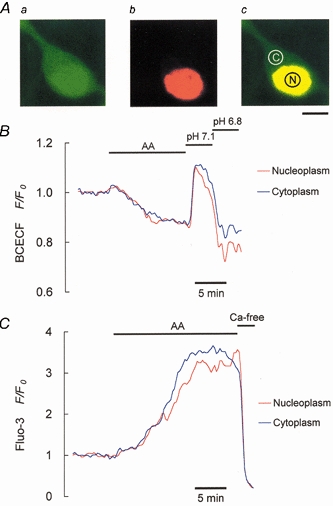
A, images of the cytosolic (a) and nuclear (b) compartments in a single neurone were obtained using 2′,7′-bis(carboxyethyl)-5,6-carboxyfluorescein acetoxymethyl ester (BCECF) and propidium iodide (PI). The horizontal bar under Ac represents 5 μm. Identical areas (5.9 μm2) of regions of interest were chosen in the cytoplasm and nucleoplasm for off-line analyses, shown in B and C. B, AA-induced pH changes in the cytosol and nucleus. C, [Ca2+]i changes were seen in the cytosol and nucleus following addition of AA.
An AA-induced pHi decrease (Fig. 9B) and [Ca2+]i increase (Fig. 9C) were seen in both the cytosol and the nucleus. Since the basal (resting state) fluorescence intensities for BCECF or fluo-3 in the cytosol and nucleoplasm were different (Fig. 9A), we used the F/F0 ratio (F0 is the resting state intensity) to normalize this difference and to represent relative changes in the pH and Ca2+ levels. On addition of AA, changes in H+ and Ca2+ levels were seen in both compartments (Fig. 9B and C). The pH in the cytosol and nucleus was lowered by 18 ± 5 and 20 ± 5 %, respectively (n = 8 cells), compared to resting levels, while [Ca2+]i in the cytosol and nucleus was increased to 320 ± 10 and 305 ± 15 % of resting levels, respectively (n = 6 cells). Moreover, the AA-evoked nuclear pH changes still occurred in calcium-free medium (data not shown). At the end of the experiment, pH 7.1 and 6.8 solutions (containing 140 mm K+-3 μm valinomycin to equalize the pHo and pHi) or calcium-free medium (containing 5 μm ionomycin, to equalize [Ca2+]o and [Ca2+]i) was used to confirm that the observed pH and Ca2+ changes were real.
DISCUSSION
Both heterogeneous and homogeneous distributions of Ca2+ between the cytoplasm and nucleoplasm have been reported in neurones (Al-Mohanna et al. 1994; O'Malley, 1994). In terms of inter-organellar H+ distribution, there are only two reports of basal [H+] levels in the cytoplasm and nucleoplasm, both of which showed the distribution to be heterogeneous (Seksek & Bolard, 1996; Weinlich et al. 1998). As far as we are aware, our study is the first to show that AA causes a simultaneous increase in H+ and Ca2+ levels in both the nucleus and cytosol (Fig. 8B and C). The simplest explanation is that H+ and Ca2+ ions diffuse freely through the nuclear pores, which may be important when considering neuronal gene expression. Moreover, it is also the first to show that essential FAs (including linoleic acid, AA, α-linolenic acid and DHA), which are important in neuronal development (Simopoulos, 1996), can diffuse freely into neurones.
To produce an acidosis of similar magnitude to that seen with 10 μm AA (≈0.32 pH units), a much higher concentration (40 mm) of another weak acid, propionic acid, was required (Fig. 6C). It is intriguing that this profound acidosis induced in neurones by such a low concentration of AA or other FAs can be explained by a simple diffusion mechanism, as proposed by Kamp & Hamilton (1992) for artificial phospholipid bilayers (the ‘flip-flop’ model). The ‘flip-flop’ model is explained in more detail below.
