

Abstract
The existence of adenosine transporters in plasma membrane giant vesicles from rat skeletal muscles and in primary skeletal muscle cell cultures was investigated. In addition, the contribution of intracellularly or extracellularly formed adenosine to the overall extracellular adenosine concentration during muscle contraction was determined in primary skeletal muscle cell cultures.
In plasma membrane giant vesicles, the carrier-mediated adenosine transport demonstrated saturation kinetics with Km= 177 ± 36 μm and Vmax= 1.9 ± 0.2 nmol ml−1 s−1 (0.7 nmol (mg protein)−1 s−1). The existence of an adenosine transporter was further evidenced by the inhibition of the carrier-mediated adenosine transport in the presence of NBMPR (nitrobenzylthioinosine; 72 % inhibition) or dipyridamol (64 % inhibition; P < 0.05).
In primary skeletal muscle cells, the rate of extracellular adenosine accumulation was 5-fold greater (P < 0.05) with electrical stimulation than without electrical stimulation. Addition of the adenosine transporter inhibitor NBMPR led to a 57 % larger (P < 0.05) rate of extracellular adenosine accumulation in the electro-stimulated muscle cells compared with control cells, demonstrating that adenosine is taken up by the skeletal muscle cells during contractions.
Inhibition of ecto-5′-nucleotidase with AOPCP in electro-stimulated cells resulted in a 70 % lower (P < 0.05) rate of extracellular adenosine accumulation compared with control cells, indicating that adenosine to a large extent is formed in the extracellular space during contraction.
The present study provides evidence for the existence of an NBMPR-sensitive adenosine transporter in rat skeletal muscle. Our data furthermore demonstrate that the increase in extracellular adenosine observed during electro-stimulation of skeletal muscle is due to production of adenosine in the extracellular space of skeletal muscle and that adenosine is taken up rather than released by the skeletal muscle cells during contraction.
The purine compound adenosine regulates various physiological functions in many organs including the cardiovascular system (Berne et al. 1983; Olsson & Pearson, 1990) and skeletal muscle tissue (Hespel & Richter, 1998; Radegran & Hellsten, 2000). In skeletal muscle adenosine has been proposed to be involved in the regulation of both blood flow (Berne et al. 1971) and glucose metabolism (Espinal et al. 1983; Vergauwen et al. 1994; Han et al. 1998). Adenosine exerts several of its actions by binding to adenosine receptors located on the surface of vascular and skeletal muscle cells (Proctor & Duling, 1982; Han et al. 1998; Wunsch et al. 2000; Lynge & Hellsten, 2000), and it is thus in the interstitium that adenosine exerts its action. An increase in interstitial adenosine concentration has been shown in contracting human muscle (Hellsten et al. 1998) and adenosine has also been shown to increase in the extracellular medium of contracting primary rat skeletal muscle cells (Hellsten & Frandsen, 1997). The major source of adenosine formation in skeletal muscle is adenosine monophosphate (AMP) 5′-nucleotidase, which dephosphorylates AMP. Adenosine monophosphate 5′-nucleotidase exists both as a soluble cytosolic enzyme and as an ecto-enzyme in skeletal muscle (Newby et al. 1975; Hellsten, 1999). Previous studies have suggested that adenosine is formed from nucleotide degradation within the skeletal muscle cells and released to the interstitium in response to contractions (Berne et al. 1971; Achike & Ballard, 1993). On the other hand, more recent studies have indicated that the formation of adenosine by ecto-5′-nucleotidase is of importance for the increase in extracellular adenosine (Hellsten, 1999; Cheng et al. 2000). The actual contribution of intracellularly or extracellularly formed adenosine to the overall extracellular adenosine concentration during muscle contraction is, however, not known.
Adenosine is a hydrophilic molecule and requires specialized transporter proteins for permeation of the muscle membrane. Two types of nucleoside transport processes have been described: equilibrative and concentrative. For the equilibrative process, the direction of transport is dependent upon the direction of the nucleoside concentration gradient across the membrane, whereas the concentrative process allows nucleosides to be transported into the cell against their concentration gradients using the sodium gradient that exists across the plasma membrane (Baldwin et al. 1999; Wang et al. 1997a). The equilibrative transporters, which are widely distributed in different cell types (Griffith & Jarvis, 1996), are subdivided into two types based on their sensitivity to inhibition by nitrobenzylthioinosine (NBMPR). Transporters of the sensitive (es) type are potently inhibited by NBMPR (Ki 0.1-10 nm). In contrast, transporters of the equilibrative insensitive (ei) type are little affected by concentrations of NBMPR < 1 μm. Both transporter types display broad substrate specificity for pyrimidine and purine nucleosides, including adenosine (Baldwin et al. 1999). The concentrative transporters have a more limited tissue distribution and have primarily been described in specialized cells, such as intestinal and renal epithelia and liver cells. They are typically insensitive to NBMPR and can be subdivided into three main types based on their substrate selectivity (Wang et al. 1997a).
Adenosine transporters have not been characterized in skeletal muscle, but in human skeletal muscles both the equilibrative and concentrative nucleoside transporter mRNAs have been detected (Wang et al. 1997b; Crawford et al. 1998). Whether these findings reflect the presence of physiologically significant amounts of transporter protein remains to be determined for skeletal muscle. The aims of the present study were: (1) to examine the contribution of intracellular and extracellular adenosine formation to extracellular adenosine accumulation in muscle tissue and (2) to establish the existence of and characterize a NBMPR-sensitive adenosine transporter in skeletal muscle. The plasma membrane giant vesicle preparation was used to characterize the adenosine transporters, as this preparation has been proven suitable for characterizing lactate and glucose transporters in skeletal muscle (Juel, 1991; Ploug et al. 1993). Furthermore, primary skeletal muscle cell cultures were used to examine the site of origin of adenosine production.
Some of the described data in this paper were presented at the Frontiers in Physiology, Scandinavian Physiological Society and American Physiological Society joint meeting in Stockholm, Sweden, August 16-19 2000 (Lynge et al. 2000).
