

Abstract
Limb vascular beds exhibit a graded dilatation in response to hypoxia despite increased sympathetic vasoconstrictor nerve activity. We investigated the extent to which sympathetic vasoconstriction can mask hypoxic vasodilatation and assessed the relative contributions of β-adrenergic and nitric oxide (NO) pathways to hypoxic vasodilatation.
We measured forearm blood flow responses (plethysmography) to isocapnic hypoxia (arterial saturation ∼85 %) in eight healthy men and women (18-26 years) after selective α-adrenergic blockade (phentolamine) of one forearm. Subsequently, we measured hypoxic responses after combined α- and β-adrenergic blockade (phentolamine and propranolol) and after combined α- and β-adrenergic blockade coupled with NO synthase inhibition (NG-monomethyl-l-arginine, l-NMMA).
Hypoxia increased forearm vascular conductance by 49.0 ± 13.5 % after phentolamine (compared to +16.8 ± 7.0 % in the control arm without phentolamine, P < 0.05). After addition of propranolol, the forearm vascular conductance response to hypoxia was reduced by ∼50 %, but dilatation was still present (+24.7 ± 7.0 %, P < 0.05 vs. normoxia). When l-NMMA was added, there was no further reduction in the forearm vascular conductance response to hypoxia (+28.2 ± 4.0 %, P < 0.05 vs. normoxia).
Thus, selective regional α-adrenergic blockade unmasked a greater hypoxic vasodilatation than occurs in the presence of functional sympathetic nervous system responses to hypoxia. Furthermore, approximately half of the hypoxic vasodilatation in the forearm appears to be mediated by β-adrenergic receptor-mediated pathways. Finally, since considerable dilatation persists in the presence of both β-adrenergic blockade and NO synthase inhibition, it is likely that an additional vasodilator mechanism is activated by hypoxia in humans.
Hypoxia can have profound influences on vascular tone. The balance between the local effects of hypoxia and changes in the neural control of vascular tone determines whether net vasoconstriction or vasodilatation occurs in a vascular bed (Mancia, 1975; Heistad & Abboud, 1980). Several studies indicate that this balance is dependent upon species, vascular region and the degree of hypoxia. In humans, the degree to which changes in sympathetic nerve activity mask hypoxic vasodilatation has never been assessed, the factors that mediate this dilatation are poorly defined, and the extent to which the cutaneous circulation is involved in these responses has been largely ignored.
Sympathetic activation during hypoxia occurs without measurable increases in plasma noradrenaline, suggesting that hypoxia alters noradrenaline kinetics (release or re-uptake). In fact, Leuenberger et al. (1991) found that hypoxia increases noradrenaline re-uptake in humans. Despite these changes in noradrenaline kinetics, it has been shown that in humans, sympathetic vasoconstrictor responses are intact, although blunted. Since hypoxic vasodilatation is observed in human limbs despite a significant rise in sympathetic vasoconstrictor nerve activity, an interesting issue that has yet to be addressed is to what degree these increases in sympathetic nerve activity mask hypoxic vasodilatation.
In humans, the whole-limb vasodilatation seen during severe hypoxia can be reduced by β-adrenergic blockade (Richardson et al. 1967; Blauw et al. 1995), suggesting that it is mediated by a β-adrenergic pathway. However, the vasodilatation can also be reduced by nitric oxide (NO) synthase inhibitors (Blitzer et al. 1996), suggesting that it is mediated by NO. Along these lines, previous studies have demonstrated that the vasodilatation that occurs during several sympathoexcitatory manoeuvres is dependent upon an interaction between increased NO production and β-adrenergic receptor-mediated vasodilatation (Dietz et al. 1994; Halliwill et al. 1997; Reed et al. 2000). A plausible scenario is that NO functions in part as a final pathway for vasodilatation that is secondary to the activation of β-adrenergic receptors by circulating adrenaline (Dawes et al. 1997).
In humans, limb vascular beds show a graded dilatation in response to moderate and severe hypoxia (despite increased sympathetic vasoconstrictor nerve activity), while vasoconstriction is seen in the hand (Heistad & Wheeler, 1970; Sagawa et al. 1986). The fact that in humans, hand blood flow reflects blood flow to the skin rather than muscle, supports the belief that in the limb vasculature, only the skeletal muscle vascular beds (and not skin) contribute to hypoxic vasodilatation. However, as hypoxic vasodilatation represents the balance between the local effects of hypoxia and changes in the neural control of vascular tone, it is possible that the hypoxic vasoconstrictor response in skin overcomes hypoxic vasodilator influences.