The apparent pK for long-chain FAs in lipid bilayers is high (≈7.6, as assessed by 13C-NMR imaging; Kamp et al. 1995). One explanation for this is that the charged surface of the membrane affects the ionization of FAs (FA−), thus increasing the apparent pK and the formation of the non-ionized form (FA-H). Since FAs have very high oil-water partition coefficients (i.e. high lipid solubility), almost all added FA molecules bind to the lipid bilayer. At a pHo of 7.4 and with a pK of 7.6, the FA bound in the outer leaflet consists of almost equal amounts of FA-H and FA− (Henderson-Hasselbalch equation). FA− diffuses slowly (a t½ of minutes) and, consequently, cyclic transport of H+ does not occur in the bilayer. However, 50 % of the FA-H diffuses rapidly (a t½ of seconds) from the outer to the inner leaflet (‘flip’), where about half dissociates into FA− and H+ (i.e. ≈25 % of the total amount of FA may donate H+ ions to the cytoplasm; Kamp & Hamilton, 1992). On the addition of BSA, FA-H leaves the outer leaflet to bind to BSA and is replaced by FA-H moving from the inner to the outer leaflet (‘flop’) of the membrane, thus transporting H+ from the cytosol to the external medium (Fig. 6 and Fig. 7). Since propionic acid does not accumulate in the lipid bilayer, intracellular protonation depends upon the amount of propionic acid (HA) that dissolves in, and fluxes from, the external solution, resulting in the release of H+ into the cytosol. Since in both the external and internal solutions, propionate has a much lower pK (≈4.7; see Szatkowski & Thomas, 1989) than AA (pK for FAs ≈7.6), almost a 1000-fold higher concentration of propionate (millimolar range; Henderson-Hasselbalch equation) would be required to produce sufficient protonation and acidosis.
One interesting observation that is not seen in any other cell type needs to be discussed. As the outwardly directed H+ gradient increased (pHo - pHi > 0, treatments 1-4, treatment 2 is the control, see Table 2), the AA-induced acidosis decreased (ΔpHi). Compared with treatment 1 (pHo - pHi=+0.15 pH units, an outward proton gradient), however, when an inward proton gradient was imposed (pHo - pHi= -0.1 pH units, treatment 5 in Table 2), the AA-induced acidosis decreased, rather than increased. Thus, the transmembrane H+ gradient seems not to play a major role in the acidosis. One alternative explanation for the above phenomenon is the flip-flop model using the Henderson-Hasselbalch equation: AA-Hi (= AA-Ho) in the inner leaflet can dissociate into equal amounts of and , but, as pHi decreases, the amount of (= ) formed is less (treatment 3 vs. treatment 2 in Table 2) than in intracellular alkalosis (treatment 1 vs. treatment 2). Compared with pHo 7.4 (treatments 1-3), however, when pHo increased to 7.8 (treatment 4), the amount of [AA-H]o (=[AA-H]i) formed in the outer leaflet was also less, resulting in less [AA−]i (=[H+]i) being formed. Since, at different values of pHo and pHi, many factors, including acid extrusion, βi, and/or AA metabolism, can affect or modulate the AA-induced acidosis, other possibilities cannot be ruled out at present.
Factors common to both neurones and artificial bilayers are that: (1) FAs with a -COOH group induce intracellular acidosis, but a FA with a -COOCH3 group has little effect on the pHi, (2) a FA amine induces intracellular alkalosis, and (3) the AA-/FAs-induced pHi changes are reversed by 0.3 % BSA (Fig. 6 and Fig. 7).
Other important evidence from the present study in support of a simple diffusion mechanism, rather than a membrane protein carrier, is as follows.