METHODS
Materials and abbreviations
Hepes, erythro-9-(2-hydroxy-3-nonyl)adenine (EHNA), adenosine 5′-triphosphate (ATP), adenosine 5′-diphosphate (ADP), adenosine 5′-monophosphate (AMP), β-glycerophosphate, 3-(N-morpholino)propanesulphonic acid (Mops), trazylol, DNase, ethylenediaminetetraacetic acid (EDTA), collegenase (type VII), perchloric acid (PCA), Percoll, dipyridamol, S-(P-nitrobenzyl)-6-thioinosine (6-[(4-nitrobenzyl)thio]-9-B-d-ribofuranosylpurine; NBMPR), α,β-methylene-ADP (AOPCP) and adenosine were all products from Sigma, USA. [14C]Adenosine (20 μCi ml−1) and [3H]mannitol (1 mCi ml−1) were from Amersham Pharmacia Biotech, Sweden. Nycodenz was from Nycomed, Norway. Trypsin and adenosine deaminase (ADA) were from Boehringer Mannheim GmbH, Germany. NBMPR and dipyridamol were dissolved in DMSO (final concentration of DMSO was 5 %).
Isolation of giant vesicles
All experiments complied with the European Convention for the Protection of Vertebrate Animals Used for Experiments or Other Scientific Purposes (Council of Europe No. 123, Strasbourg, France, 1985). The isolation of plasma membrane giant vesicles was performed as previously described (Juel, 1991; Ploug et al. 1993). In brief, Wistar rats were killed by inhalation of carbon dioxide and hindlimb muscles from the thigh and calf were removed and split lengthwise. The muscles were incubated for 1 h at 34 °C with 140 mm KCl, 10 mm Mops (pH 7.4), 150 units ml−1 collagenase (type VII) and 0.01 mg ml−1 of the protease inhibitor trazylol. The muscles were then washed with 140 mm KCl-10 mm Mops buffer containing 10 mm EDTA. Percoll (final concentration 16 %) and trazylol (final concentration 0.01 mg ml−1) were added to the suspension of vesicles and cell debris. A three-step density gradient was used to isolate the vesicles. The upper layer consisted of 1 ml KCl-Mops. The middle layer consisted of 3 ml 4 % nycodenz in KCl-Mops. The vesicle suspension was placed at the bottom of the tube. After centrifugation (50 g for 45 min) at room temperature the vesicles were collected from the interface of the two upper layers. The vesicles were then diluted with KCl-Mops and spun down (830 g for 30 min).
Incubation
Before the efflux experiments vesicles were loaded with unlabelled adenosine at concentrations ranging from 10 μm to 1000 μm and labelled [14C]adenosine and [3H]mannitol. The latter was used as an extravesicular marker. After 35 min of incubation the vesicles were spun down and the sedimented vesicles were ready for zero-trans (adenosine present only on one side of the membrane) or equilibrium exchange studies (adenosine present on both sides of the membrane in equal concentrations).
Efflux experiments
Experiments were carried out with concentrations of adenosine ranging from 10 to 1000 μm. Carrier-mediated adenosine efflux was also examined in the presence of 100 μm NBMPR or 500 μm dipyridamol to inhibit the adenosine transporter. In addition, a series of efflux experiments were performed with 5 μm EHNA added to the vesicle preparation to prevent adenosine deamination (Jacobs et al. 1988). The efflux was initiated when 100 μl of vesicles were transferred to 25 ml of efflux medium (KCl-Mops) with either adenosine (equilibrium exchange) or no adenosine (zero-trans) present in the efflux medium. During the 10 min efflux period, 13 vesicle-free 500 μl samples were obtained using a syringe mounted with a 0.25 μm filter. Samples were obtained 5, 10, 15, 20, 35, 60, 80, 120, 180, 240, 300 and 600 s, after the start of efflux. The extravesicular mannitol space was calculated from 3H-counts. The vesicular adenosine space was then calculated as the difference between the total (internal and external) adenosine space and the extravesicular adenosine space, which was assumed to be identical to the extravesicular mannitol space. 3H- and 14C-activities were determined with a Tri-Carb 2000CA liquid scintillation counter.
Conversion of adenosine to inosine in vesicles
Vesicle experiments were performed to determine the quantity of adenosine converted to inosine by the adenosine deaminase reaction. 30 μl of vesicles were added to 10 μl of 140 mm KCl/10 mm Mops buffer containing either 0 or 5 mm adenosine. One, 5, 15, 30 and 60 min after the addition of adenosine, 4 μl of 5.5 m PCA was added to stop the conversion process. Then 940 μl of the KCl-Mops buffer was added to the samples and the pH was adjusted to 7.4 with 2 m KOH. Adenosine and inosine were determined by reverse-phase HPLC (see below).
Calculations
Because the efflux rate is dependent on the ratio between the vesicular surface and volume, the efflux from vesicles with different diameters is not mono-exponential. The efflux curve is the sum of mono-exponential functions:
where Y is the external accumulated adenosine, t is time, Vd is vesicle volume, Sd is surface, Fd is frequency of vesicles with the diameter d and k is the rate constant; Juel, 1991). The sum of exponential functions was fitted to the experimental data by means of the non-linear least-squares regression method. The initial efflux rate was calculated from the curve fit. With this method the initial efflux rate is based on all samples (13) instead of the smaller number of (2-3) samples, which can be obtained within the first approximately linear part of the efflux curve. A regression line (y = (ax/b+x) +cx), which is based on Michaelis-Menten kinetics, is then calculated. Y = adenosine transport rate, a =Vmax, b =Km, and the term cx represents simple diffusion of adenosine. The carrier-mediated adenosine transport is achieved by subtracting the calculated diffusion from the total adenosine efflux, and Vmax and Km were obtained from the regression line.
Primary cell culture
Isolation and culture of rat primary skeletal muscle cells
All experiments complied with the European Convention for the Protection of Vertebrate Animals Used for Experiments or Other Scientific Purposes (Council of Europe No. 123, Strasbourg, France, 1985). Pregnant rats were killed by inhalation of carbon dioxide. The fetuses were immediately removed. The hindlimbs of the 21-day-old rat fetuses were used to prepare muscle cell cultures (Daniels, 1990). In brief, muscle tissue was dissected out from hindlimbs under a microscope and the tissue was minced into small pieces with scissors. The suspension was digested with 0.1 % (w/v) collagenase, 0.2 % trypsin and 0.1 % DNase at 37 °C for 30 min. DMEM, containing 10 % fetal calf serum and 10 % horse serum (growth medium), was added to the suspension and the remaining tissue fragments were dissociated by triturating with a 10 ml pipette. The cell suspension was centrifuged at 300 g for 8 min and the supernatant was discarded. Twenty millilitres of growth medium was added and the suspension was again triturated with a 10 ml wide-bore pipette to dissociate aggregated cells. The suspension was filtered through a 70 μm nylon mesh and the cells were seeded onto two 90 mm Petri dishes for 45 min during which time fibroblasts attached to the bottom of the dish whereas the myoblasts remained in suspension. The medium was removed from the dishes and the dishes were discarded. Cells were counted and seeded out onto 90 mm dishes coated with 0.1 % gelatine. For the purpose of further purification of the myocytes, fibroblasts and other contaminating cell types were removed by dispase treatment 48 h after seeding, as described by Daniels (1990). All experiments were performed on first passage cells 11-12 days after dispase re-plating.