Given this background, we hypothesized that: (1) selective regional α-adrenergic receptor blockade would unmask a substantial hypoxic vasodilator response in the vascular beds of the human forearm; (2) this vasodilator response would be associated with a rise in circulating adrenaline and would be blunted by selective regional β-adrenergic receptor blockade; (3) the addition of NO synthase inhibition would be unable to further blunt this vasodilatation; and (4) regional α-adrenergic receptor blockade would uncover a previously unappreciated cutaneous component of this vasodilator response.
METHODS
This study was approved by the Institutional Review Board of the Mayo Clinic and Foundation and conforms with the Declaration of Helsinki. Prior to the beginning of the study, each subject gave his or her written informed consent to participate.
Subjects
Eight healthy, non-smoking, normotensive subjects (3 women, 5 men) between the ages of 18 and 26 years participated in this study (mean ±s.d.: height 177.8 ± 3.4 cm, body mass 78.1 ± 5.1 kg, body mass index 24.6 ± 1.0 kg m−2). None of the subjects were taking medications other than oral contraceptives, and none had been at altitude (> 1500 m) for at least 5 months. Average haematocrit was 38.6 ± 1.8 % and haemoglobin was 13.2 ± 0.6 g dl−1. All female subjects had a negative serum pregnancy test within 12 h prior to participation, but were not scheduled to participate during any specific phase of their menstrual cycle or oral contraceptive use.
Experimental protocol
Subjects were instrumented in the supine position and then underwent three separate hypoxia trials, as shown in Fig. 1. During each trial, the subject breathed a normoxic gas mixture (air) for 5 min, followed by a hypoxic gas mixture for 6 min. The hypoxic gas mixture was titrated to establish and maintain a target arterial O2 saturation of 85 % (see below). Arterial blood samples were collected at the end of each gas period (i.e. end-normoxia and end-hypoxia) for blood gas analysis and measurement of catecholamines. Trial 1 was performed under selective regional α-adrenergic blockade. Trial 2 was performed under combined selective regional α- and β-adrenergic blockade. Trial 3 was performed under combined selective regional α- and β-adrenergic blockade and regional NO synthase inhibition. Loading doses of the appropriate drugs were given i.a. prior to the trials. During the trials, low-dose i.a. infusions of drugs were used to control for drug washout. Throughout the trials, we measured heart rate, ventilation, arterial pressure, arterial saturation, forearm blood flow and cutaneous blood flow. There were 15 min rest periods between each trial. After the third trial, subjects underwent a local heating protocol (see below).
Figure 1. Time-line for the protocol.
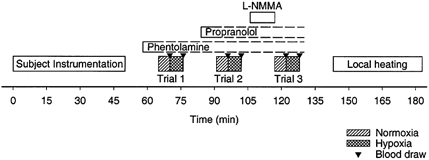
After instrumentation, subjects underwent three trials of measurements during normoxia and hypoxia. Blood draws were performed at the end of each gas period, as indicated by triangles. Local heating of laser Doppler measurement sites was performed at the end of the study to determine maximal cutaneous vascular conductance.
Interventions
Selective regional α-adrenergic blockade
Phentolamine mesylate (CIBA Pharmaceutical, Summit, NJ, USA) was administered i.a. to inhibit α-adrenergic receptors in the left forearm (100 μg min−1 for 5 min; total loading dose, 500 μg; continued infusion at 25 μg min−1 for the duration of the study; Eklund & Kaijser, 1976).
Selective regional β-adrenergic receptor blockade
Propranolol hydrochloride (SoloPak Laboratories, Elk Grove Village, IL, USA) was administered i.a. to inhibit β-adrenergic receptors in the left forearm (200 μg min−1 for 5 min; total loading dose, 1 mg; continued infusion at 25 μg min−1 for the duration of study). Doses of 0.5 mg have been shown to block the forearm vasodilator response to the β-adrenergic agonist isoproterenol (isoprenaline; Johnsson, 1967; Eklund & Kaijser, 1976).
Selective regional NO synthase inhibition
NG-monomethyl-l-arginine (l-NMMA), a NO synthase inhibitor, was administered i.a. to inhibit NO production in the left forearm (5 mg min−1 for 10 min; total dose 50 mg; Dietz et al. 1994; Engelke et al. 1996). Due to the long half-life of this drug and the order of the protocol, an ongoing infusion was not necessary.