(1) Using radiolabelled FAs, the saturation concentrations (Vmax) for the known membrane carriers in other cell types are within the range of 0.4-2 μm, and transport is sodium dependent and DIDS sensitive (Stremmel et al. 1986; Abumrad et al. 1991; Schaffer & Lodish, 1994). In contrast, in our study, the AA-induced acidosis was sodium independent (Fig. 3A) and DIDS insensitive (Fig. 8A), indicating that it is probably not mediated via a carrier. Moreover, a plot of the initial rate of acid flux (JH) versus [AA]o (2-100 μm) was linear and did not show saturation (Vmax) at an [AA]o of 100 μm (Fig. 1D), suggesting that a simple diffusion model may be involved (Fick's first law). The idea that at high FA concentrations (micromolar), uptake involves the permeation of the protonated species across the membrane by passive diffusion is supported by the results of Hamilton and co-workers (1994). Since no pHi change was seen when [AA]o was less than 2 μm, and since a detergent effect was seen on the membrane (Farooqui et al. 1997) when [AA]o was greater than 100 μm, the involvement of a very low- or very high-capacity protein carrier in the plasma membrane of granule cells cannot be completely ruled out.
(2) Such a membrane carrier would probably bind the FA anion (e.g. AA−) and, by attracting H+ from the cytosol, cause intracellular alkalinization rather than acidosis. Moreover, no known carrier binds both the positively charged FA amine (e.g. tetradecylamine+, Fig. 7E) and the negatively charged FA anion (e.g. AA−). A putative protein carrier acting as a FA−-H+ co-transporter is also unlikely because of the observed pHi effects of many FA analogues (e.g. Fig. 7).
(3) Although we did not use AA metabolic inhibitors (which result in acid load) to test directly the effect of AA metabolites on the acidosis, our results suggest that the acidosis is unlikely to be caused by metabolites, since (1) in addition to AA itself, AA metabolic pathway-unrelated FAs (e.g. α-linolenic acid and DHA) also produced acidosis (Figs 7B and C), while FA amine (Fig. 7E) produced alkalosis, and (2) SHAM, a Δ6 desaturase inhibitor used to block the metabolism of linoleic acid, the AA precursor, had little effect (Figs 8B and C).
From the above and the following evidence, the lipid bilayer flip-flop model may be modified in living cells:
(1) The AA-induced acidosis is voltage independent (120 mm K+ and voltage-clamp experiments, Fig. 3C and Fig. 5A), suggesting that AA transport is electroneutral.
(2) The involvement of electrogenic H+ conductance (Fig. 4 and Fig. 5) and all known ion (Ca2+, Na+, K+ or Cl−)-coupled, electroneutral/electrogenic membrane H+ carriers was ruled out (Figs 2D, 3A–C and 8A).
(3) The FA-/AA-induced intracellular protonation in granule cells is marked (ΔpHi×βi, ≈mm). If the back flop of FA−/AA− in the inner leaflet is slow, a large amount of FA−/AA− (≈mm) could accumulate in the inner leaflet; however, this seems inherently unlikely.
(4) Cytoplasmic FA− binding proteins (FABPs), have been found in many types of tissue (Ockner et al. 1972), including the cerebellum (Sellner et al. 1995).
From points (1)–(4) above, it is feasible that AA−/FA− in the inner leaflet could be continuously transported by FABPs and metabolized in the mitochondria (this may act as a H+ sink), resulting in the marked protonation. In other words, the driving force for FA-induced intracellular acidosis would be an inwardly directed FA gradient.
The acidic steady state seen when AA was added for about 15 min (Fig. 1A–C) is presumably due to a balance between acid influx and acid extrusion/buffering, the latter probably involving the Na+-H+ exchanger, and βi, FABPs, and FA metabolism. It is not clear why the plateau of the dose-response curve for AA-induced acidosis (ΔpHi) occurred at a concentration as low as 10 μm (Fig. 1C). Possibly, the activity of the Na+-H+ exchanger increases as pHi decreases (Wu & Tseng, 1993). An increase in the activity of cytoplasmic FABPs and/or in the rate of metabolism of FAs as [AA]o increases should also be considered.