Effect of NBMPR on adenosine uptake in muscle cell cultures
The effect of the adenosine transport inhibitor NBMPR was examined in non-stimulated muscle cells. The medium used in these experiments was Krebs-Ringer buffer containing (mm): 118.5 NaCl, 24.6 NaHCO3, 4.74 KCl, 1.18 MgSO4.7H2O, 0.71 KH2PO4.3H2O, 3.36 CaCl.2H2O, 25 bicarbonate, 25 Hepes, 1 % d-glucose, 0.005 EHNA and 50 β-glycerophosphate; pH 7.4 and 32 °C. Dishes with muscle cells were rinsed twice with buffer. Buffer containing either 100 μm NBMPR dissolved in DMSO or the equivalent amount of DMSO (control) was added to each dish and incubated for 10 min at 32 °C, allowing the inhibitor to bind to the transporter. Then the buffer was replaced with 600 μl of equivalent buffer containing 2.5 mm unlabelled adenosine and labelled [14C]adenosine (final concentration of 20 nCi ml−1) and incubated for either 30, 60 or 180 s at 32 °C. Dishes with cells were placed on ice, then rinsed with ice-cold PBS and the muscle cells were removed with a rubber policeman. The muscle cell samples were sonicated and 200 μl were used for protein determination and 200 μl were used for measuring the 14C-activities in a Tri-Carb 2000CA liquid scintillation counter. The rate of adenosine uptake was calculated as the amount of adenosine taken up (c.p.m.) divided by the protein concentration. Values are expressed as c.p.m. (μg protein)−1. Experiments were conducted pair-wise with and without NBMPR.
Effect of muscle contraction on adenosine formation with or without NBMPR and/or AOPCP
Muscle contraction was achieved by placing silver electrodes in 35 mm cell culture dishes and applying current to the muscle cells. In order to elicit ATP degradation a strong stimulation protocol was used. The strong stimulation protocol consisted of 0.7 s trains with 0.3 s pauses between trains; the trains consisted of stimuli of 1 ms pulse width and 0.01 s pulse interval delivered at 50 V. The effect of muscle contraction on the rate of adenosine accumulation in the extracellular space was studied in primary rat skeletal muscle cells. Adenosine, AMP, ADP and ATP were measured in the extracellular fluid of non-stimulated or electro-stimulated cells without and with the addition of either NBMPR and/or AOPCP to inhibit adenosine transport and ecto-5′-nucleotidase, respectively. These experiments were performed with the same Krebs-Ringer buffer as described above in the previous paragraph. Dishes with muscle cells were rinsed twice with buffer. Buffer containing either 100 μm NBMPR (NBMPR) dissolved in DMSO (5 %) or the equivalent amount of DMSO or 50 μm AOPCP or both (NBMPR/AOPCP) was added to each dish and the cells were incubated for 10 min at 32 °C; this time point is defined as -10 min in the results. Samples were obtained from the buffer at 0, 10 and 30 min. Stimulation was initiated at 0 min and continued for 30 min. Dishes with cells were placed on ice, then rinsed with ice-cold PBS and the muscle cells were removed with a rubber policeman. The samples were sonicated and 200 μl were used for protein determination. Samples were stored at -80 °C until further analysis. Concentrations of extracellular adenosine, AMP, ADP and ATP were determined during the 30 min period when the muscle cells were either not stimulated or electro-stimulated.
Analysis
Media nucleotide and nucleoside concentrations were determined by reverse-phase HPLC, as previously described (Tullson et al. 1990). Cell protein concentrations were determined with BCA protein assay (Pierce, Rockford, IL, USA).
Statistics
All data are presented as means ±s.e.m. In the plasma membrane experiments, the mean values of the carrier-mediated adenosine effluxes using the inhibitors NBMPR and dipyridamol were compared with carrier-mediated adenosine effluxes in control conditions, at the same adenosine concentrations, using Student's t test for unpaired samples. In the primary skeletal muscle cells, the extracellular adenosine and AMP concentrations, for the various conditions, were compared using two-way ANOVA with repeated measurements and the Student-Newman-Keuls test as post hoc analysis. The rates of extracellular adenosine and AMP accumulation, determined during 30 min, for the various conditions were compared using one-way ANOVA and the Student-Newman-Keuls test as post hoc test. P < 0.05 was considered significant.
RESULTS
Efflux experiments
An example of the time course for zero-trans efflux of adenosine (20 μm) from giant vesicles is shown in Fig. 1A. The initial rate of total adenosine efflux is plotted as a function of initial vesicle adenosine concentration in Fig. 1B. The calculated carrier-mediated adenosine efflux is shown in Fig. 1C. The carrier-mediated efflux demonstrated saturation kinetics with Km= 177 ± 36 μm and Vmax= 1.9 ± 0.2 μm s−1 (mean ±s.d.). Vmax, expressed as the maximal transport capacity per amount of membrane protein in the vesicles, was 0.7 nmol (mg protein)−1 s−1. Equilibrium exchange experiments (100 μm) showed no differences (P > 0.05) in initial adenosine efflux (0.91 ± 0.07 μm s−1) compared with zero-trans experiments (1.21 ± 0.04 μm s−1). In the presence of NBMPR or dipyridamol the carrier-mediated adenosine efflux (vesicles loaded with 50 μm adenosine) was inhibited (P < 0.05) by 72 ± 12 % and 64 ± 18 %, respectively (number of observations was eight in control, six in NBMPR and four in dipyridamol). When EHNA was added to the vesicle preparation to prevent adenosine deamination, the adenosine efflux (vesicles loaded with 20 μm adenosine) was 0.28 ± 0.02 μm s−1 and this was not significantly different (P > 0.05) from control (0.22 ± 0.03 μm s−1). To exclude the possibility that adenosine transport measurements included inosine transport, both adenosine and inosine were assessed in the vesicle preparation during a 60 min incubation period. These measurements revealed no detectable increase in inosine concentration during this time period (data not shown).