All doses of each drug have been proven effective in previous studies (Johnsson, 1967; Eklund & Kaijser, 1976; Dietz et al. 1994; Engelke et al. 1996).
Hypoxia
In order to establish and maintain isocapnic hypoxia, we adapted the ‘alveolar ventilation clamp’ method developed by Banzett et al. (2000). This method uses a self-regulating partial-rebreathe system to maintain constant alveolar fresh-air ventilation, independent of changes in breathing frequency or tidal volume. During the protocol, we ‘clamped’ alveolar ventilation such that end-tidal CO2 would be constant despite large changes in minute ventilation. Using this system, the level of O2 provided in the inspiratory gas was manipulated by mixing N2 with air via a medical gas blender. For hypoxic conditions, the level of O2 was titrated to achieve an arterial O2 saturation of 85 % (as assessed by pulse oximetry). Subjects breathed through a scuba mouthpiece while nasal breathing was prevented with a nose-clip. Gas concentrations were monitored at the mouthpiece by using a respiratory mass spectrometer (Perkin-Elmer MGA 1100). Ventilation was measured via a pneumotachograph (model VMM-2a, Interface Associates, Laguna Niguel, CA, USA).
Measurements
During the study, heart rate was monitored using a five-lead electrocardiogram. Arterial pressure measurements and drug infusions were performed using a 5 cm, 20 gauge brachial artery catheter placed in the non-dominant arm (in all cases, the left arm) using sterile techniques after local anaesthesia (1-2 ml of 1 % lidocaine (lignocaine) hydrochloride; Fujisawa USA, Deerfield, IL, USA). A three-port connector was placed in series with the catheter-transducer system so that arterial pressure could be measured via one port while the drugs were infused through the other two ports (Dietz et al. 1994; Engelke et al. 1996). The catheter was continuously flushed (3 ml h−1) with heparinized (2 units ml−1) saline. Throughout the protocol, arterial O2 saturation was assessed by pulse oximetry of the ear lobe (Biox 3740 Pulse Oximeter, Ohmeda, Boulder, CO, USA).
Forearm whole-limb blood flow
Using the standard approach (Greenfield et al. 1963; Dietz et al. 1994; Engelke et al. 1996), forearm blood flow was estimated by venous occlusion plethysmography with mercury-in-Silastic strain gauges. During measurements, an arterial occlusion cuff around the wrist was continuously inflated to suprasystolic pressure (250 mmHg), while a venous occlusion cuff around the upper arm was inflated to 50 mmHg for 7.5 s out of every 15 s, providing one blood flow measurement every 15 s (reported as ml (dl of tissue)−1 min−1). In addition, forearm vascular conductance was calculated as (100 × blood flow)/mean arterial pressure, and is expressed as arbitrary conductance units.
Blood flow was measured simultaneously in both the dominant arm (control arm) and the non-dominant arm (experimental arm) in which infusions were made. This allowed direct comparison of the hypoxic vasodilator response in the control arm to that in the experimental arm undergoing various pharmacological interventions. In pilot studies, we established that the forearm vascular response to hypoxia was bilateral and symmetrical.
Forearm cutaneous blood flow
Using integrated probes that contain seven transmitter-receiver fibre pairs (Probe 413, PeriFlux System 5000, Perimed, Stockholm, Sweden), forearm cutaneous blood flow was estimated by laser Doppler flowmetry in both the control and the experimental arm (in which infusions were made) in six of the eight subjects. This allowed a direct comparison of the cutaneous hypoxic vasodilator response in the control arm to that in the experimental arm undergoing various pharmacological interventions. Cutaneous vascular conductance was calculated as cutaneous red blood cell flux/mean arterial pressure and normalized to the maximum conductance achieved during 40 min of local heating to 43 °C at the end of the protocol (Kellogg et al. 1998).
Blood gas and catecholamine analysis
Brachial artery blood samples were analysed with a clinical blood gas analyser (I-STAT Portable Clinical Analyser, I-STAT, Princeton, NJ, USA) for partial pressures of O2 and CO2 (PO2 and PCO2), pH, O2 saturation, haemoglobin and haematocrit. In addition, brachial artery plasma catecholamine (adrenaline and noradrenaline) levels were determined by HPLC with electrochemical detection.