Another interesting finding is that AA also induced a marked Ca2+ influx in both the cytoplasm and nucleoplasm (Fig. 9). Our results suggest that NMDA channels (Fig. 2B) and the Na+-Ca2+ exchanger (Fig. 2C) are not involved. Although the exact mechanism is still unclear, the pathway involved in the Ca2+ increase appears to differ from that proposed for acidosis, since Ni2+ blocked the influx of Ca2+ (Fig. 2B and C), but not that of H+ ions (Fig. 2E; it should be noted that 1 mm Ni2+ did not interfere with the signals at either 340/380 or 490/440 nm when mixed with fura-2 or BCECF free acid in the cell bath). It is possible that AA reversibly opens or ‘creates’ non-selective nickel-sensitive channels or pores that allow the passage of Ca2+. The effect of AA on H+ and Ca2+ levels was not completely removed by simple wash-off of AA (AA-free media), complete and fast recovery of these ion levels only being seen in the presence of BSA or Ni2+ (Figs 2, 6 and 7). Thus, under normal conditions, AA produces a long-lasting response. One possible explanation for this is its high lipid solubility.
The AA-induced H+ and Ca2+ increase seen in both the cytoplasm and nucleoplasm is of interest, since lowered pHi and increased [Ca2+]i levels in the nucleus may have a marked effect on immediate-early gene expression or nuclear protein phosphorylation (for review, see Finkbeiner & Greenberg, 1998). Moreover, different patterns of [Ca2+]i increase (Ca2+ spikes, Ca2+ oscillations, and a sustained Ca2+ increase) may result in the expression of different sets of genes (see Finkbeiner & Greenberg, 1998 for review). The present study shows that treatment with AA produced increased nuclear concentrations of both H+ and Ca2+ that were sustained for at least 15-20 min. Since AA is thought to be involved in maintaining LTP (Schaechter & Benowitz, 1993) and in the expression of many growth-associated immediate-early genes (Rao et al. 1993; Danesch et al. 1994), it would be interesting to determine whether this AA-induced long-term increase in H+ and Ca2+ levels has a more direct effect on gene expression.
It is also feasible that if large amounts of AA accumulate at, or are not removed from, the synapse for a long period, this may result in pathological effects (e.g. glutamate-induced neurotoxicity; Choi, 1988; Umemura et al. 1992). Extensive activation of NMDA channels, resulting in AA accumulation, is suggested to be involved in glutamate-induced neurotoxicity (Rothman et al. 1993), and during brain ischaemia, seizures, and trauma, massive amounts of AA and other FAs are released (Umemura et al. 1992) and may cause neuronal damage (Katsuki & Okuda, 1995; Farooqui et al. 1997). However, more studies are required to clarify this point.
Acknowledgments
We gratefully acknowledge the technical help of Mr C.-C. Chan. This work was supported by grant NSC 89-2320-B002-168 to M.-L.W. and grants NTUH90-1000-10 and NSC 89-2320-B-002-172 to W.-H.C.
W.-H. Chen and C.-R. Chen contributed equally to this work.
References
- Abumrad NA, Park JH, Park CR. Permeation of long-chain fatty acid into adipocytes. Journal of Biological Chemistry. 1991;259:8945–8953. [PubMed] [Google Scholar]
- Al-Mohanna FA, Caddy KWT, Bolsover SR. The nucleus is insulated from large cytosolic calcium ion changes. Nature. 1994;367:745–750. doi: 10.1038/367745a0. [DOI] [PubMed] [Google Scholar]
- Chen WH, Chu KC, Wu SJ, Wu JC, Shui HA, Wu ML. Early metabolic inhibition-induced intracellular sodium and calcium increase in rat cerebellar granule cells. Journal of Physiology. 1999;515:133–146. doi: 10.1111/j.1469-7793.1999.133ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Wu ML. Protein kinase C isoform δ is involved in the stimulation of the Na-H exchanger in C6 glioma cells. Molecular Pharmacology. 1995;48:995–1003. [PubMed] [Google Scholar]