Figure 1. Adenosine transport in rat skeletal muscle plasma membrane giant vesicles.
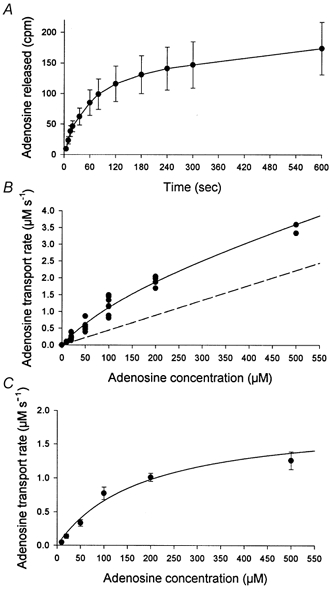
Experiments were carried out with vesicles loaded with adenosine concentrations ranging from 10 to 1000 μm. Released adenosine was determined in vesicle-free samples obtained from 5 to 600 s after the transfer of vesicles to efflux medium. A, example of the total released adenosine plotted as a function of time. The vesicles were loaded with 20 μm adenosine. Values are corrected for extravesicular adenosine with [3H]mannitol. Values are means ±s.e.m. of seven vesicle preparations, with each time point measured in duplicate. B, rates of total initial adenosine transport plotted as a function of the vesicle adenosine concentrations. Each point represents total initial efflux rate obtained in independent experiments of the type depicted in A. The continuous line is the best curve fit to the Michaelis-Menten equation. The dashed line represents calculated diffusion of adenosine. C, the carrier-mediated adenosine transport rates plotted as a function of vesicle adenosine concentrations. The carrier-mediated adenosine transport is achieved by subtracting the calculated diffusion from the total adenosine efflux.
Primary skeletal muscle cell experiments
Effect of NBMPR on adenosine uptake
To establish whether a NBMPR-sensitive adenosine transporter was present in primary skeletal muscle cells adenosine uptake was measured in cells without and with prior NBMPR treatment. Addition of NBMPR to skeletal muscle cells decreased (P < 0.05) adenosine uptake in muscle cells by 66 % and 54 % after 60 and 180 s of incubation, respectively (Fig. 2).
Figure 2. The effect of NBMPR on adenosine uptake in primary skeletal muscle cells.
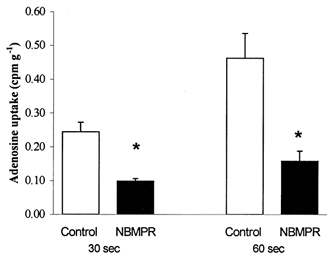
Dishes with muscle cells were incubated with and without NBMPR (100 μm) for 10 min at 32 °C, allowing the inhibitor to bind to the transporter. Then the buffer was replaced with equivalent buffer containing 2.5 mm unlabelled and labelled [14C]adenosine and incubated for either 30 or 60 s at 32 °C. Muscle cells were removed and adenosine uptake and protein content were determined. □, control, adenosine uptake without the inhibitor; ▪, NBMPR, adenosine uptake with the inhibitor NBMPR. Values are means ±s.e.m. of 6-9 cell dishes. * Significantly (P < 0.05) different from control.
Extracellular adenosine
The rate of extracellular adenosine accumulation was greater (P < 0.05) in electrically stimulated than in non-stimulated muscle cell cultures (Table 1, Fig. 3A and B). Addition of the adenosine transport inhibitor NBMPR led to a 57 % larger (P < 0.05) rate of extracellular adenosine accumulation in electro-stimulated muscle cell cultures compared with matched non-treated muscle cell cultures (Table 1, Fig. 3A and B). In order to examine whether addition of EHNA to the medium affected the gradient of adenosine across the plasma membrane, extracellular adenosine was measured during adenosine transport inhibition (NBMPR) without EHNA. The adenosine accumulation in the medium of the muscle cell culture was found to be similar (P > 0.05) with and without EHNA. Inhibition of the ecto-5′-nucleotidase with AOPCP resulted in a 70 % lower (P < 0.05) rate of accumulation of extracellular adenosine in the electro-stimulated cell cultures compared with non-treated electro-stimulated cell cultures (Table 1, Fig. 3A and B). Addition of both AOPCP and NBMPR to the stimulated muscle cells resulted in a 68 % lower (P < 0.05) rate of extracellular adenosine accumulation than in non-treated stimulated muscle cell cultures (Table 1, Fig. 3A and B). The mean protein content of the muscle cells was determined to be 832 ± 26 μg protein per dish. There was no difference (P < 0.05) in protein content between any of the groups.
Table 1.
The rate of accumulation of extracellular adenosine and AMP in primary skeletal muscle cells at rest and during contraction: effect of NBMPR and AOPCP
| Groups | Adenosine (fmol (μg protein)-1 min-1) | AMP (fmol (μg protein)-1 min-1) |
|---|---|---|
| Control non-stimulated | 6.0 ± 2.5 (17) | −11.8 ± 2.9 (16) |
| NBMPR non-stimulated | 14.3 ± 1.6 (7) | −9.9 ± 1.9 (8) |
| AOPCP non-stimulated | 1.5 ± 0.3 (6) | −1.6 ± 2.0 (8) |
| Control electro-stimulated | 32.5 ± 4.2* (17) | −2.2 ± 2.4 (16) |
| NBMPR electro-stimulated | 50.9 ± 3.9† (13) | −1.7 ± 2.8 (13) |
| AOPCP electro-stimulated | 9.9 ± 2.2† (9) | 16.0 ± 2.3† (8) |
| NBMPR/AOPCP electro-stimulated | 10.3 ± 2.8† (8) | 23.8 ± 3.9† (8) |
Effect of NBMPR and AOPCP on the rate of accumulation of extracellular adenosine and AMP in the medium in the non-stimulated and electrically stimulated primary skeletal muscle cells. Samples were collected at times 0, 10 and 30 min and the rate of accumulation was determined as the average rate of accumulation between 0 and 30 min. The values are means ±s.e.m., with the number of dishes given in parentheses. * and † indicate significant (P < 0.05) difference from non-stimulated control and electrically stimulated control, respectively.