Data analysis
All physiological signals were digitized and stored on computer at 250 Hz, and data were analysed off-line with signal processing software (WinDaq, Dataq Instruments, Akron, OH, USA). Mean arterial pressure was derived from the arterial pressure waveform. Forearm blood flow was determined from the derivative of the forearm plethysmogram. For each experimental condition (α-blockade; α- and β-blockade; α- and β-blockade, and NO synthase inhibition), steady-state data for heart rate, arterial pressure, forearm blood flow and cutaneous blood flow were averaged across the final minute of normoxia and across the final minute of hypoxia.
Statistics
Since there were no discernable differences between men and women, data from the two groups were combined for statistical analysis. The results were analysed with a two-way repeated-measures ANOVA (blockade state vs. normoxia/hypoxia). Significant effects were further tested with Fischer's LSD test, and differences were considered significant when P < 0.05. All values are reported as means ±s.e.m. unless otherwise indicated.
RESULTS
Effects of hypoxia on blood gas levels
The baseline (normoxia) arterial O2 saturation (based on pulse oximetry) for the first trial was 96.0 ± 0.4 %, and decreased to 85.1 ± 0.8 % in response to hypoxia (P < 0.05). Trial 2 showed a similar decrease, with a baseline saturation of 95.7 ± 0.4 % decreasing to 83.5 ± 0.4 % (P < 0.05). In the third trial, arterial O2 saturation decreased from 95.5 ± 0.4 % to 84.0 ± 0.6 % (P < 0.05). No differences in baseline saturation were evident throughout the three trials, but during trial 2, hypoxic saturation was slightly lower than that during trial 1 (P < 0.05).
Table 1 shows the effects of hypoxia on several blood gas variables. Hypoxia consistently decreased PO2 and arterial O2 saturation (blood gas estimate). While hypoxia had no effect on PCO2, there were small changes in end-tidal CO2 and pH. No differences were evident between trials within the same condition (i.e. blood gas changes during hypoxia were consistent across trials).
Table 1.
Effects of hypoxia on blood PO2, PCO2 and pH
| Condition | Trial 1: phentolamine | Trial 2: phentolamine and propranolol | Trial 3: phentolamine propranolol and l-NMMA | |
|---|---|---|---|---|
| PO2 (mmHg) | Normoxia | 88.9 ± 1.9 | 86.4 ± 2.2 | 89.1 ± 3.7 |
| Hypoxia | 42.2 ± 1.4 * | 43.8 ± 2.0 * | 44.1 ± 1.8 * | |
| PCO2 (mmHg) | Normoxia | 40.8 ± 1.2 | 35.8 ± 5.0 | 39.8 ± 1.4 |
| Hypoxia | 37.6 ± 1.3 | 37.7 ± 1.2 | 37.5 ± 1.8 | |
| End-tidal CO2 (%) | Normoxia | 6.2 ± 0.4 | 6.2 ± 0.4 | 6.1 ± 0.4 |
| Hypoxia | 5.8 ± 0.4 * | 5.8 ± 0.4 * | 5.8 ± 0.4 * | |
| pH | Normoxia | 7.39 ± 0.01 | 7.38 ± 0.01 | 7.38 ± 0.01 |
| Hypoxia | 7.41 ± 0.01 * | 7.42 ± 0.01 * | 7.41 ± 0.01 * | |
| Hb saturation (%) | Normoxia | 96.6 ± 0.3 | 96.1 ± 0.2 | 96.5 ± 0.6 |
| Hypoxia | 77.9 ± 1.7 * | 79.8 ± 2.0 * | 80.3 ± 2.0 * |
Hb, haemoglobin.
P <0.005, Hypoxia vs. Normoxia.
Effects of hypoxia on ventilation, heart rate and arterial pressure
Table 2 shows the effects of hypoxia on minute ventilation, heart rate and mean arterial pressure. Minute ventilation doubled during hypoxia in trials 1 and 2, but did not increase significantly during trial 3 due to an elevated baseline ventilation in that trial. Hypoxia consistently increased heart rate across trials, but had no effect on arterial pressure.
Table 2.