- Choi DW. Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends in Neurosciences. 1988;11:465–469. doi: 10.1016/0166-2236(88)90200-7. [DOI] [PubMed] [Google Scholar]
- Civelek VN, Hamilton JA, Tornheim K, Kelly KL, Corkey BE. Intracellular pH in adipocytes: effects of free fatty acid diffusion across the plasma membrane, lipolytic agonists, and insulin. Proceedings of the National Academy of Sciences of the USA. 1996;93:10139–10144. doi: 10.1073/pnas.93.19.10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danesch U, Weber PC, Sellmayer A. Arachidonic acid increases c-fos and Egr-1 mRNA in 3T3 fibroblasts by formation of prostaglandin E2 and activation of protein kinase C. Journal of Biological Chemistry. 1994;269:27258–27263. [PubMed] [Google Scholar]
- Dubinsky JM. Excitotoxicity as a stochastic process. Clinical and Experimental Pharmacology and Physiology. 1995;22:297–298. doi: 10.1111/j.1440-1681.1995.tb02001.x. [DOI] [PubMed] [Google Scholar]
- Dumuis A, Sebben M, Haynes SL, Pin JP, Bockaert J. NMDA receptors activate the arachidonic acid cascade system in striatal neurons. Nature. 1988;336:68–70. doi: 10.1038/336068a0. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Yang HC, Rosenberger TA, Horrocks LA. Phospholipase A2 and its role in brain tissue. Journal of Neurochemistry. 1997;69:889–901. doi: 10.1046/j.1471-4159.1997.69030889.x. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Greenberg ME. Ca2+ channel-regulated neuronal gene expression. Journal of Neurobiology. 1998;37:171–189. [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hamilton JA, Civelek VN, Kamp F, Tornheim K, Corkey BE. Changes in internal pH caused by movement of fatty acids into and out of clonal pancreatic β-cells (HIT) Journal of Biological Chemistry. 1994;269:20852–20856. [PubMed] [Google Scholar]
- Henderson LM, Banting G, Chappell JB. The arachidonate-activable, NADPH oxidase-associated H+ channel. Evidence that gp91-phox functions as an essential part of the channel. Journal of Biological Chemistry. 1995;270:5909–5916. [PubMed] [Google Scholar]
- Kamp F, Hamilton JA. pH gradients across phospholipid membrane caused by fast flip-flop of un-ionized fatty acids. Proceedings of the National Academy of Sciences of the USA. 1992;89:11367–11370. doi: 10.1073/pnas.89.23.11367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp F, Hamilton JA. Movement of fatty acids, fatty acid analogues, and bile acids across phospholipid bilayers. Biochemistry. 1993;32:11074–11086. doi: 10.1021/bi00092a017. [DOI] [PubMed] [Google Scholar]
- Kamp F, Zakim D, Zhang F, Noy N, Hamilton JA. Fatty acid flip-flop in phospholipid bilayers is extremely fast. Biochemistry. 1995;34:11928–11937. doi: 10.1021/bi00037a034. [DOI] [PubMed] [Google Scholar]
- Kapus A, Romanek R, Grinstein S. Arachidonic acid stimulates the membrane H+ conductance of macrophages. Journal of Biological Chemistry. 1994;269:4736–4745. [PubMed] [Google Scholar]
- Katsuki H, Okuda S. Arachidonic acid as a neurotoxic and neurotrophic substance. Progresses in Neurobiology. 1995;46:607–636. doi: 10.1016/0301-0082(95)00016-o. [DOI] [PubMed] [Google Scholar]
- Khozin-Goldberg I, Bigogno C, Cohen Z. Salicylhydroxamic acid inhibits Δ6 desaturase in the microalga Porphyridium Cruentum. Biochimica et Biophysica Acta. 1999;1439:384–394. doi: 10.1016/s1388-1981(99)00107-9. [DOI] [PubMed] [Google Scholar]