Figure 3. Adenosine and AMP accumulation in medium of non-stimulated and electro-stimulated primary skeletal muscle cells: effect of NBMPR and AOPCP.
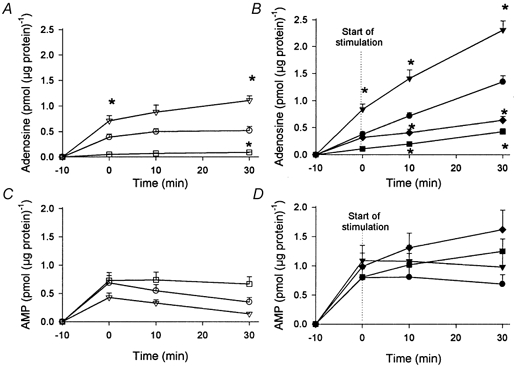
Muscle cells were incubated with medium alone (circles) or with medium containing either 100 μm NBMPR (triangles) or 50 μm AOPCP (squares) or both (NBMPR/AOPCP) (diamonds) for a total of 40 min at 32 °C. This was performed either without electro-stimulation (A and C; open symbols) or with electro-stimulation (B and D; filled symbols) of the muscle cells. Stimulation was initiated at 0 min and continued for 30 min. Adenosine and AMP were determined from medium samples collected at time 0, 10 and 30 min. The muscle cells were removed and protein concentration was determined. Values are means ±s.e.m. of 6-17 cell dishes. * Significantly different (P < 0.05) from control value at the same point of time.
AMP, ADP and ATP accumulation in the extracellular space
There was no difference in rate of accumulated extracellular AMP concentration between non-stimulated and electro-stimulated muscle cell cultures (Table 1, Fig. 3C and D). Addition of NBMPR or AOPCP to the non-stimulated muscle cells resulted in a similar rate of extracellular AMP accumulation in the non-stimulated matched control muscle cell cultures (Table 1, Fig. 3C and D). Addition of AOPCP and a combination of NBMPR and AOPCP to the muscle cells resulted in a greater (P < 0.05) rate of extracellular AMP accumulation than in electro-stimulated matched control muscle cell cultures (Table 1, Fig. 3C and D). There was no significant accumulation of extracellular ADP and ATP in any of the experiments described above (data not shown).
DISCUSSION
In the present study we investigated the existence of an adenosine transporter in skeletal muscle and the site of origin of extracellular adenosine in muscle tissue. The results show the existence of an adenosine transporter in rat skeletal muscle, as evidenced by Michaelis-Menten kinetics and inhibition by NBMPR and dipyridamol. We also demonstrated that extracellular adenosine, formed during contractions in cell cultures, was predominantly formed by ecto-5′-nucleotidase and that there was little or no contribution from intracellular sources; instead, adenosine was taken up by skeletal muscle cells via the adenosine transporter.
Adenosine transport
Adenosine transport was investigated using the plasma membrane giant vesicle preparation from rat skeletal muscle. The carrier-mediated adenosine transport in giant vesicles displayed Michaelis-Menten kinetics, suggesting the existence of an adenosine transporter protein. The transport kinetics were: Km= 177 ± 36 μm and Vmax= 1.9 ± 0.2 nmol ml−1 s−1 (0.7 nmol (mg protein)−1 s−1). The kinetic constants are determined for the overall carrier-mediated adenosine transport, without differentiating between the various nucleoside transporters. Michaelis-Menten kinetic constants for adenosine transport have been determined in erythrocytes and in cardiac myocytes. These results show that Km varies in the micromolar range (4-150 μm) (Mustafa et al. 1975; Young, 1978; Ford et al. 1985; Ford & Rovetto, 1987). One reason for the observed range in Km values could be the use of different methods or different transporter affinity for adenosine in various tissues and animal species (Young, 1978; Ford et al. 1985). The Vmax value determined in the present study is within the range of the values determined for erythrocytes (0.4-0.9 nmol ml−1 s−1 (Young, 1978; Ford et al. 1985) and cardiac cells (0.07-9.6 nmol (mg protein)−1 s−1 (Mustafa et al. 1975; Ford & Rovetto, 1987). The differences in Vmax values reported in the literature are probably due to variation in the total number of transporters in various tissues and species (Jarvis et al. 1982).
The existence of an adenosine transporter protein in muscle membrane vesicles was also evidenced by inhibition of the carrier-mediated efflux of adenosine by NBMPR (72 % inhibition) and dipyridamol (64 % inhibition). In rat primary skeletal muscle cell culture, addition of NBMPR inhibited the total adenosine uptake by 60 %. Incomplete inhibition by NBMPR of adenosine transport is in accordance with previous observations in guinea-pig cardiac myocytes (Conant & Jarvis, 1994), rat erythrocytes, normal rat kidney and human HTC hepatoma cell lines, and has been attributed in part to NBMPR-insensitive transporters (Plagemann & Woffendin, 1988). The lesser inhibition of adenosine transport, in the primary skeletal muscle cells, is probably a combined effect of simple diffusion of adenosine and the existence of NBMPR-insensitive transporters.
It may be debated whether the incomplete inhibition of adenosine transport was due to the concentration of NBMPR used. In most tissues and cell types from different species inhibition of nucleoside transport by either NBMPR or dipyridamol has been performed with concentrations of the inhibitors in the nanomolar range. However, there is a 100- to 1000-fold lower sensitivity for these inhibitors in rat cells than in cells from several other species (Williams et al. 1984; Plagemann & Woffendin, 1988). In the present study the concentrations of these inhibitors were therefore in the micromolar range for NBMPR (100 μm) and dipyridamol (500 μm). Both the NBMPR-sensitive and -insensitive equilibrative adenosine transporters can be inhibited by NBMPR in concentrations exceeding 1 μm (Jarvis & Young, 1986; Lee & Jarvis, 1988); thus it is likely that a sufficient concentrations of NBMPR and dipyridamol were used in the present study.