- Leem C-H, Vaughan-Jones RD. Sarcolemmal mechanisms for pH recovery from alkalosis in the guinea-pig ventricular myocyte. Journal of Physiology. 1998;509:487–496. doi: 10.1111/j.1469-7793.1998.487bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi AJ, Spitzer KW, Kohmoto O, Bridge JH. Depolarization-induced Ca2+ entry via Na-Ca exchange triggers SR release in guinea pig cardiac myocytes. American Journal of Physiology. 1994;266:H1422–1433. doi: 10.1152/ajpheart.1994.266.4.H1422. [DOI] [PubMed] [Google Scholar]
- Liaw YS, Yang PC, Yu CJ, Kuo SH, Luh KT, Lin YJ, Wu ML. PKC activation is required by EGF-stimulated Na-H exchanger in human pleural mesothelial cells. American Journal of Physiology. 1998;274:L665–672. doi: 10.1152/ajplung.1998.274.5.L665. [DOI] [PubMed] [Google Scholar]
- Lipp P, Thomas D, Berridge MJ, Bootman MD. Nuclear calcium signalling by individual cytoplasmic calcium puffs. EMBO Journal. 1997;16:7166–7173. doi: 10.1093/emboj/16.23.7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meech RW, Thomas RC. Voltage-dependent intracellular pH in Helix aspersa neurons. Journal of Physiology. 1987;390:433–452. doi: 10.1113/jphysiol.1987.sp016710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B, Sarantis M, Traynelis SF, Attwell D. Potentiation of NMDA receptor currents by arachidonic acid. Nature. 1992;355:722–725. doi: 10.1038/355722a0. [DOI] [PubMed] [Google Scholar]
- Nanda A, Romanek R, Curnutte JT, Grinstein S. Assessment of the contribution of the cytochrome b moiety of the NADPH oxidase to the transmembrane H+ conductance of leukocytes. Journal of Biological Chemistry. 1994;269:27285–27289. [PubMed] [Google Scholar]
- Ockner RK, Manning JA, Poppenhausen RB, Ho WKL. A binding protein for fatty acids in cytosol of intestinal mucosa, liver, myocardium, and other tissues. Science. 1972;177:56–58. doi: 10.1126/science.177.4043.56. [DOI] [PubMed] [Google Scholar]
- O'Malley DM. Calcium permeability of the neuronal nuclear envelope: evaluation using confocal volumes and intracellular perfusion. Journal of Neuroscience. 1994;14:5741–5758. doi: 10.1523/JNEUROSCI.14-10-05741.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, G N, Lassègue B, Griendling KK, Alexander RW. Hydrogen peroxide stimulates transcription of c-jun in vascular smooth muscle cells: role of arachidonic acid. Oncogene. 1993;8:2759–2764. [PubMed] [Google Scholar]
- Rink TJ, Tsien RY, Pozzan T. Cytoplasmic pH and free Mg2+ in lymphocytes. Journal of Cell Biology. 1982;95:189–196. doi: 10.1083/jcb.95.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM, Yamada KA, Lancaster N. Nordihydroguaiaretic acid attenuates NMDA neurotoxicity – action beyond the receptor. Neuropharmacology. 1993;32:1279–1288. doi: 10.1016/0028-3908(93)90022-u. [DOI] [PubMed] [Google Scholar]
- Schaechter JD, Benowitz LI. Activation of protein kinase C by arachidonic acid selectively enhances the phosphorylation of GAP-43 in nerve terminal membranes. Journal of Neuroscience. 1993;13:4361–4371. doi: 10.1523/JNEUROSCI.13-10-04361.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer JE, Lodish HF. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell. 1994;79:427–436. doi: 10.1016/0092-8674(94)90252-6. [DOI] [PubMed] [Google Scholar]
- Seksek O, Bolard J. Nuclear pH gradient in mammalian cells revealed by laser microspectrofluorimetry. Journal of Cell Science. 1996;109:257–262. doi: 10.1242/jcs.109.1.257. [DOI] [PubMed] [Google Scholar]
- Sellner PA, Chu W, Glatz JFC, Berman NEJ. Developmental role of fatty acid-binding proteins in mouse brain. Developmental Research. 1995;89:33–46. doi: 10.1016/0165-3806(95)00099-y. [DOI] [PubMed] [Google Scholar]