It is possible that the radiolabelled adenosine loaded inside the plasma membrane giant vesicles can be degraded to inosine. To be sure that we were measuring adenosine and not inosine transport, we quantified conversion of adenosine to inosine in the plasma membrane giant vesicles. The results showed that there was no significant decrease in adenosine or significant increase in inosine in the vesicles during 60 min. We also examined where the nucleoside transporters exhibited differential mobility of empty and loaded carrier. A higher mobility of substrate-loaded carrier than empty carrier is readily detectable as higher rates of equilibrium exchange than zero-trans flux in the vesicles. The experiment demonstrated no difference in adenosine transport capacity during zero-trans and equilibrium experiments. This implies that the nucleoside transporter translocates with the same speed whether it is loaded or unloaded.
Origin of extracellular adenosine
During muscle contractions there is an increase in extracellular adenosine in skeletal muscle, as demonstrated by in vivo human experiments (Hellsten et al. 1998) and in vitro animal experiments (Achike & Ballard, 1993). This finding was confirmed in the present study, which showed a 5-fold increase in the rate of extracellular adenosine accumulation during electrical stimulation of muscle cells. To examine the contribution of adenosine from intracellular versus extracellular sources, we determined the rate of extracellular adenosine accumulation in primary muscle cells with and without adenosine transport inhibition. If adenosine is mainly formed in the cytosol, then membrane adenosine transport inhibition should decrease net cellular adenosine release and consequently the rate of extracellular adenosine accumulation. If, however, adenosine is formed mainly extracellularly, no change or even an increase in the rate of extracellular adenosine accumulation is to be expected. Upon inhibition of the adenosine transporters in cells, we observed an increase in the rate of extracellular adenosine accumulation during contraction, indicating that adenosine does not originate from intracellular sources and that extracellular adenosine is taken up rather than released by muscle cells during contraction. This observation cannot be explained by a lack of ATP degradation in the skeletal muscle as we have previously shown that the exact same stimulation protocol results in a close to 20 % reduction of ATP (Hellsten & Frandsen, 1997).
To investigate whether adenosine was formed outside the muscle cells we incubated the cells with the ecto-5′-nucleotidase inhibitor AOPCP. Addition of AOPCP reduced the rate of extracellular adenosine accumulation by 70 % during contraction, confirming that adenosine is formed predominantly outside the muscle cells. Addition of a combination of NBMPR and AOPCP resulted in a similar (P > 0.05) rate of extracellular adenosine accumulation in the medium as observed with AOPCP alone, again supporting a lack of release of adenosine from contracting muscle. Combined, our results demonstrate that adenosine is formed outside the muscle cells and that there is little or no contribution from intracellular sources; instead there appears to be an uptake of adenosine from the extracellular space. This finding is in agreement with the conclusion of Cheng et al. (2000) that adenosine is formed in the extracellular space via ecto-5′-nucleotidase in perfused rat gracilis muscle. Their conclusion was derived from measurements of the activities of the adenosine-forming (5′-nucleotidase and non-specific-phosphatases) and adenosine-removing (adenosine kinase and adenosine deaminase) enzymes in the cytosolic and membrane fractions of muscle homogenate and values of intracellular and extracellular AMP concentrations in skeletal muscle given in the literature. In accordance with our observation of an uptake of extracellular adenosine by cells, Deussen et al. (1999) observed that, although extracellular adenosine formation only accounted for 10 % of the total adenosine produced in guinea-pig heart, an uptake of adenosine into the cardiac cells was observed due to the high rate of intracellular adenosine metabolism.
The observation that adenosine is mainly formed by ecto-5′-nucleotidase raises the question of the origin of the substrate AMP. The concentrations of ATP and ADP were found to be below the detection limit in the current study, possibly due to a rapid conversion of the released nucleotides. However, AMP was detectable in the extracellular medium and must have originated from the skeletal muscle cells. The rate of extracellular AMP accumulation in electro-stimulated muscle cell cultures was not significantly different from that of non-stimulated muscle cells in culture. However, when the formation of adenosine from AMP was inhibited by the addition of AOPCP to the electro-stimulated cells there was a significant accumulation of AMP extracellularly, suggesting that in the non-treated condition there was a rapid conversion of AMP to adenosine. Consequently, if it is assumed that all extracellular adenosine originates from extracellular AMP, the rate of extracellular adenosine and AMP accumulation in the non-stimulated muscle cells (-5.8 ± 3.8 fmol (μg protein)−1 min−1) can be compared with the rate of extracellular adenosine and AMP accumulation in the electro-stimulated muscle cells (30.3 ± 2.4 fmol (μg protein)−1 min−1). Such a comparison shows that there is a significant difference in AMP between the two conditions, indicating that nucleotides have been released from muscle during contractions. In support of this proposition is the previous observation, from microdialysis experiments, of a 3- to 6-fold increase in ATP, ADP and AMP in contracting human skeletal muscle (Hellsten et al. 1998). In vivo sources of adenine nucleotides other than skeletal muscle cells have been demonstrated. A selective release of ATP can occur from vascular cells (Pearson & Gordon, 1979) and from myocardial cells (Forrester & Williams, 1977; Borst & Schrader, 1991). Adenine nucleotides have moreover been observed to be released from both nerve terminals and muscle fibres upon electrical stimulation of the innervated skeletal muscle of the frog (Cunha & Sebastiao, 1993). It has also been suggested that erythrocytes release ATP during passage through the capillaries, which could be degraded to adenosine in the interstitium. In vitro experiments have shown that erythrocytes release ATP both during conditions of lowered oxygen tension and during compression (Ellsworth et al. 1995). In vivo, there are thus several potential sources of AMP in skeletal muscle tissue.
In conclusion, the present study provides evidence for the existence of an NBMPR- and dipyridamol-inhibitable adenosine transporter with saturation kinetics in rat skeletal muscle. Our data also demonstrate that electrical stimulation of skeletal muscle leads to an increase in extracellular adenosine. This increase in extracellular adenosine is predominantly due to formation of adenosine by ecto-5′-nucleotidase, whereas the main function of the adenosine transporter is not to release but rather to take up adenosine into the muscle cells during muscle contraction, probably for preservation of intracellular nucleotides.
Acknowledgments
The project was supported by The Danish National Research Foundation (504-14) and the project and Jan Lynge were supported by Team Denmark, Danish Sports Research Council, The Danish Research Academy. We would like to thank Merete Vannby and Karina Olsen of the Institute of Exercise and Sport Sciences, August Krogh Institute, Copenhagen University, Denmark for expert technical assistance.
References
- Achike FI, Ballard HJ. Influence of stimulation parameters on the release of adenosine, lactate and CO2 from contracting dog gracilis muscle. Journal of Physiology. 1993;463:107–121. doi: 10.1113/jphysiol.1993.sp019586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin SA, Mackey JR, Cass CE, Young JD. Nucleoside transporters: molecular biology and implications for therapeutic development. Molecular Medicine Today. 1999;5:216–224. doi: 10.1016/S1357-4310(99)01459-8. [DOI] [PubMed] [Google Scholar]
- Berne RM, Knabb RM, Ely SW, Rubio R. Adenosine in the local regulation of blood flow: a brief overview. Federation Proceedings. 1983;42:3136–3142. [PubMed] [Google Scholar]
- Berne RM, Rubio R, Dobson JG, Jr, Curnish RR. Adenosine and adenine nucleotides as possible mediators of cardiac and skeletal muscle blood flow regulation. Circulation Research. 1971;28(suppl.1):115. [PubMed] [Google Scholar]
- Borst MM, Schrader J. Adenine nucleotide release from isolated perfused guinea pig hearts and extracellular formation of adenosine. Circulation Research. 1991;68:797–806. doi: 10.1161/01.res.68.3.797. [DOI] [PubMed] [Google Scholar]
- Cheng B, Essackjee HC, Ballard HJ. Evidence for control of adenosine metabolism in rat oxidative skeletal muscle by changes in pH. Journal of Physiology. 2000;522:467–477. doi: 10.1111/j.1469-7793.2000.t01-1-00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant AR, Jarvis SM. Nucleoside influx and efflux in guinea-pig ventricular myocytes. Inhibition by analogues of lidoflazine. Biochemical Pharmacology. 1994;48:873–880. doi: 10.1016/0006-2952(94)90357-3. [DOI] [PubMed] [Google Scholar]
- Crawford CR, Patel DH, Naeve C, Belt JA. Cloning of the human equilibrative nitrobenzylmercaptopurine riboside (NBMPR)-insensitive nucleoside transporter ei by functional expression in a transport-deficient cell line. Journal of Biological Chemistry. 1998;273:5288–5293. doi: 10.1074/jbc.273.9.5288. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Sebastiao AM. Adenosine and adenine nucleotides are independently released from both the nerve terminals and the muscle fibres upon electrical stimulation of the innervated skeletal muscle of the frog. Pflügers Archiv. 1993;424:503–510. doi: 10.1007/BF00374914. [DOI] [PubMed] [Google Scholar]
- Daniels MP. Localization of actin, beta-spectrin, 43 × 10(3) Mr and 58 × 10(3) Mr proteins to receptor-enriched domains of newly formed acetylcholine receptor aggregates in isolated myotube membranes. Journal of Cell Science. 1990;97:615–626. doi: 10.1242/jcs.97.4.615. [DOI] [PubMed] [Google Scholar]
- Deussen A, Stappert M, Schafer S, Kelm M. Quantification of extracellular and intracellular adenosine production: understanding the transmembranous concentration gradient. Circulation. 1999;99:2041–2047. doi: 10.1161/01.cir.99.15.2041. [DOI] [PubMed] [Google Scholar]
- Ellsworth ML, Forrester T, Ellis CG, Dietrich HH. The erythrocyte as a regulator of vascular tone. American Journal of Physiology. 1995;269:H2155–2161. doi: 10.1152/ajpheart.1995.269.6.H2155. [DOI] [PubMed] [Google Scholar]
- Espinal J, Challiss RA, Newsholme EA. Effect of adenosine deaminase and an adenosine analogue on insulin sensitivity in soleus muscle of the rat. FEBS Letters. 1983;158:103–106. doi: 10.1016/0014-5793(83)80685-1. [DOI] [PubMed] [Google Scholar]
- Ford DA, Rovetto MJ. Rat cardiac myocyte adenosine transport and metabolism. American Journal of Physiology. 1987;252:H54–63. doi: 10.1152/ajpheart.1987.252.1.H54. [DOI] [PubMed] [Google Scholar]
- Ford DA, Sharp JA, Rovetto MJ. Erythrocyte adenosine transport: effects of Ca2+ channel antagonists and ions. American Journal of Physiology. 1985;248:H593–598. doi: 10.1152/ajpheart.1985.248.5.H593. [DOI] [PubMed] [Google Scholar]
- Forrester T, Williams CA. Release of adenosine triphosphate from isolated adult heart cells in response to hypoxia. Journal of Physiology. 1977;268:371–390. doi: 10.1113/jphysiol.1977.sp011862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith DA, Jarvis SM. Nucleoside and nucleobase transport systems of mammalian cells. Biochimica et Biophysica Acta. 1996;1286:153–181. doi: 10.1016/s0304-4157(96)00008-1. [DOI] [PubMed] [Google Scholar]
- Han DH, Hansen PA, Nolte LA, Holloszy JO. Removal of adenosine decreases the responsiveness of muscle glucose transport to insulin and contractions. Diabetes. 1998;47:1671–1675. doi: 10.2337/diabetes.47.11.1671. [DOI] [PubMed] [Google Scholar]
- Hellsten Y. The effect of muscle contraction on the regulation of adenosine formation in rat skeletal muscle cells. Journal of Physiology. 1999;518:761–768. doi: 10.1111/j.1469-7793.1999.0761p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten Y, Frandsen U. Adensosine formation in contracting primary rat skeletal muscle cells and endothelial cells in culture. Journal of Physiology. 1997;504:695–704. doi: 10.1111/j.1469-7793.1997.695bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten Y, Maclean, D A, Rådegran G, Saltin B, Bangsbo J. Adenosine concentrations in the interstitium of resting and contracting human skeletal muscle. Circulation. 1998;98:6–8. doi: 10.1161/01.cir.98.1.6. [DOI] [PubMed] [Google Scholar]
- Hespel P, Richter EA. Role of adenosine in regulation of carbohydrate metabolism in contracting muscle. In: Richter EA, Kiens B, Galbo H, Saltin B, editors. Skeletal Muscle Metabolism in Exercise and Diabetes. NY: Plenum Press; 1998. pp. 97–105. [DOI] [PubMed] [Google Scholar]
- Jacobs AE, Oosterhof A, Veerkamp JH. Purine and pyrimidine metabolism in human muscle and cultured muscle cells. Biochimica et Biophysica Acta. 1988;970:130–136. doi: 10.1016/0167-4889(88)90171-1. [DOI] [PubMed] [Google Scholar]
- Jarvis SM, Hammond JR, Paterson AR, Clanachan AS. Species differences in nucleoside transport. A study of uridine transport and nitrobenzylthioinosine binding by mammalian erythrocytes. Biochemical Journal. 1982;208:83–88. doi: 10.1042/bj2080083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis SM, Young JD. Nucleoside transport in rat erythrocytes: two components with differences in sensitivity to inhibition by nitrobenzylthioinosine and p-chloromercuriphenyl sulfonate. Journal of Membrane Biology. 1986;93:1–10. doi: 10.1007/BF01871013. [DOI] [PubMed] [Google Scholar]
- Juel C. Muscle lactate transport studied in sarcolemmal giant vesicles. Biochimica et Biophysica Acta. 1991;1065:15–20. doi: 10.1016/0005-2736(91)90004-r. [DOI] [PubMed] [Google Scholar]
- Lee CW, Jarvis SM. Nucleoside transport in rat cerebral-cortical synaptosomes. Evidence for two types of nucleoside transporters. Biochemical Journal. 1988;249:557–564. doi: 10.1042/bj2490557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynge J, Hellsten Y. Distribution of adenosine A1, A2A and A2B receptors in human skeletal muscle. Acta Physiologica Scandinavica. 2000;169:283–290. doi: 10.1046/j.1365-201x.2000.00742.x. [DOI] [PubMed] [Google Scholar]
- Lynge J, Juel C, Hellsten Y. Adenosine transport and transporters: No release of adenosine from skeletal muscle during contraction. Frontiers in Physiology. 2000. p. A97.