- Simopoulos AP. Ω-3 Fatty acids: metabolic effects of Ω-3 fatty acids and essentiality. In: Spiller GA, editor. Lipids in Human Nutrition. Boca Raton, FL, USA: CRC Press; 1996. pp. 51–73. [Google Scholar]
- Stremmel W, Strohmeyer G, Berk PD. Hepatocellular uptake of oleate is energy dependent, sodium linked and inhibited by an antibody to a hepatocyte plasma membrane. Proceedings of the National Academy of Sciences of the USA. 1986;83:3584–3588. doi: 10.1073/pnas.83.11.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su MJ, Chang GJ, Wu MH, Kuo SC. Electrophysiological basis for the antiarrhythmic action and positive inotropy of HA-7, a furoquinoline alkaloid derivative, in rat heart. British Journal of Pharmacology. 1997;122:1285–1298. doi: 10.1038/sj.bjp.0701510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatkowski MS, Thomas RC. The intrinsic intracellular H+ buffering power of snail neurons. Journal of Physiology. 1989;409:89–101. doi: 10.1113/jphysiol.1989.sp017486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai KL, Wang SL, Chen CC, Fong TH, Wu ML. Mechanism of oxidative stress-induced intracellular acidosis in rat cerebellar astrocytes and C6 glioma cells. Journal of Physiology. 1997;502:161–174. doi: 10.1111/j.1469-7793.1997.161bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemura A, Mabe H, Nagai H, Sugino F. Action of phospholipase A2 and C on free fatty acid release during complete ischemia in rat neocortex. Effect of phospholipase C inhibitor and N-methyl-d-aspartate antagonist. Journal of Neurosurgery. 1992;76:648–651. doi: 10.3171/jns.1992.76.4.0648. [DOI] [PubMed] [Google Scholar]
- Vaughan-Jones RD, Wu M-L. pH dependence of intrinsic H+ buffering power in the sheep cardiac Purkinje fibre. Journal of Physiology. 1990;425:429–448. doi: 10.1113/jphysiol.1990.sp018112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinlich M, Thei C, Lin CT, Kinne RH. BCECF in single cultured cells. Inhomogeneous distribution but homogeneous response. Journal of Experimental Biology. 1998;201:57–62. doi: 10.1242/jeb.201.1.57. [DOI] [PubMed] [Google Scholar]
- Wu M-L, Chen CC, Su MJ. Possible mechanism(s) of arachidonic acid-induced intracellular acidosis in rat cardiac myocytes. Circulation Research. 2000;86:e55–e62. doi: 10.1161/01.res.86.3.e55. [DOI] [PubMed] [Google Scholar]
- Wu M-L, Chen JH, Chen WH, Chen YJ, Chu KC. Novel role of the Ca-ATPase in NMDA-induced intracellular acidification. American Journal of Physiology. 1999;277:C717–727. doi: 10.1152/ajpcell.1999.277.4.C717. [DOI] [PubMed] [Google Scholar]
- Wu M-L, Kao E-F, Liu I-H, Wang B-S, Lin-Shiau S-Y. The capacitative Ca2+ influx in glial cells is inhibited by glycolytic inhibitors. Glia. 1997;21:315–326. doi: 10.1002/(sici)1098-1136(199711)21:3<315::aid-glia6>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Wu M-L, Tsai ML, Tseng YZ. DIDS-sensitive pHi regulation in single rat cardiac myocytes in nominally HCO3-free conditions. Circulation Research. 1994;57:123–132. doi: 10.1161/01.res.75.1.123. [DOI] [PubMed] [Google Scholar]
- Wu M-L, Tseng YZ. The modulatory effects of endothelin-1, carbachol and isoprenaline upon Na+-H+ exchange in dog cardiac Purkinje fibres. Journal of Physiology. 1993;471:583–597. doi: 10.1113/jphysiol.1993.sp019917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Ma E, Haddad GG. Intracellular pH regulation of CA1 neurons in Na-H isoform 1 mutant mice. Journal of Clinical Investigation. 1999;104:637–645. doi: 10.1172/JCI6785. [DOI] [PMC free article] [PubMed] [Google Scholar]