- Mustafa SJ, Rubio R, Berne RM. Uptake of adenosine by dispersed chick embryonic cardiac cells. American Journal of Physiology. 1975;228:62–67. doi: 10.1152/ajplegacy.1975.228.1.62. [DOI] [PubMed] [Google Scholar]
- Newby AC, Luzio JP, Hales CN. The properties and extracellular location of 5′-nucleotidase of the rat fat-cell plasma membrane. Biochemical Journal. 1975;146:625–633. doi: 10.1042/bj1460625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson RA, Pearson JD. Cardiovascular purinoceptors. Physiological Reviews. 1990;70:761–845. doi: 10.1152/physrev.1990.70.3.761. [DOI] [PubMed] [Google Scholar]
- Pearson JD, Gordon JL. Vascular endothelial and smooth muscle cells in culture selectively release adenine nucleotides. Nature. 1979;281:384–386. doi: 10.1038/281384a0. [DOI] [PubMed] [Google Scholar]
- Plagemann PG, Woffendin C. Species differences in sensitivity of nucleoside transport in erythrocytes and cultured cells to inhibition by nitrobenzylthioinosine, dipyridamole, dilazep and lidoflazine. Biochimica et Biophysica Acta. 1988;969:1–8. doi: 10.1016/0167-4889(88)90081-x. [DOI] [PubMed] [Google Scholar]
- Ploug T, Wojtaszewski J, Kristiansen S, Hespel P, Galbo H, Richter EA. Glucose transport and transporters in muscle giant vesicles: differential effects of insulin and contractions. American Journal of Physiology. 1993;264:E270–278. doi: 10.1152/ajpendo.1993.264.2.E270. [DOI] [PubMed] [Google Scholar]
- Proctor KG, Duling BR. Adenosine and free-flow functional hyperemia in striated muscle. American Journal of Physiology. 1982;242:H688–697. doi: 10.1152/ajpheart.1982.242.4.H688. [DOI] [PubMed] [Google Scholar]
- Radegran G, Hellsten Y. Adenosine and nitric oxide in exercise-induced human skeletal muscle vasodilatation. Acta Physiologica Scandinavica. 2000;168:575–591. doi: 10.1046/j.1365-201x.2000.00705.x. [DOI] [PubMed] [Google Scholar]
- Tullson PC, Whitlock DM, Terjung RL. Adenine nucleotide degradation in slow-twitch red muscle. American Journal of Physiology. 1990;258:C258–265. doi: 10.1152/ajpcell.1990.258.2.C258. [DOI] [PubMed] [Google Scholar]
- Vergauwen L, Hespel P, Richter EA. Adenosine receptors mediate synergistic stimulation of glucose uptake and transport by insulin and by contractions in rat skeletal muscle. Journal of Clinical Investigation. 1994;93:974–981. doi: 10.1172/JCI117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Schaner ME, Thomassen S, Su SF, Piquette-Miller M, Giacomini KM. Functional and molecular characteristics of Na(+)-dependent nucleoside transporters. Pharmaceutical Research. 1997a;14:1524–1532. doi: 10.1023/a:1012113931332. [DOI] [PubMed] [Google Scholar]
- Wang J, Su SF, Dresser MJ, Schaner ME, Washington CB, Giacomini KM. Na(+)-dependent purine nucleoside transporter from human kidney: cloning and functional characterization. American Journal of Physiology. 1997b;273:F1058–1065. doi: 10.1152/ajprenal.1997.273.6.F1058. [DOI] [PubMed] [Google Scholar]
- Williams EF, Barker PH, Clanachan AS. Nucleoside transport in heart: species differences in nitrobenzylthioinosine binding, adenosine accumulation, and drug-induced potentiation of adenosine action. Canadian Journal of Physiology and Pharmacology. 1984;62:31–37. doi: 10.1139/y84-005. [DOI] [PubMed] [Google Scholar]
- Wunsch SA, Muller-Delp J, Delp MD. Time course of vasodilatory responses in skeletal muscle arterioles: role in hyperemia at onset of exercise. American Journal of Physiology. 2000;279:H1715–1723. doi: 10.1152/ajpheart.2000.279.4.H1715. [DOI] [PubMed] [Google Scholar]
- Young JD. Nucleoside transport in sheep erythrocytes: genetically controlled transport variation and its influence on erythrocyte ATP concentrations. Journal of Physiology. 1978;277:325–339. doi: 10.1113/jphysiol.1978.sp012274. [DOI] [PMC free article] [PubMed] [Google Scholar]