Effects of hypoxia on ventilation, heart rate and arterial pressure
| Condition | Trial 1: phentolamine | Trial 2: phentolamine and propranolol | Trial 3: phentolamine propranolol and l-NMMA | |
|---|---|---|---|---|
| Minute ventilation | Normoxia | 6.5 ± 0.7 | 6.7 ± 1.0 | 8.6 ± 2.3 |
| (l min-1 STPD) | Hypoxia | 13.3 ± 2.4 * | 13.1 ± 3.3 * | 12.0 ± 2.0 |
| Heart rate | Normoxia | 62 ± 5 | 58 ± 4 † | 56 ± 4 † |
| (beats min-1) | Hypoxia | 77 ± 5 * | 74 ± 5 * | 71 ± 5 *† |
| Mean arterial pressure | Normoxia | 86.6 ± 1.7 | 87.5 ± 1.8 | 90.8 ± 1.0 |
| (mmHg) | Hypoxia | 87.3 ± 1.6 | 89.7 ± 2.1 | 92.5 ± 1.6 |
STPD, standard temperature and pressure, dry.
P < 0.05, Hypoxia vs. Normoxia
P < 0.05 vs. Trial 1 within condition.
Effects of autonomic blocking agents on forearm blood flow
Administration of phentolamine (trial 1) to the experimental arm increased resting (normoxic) blood flow in the experimental arm from 2.5 ± 0.3 to 4.9 ± 0.9 ml dl−1 min−1 (P < 0.05). The administration of propranolol (trial 2) after phentolamine did not further alter resting blood flow (4.9 ± 0.8 ml dl−1 min−1) in the experimental arm. Subsequent administration of l-NMMA (trial 3) decreased resting blood flow to 2.3 ± 0.3 ml dl−1 min−1 (P < 0.05). Throughout these pharmacological interventions, no changes were evident in the control arm (2.9 ± 0.5, 2.8 ± 0.7 and 2.9 ± 0.8 ml dl−1 min−1, P > 0.05), indicating that the blocking agents had selective regional effects only. Table 3 shows the corresponding forearm vascular conductance values.
Table 3.
Effects of hypoxia on forearm vascular conductance
| Condition | Trial 1: phentolamine | Trial 2: phentolamine and propranolol | Trial 3: phentolamine propranolol and l-NMMA | |
|---|---|---|---|---|
| FVC in control arm | Normoxia | 3.34 ± 0.64 | 3.22 ± 0.84 | 3.25 ± 0.88 |
| (conductance units) | Hypoxia | 3.86 ± 0.59 * | 3.65 ± 0.86 * | 3.84 ± 0.91 * |
| FVC in experimental arm | Normoxia | 5.69 ± 1.12 | 5.62 ± 0.96 | 2.58 ± 0.39 † |
| (conductance units) | Hypoxia | 8.02 ± 1.33 * | 6.87 ± 1.03 *† | 3.30 ± 0.51 *† |
FVC, forearm vascular conductance.
P < 0.05, Hypoxia vs. Normoxia
P < 0.05 vs. prior trial within condition.
Effects of hypoxia on forearm blood flow
Figure 2 shows representative data from one subject, demonstrating the changes in forearm blood flow during hypoxia in all three trials. In this particular subject, there was little or no change in blood flow in the control arm throughout the three trials (i.e. this individual did not exhibit much hypoxic vasodilatation). In contrast, the experimental arm treated with phentolamine exhibited marked vasodilatation in response to hypoxia (trial 1). This vasodilatation was significantly attenuated by propranolol (trial 2). The addition of l-NMMA, while changing baseline blood flow, failed to further impair the hypoxic vasodilatation.
Figure 2. Representative data from one subject.
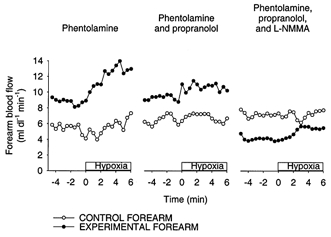
Shown are forearm blood flow values in the control arm (○) and the experimental forearm (•) before and during the hypoxia trials. This individual showed little or no dilatation in the control arm during all three trials. In contrast, the experimental arm exhibited marked hypoxic vasodilatation under α-adrenergic blockade (phentolamine). This hypoxic vasodilator response was largely blocked by propranolol, but was not further blocked by the addition of l-NMMA.
The group averages for forearm vascular conductance followed a similar trend and are shown in Table 3. Since baseline (normoxic) blood flow and conductance were altered by l-NMMA administration, we assessed the hypoxic vasodilator response in terms of absolute change and per cent change in conductance (Fig. 3). The increase in conductance caused by hypoxia was greatest with phentolamine alone, and less after phentolamine and propranolol or phentolamine and propranolol and l-NMMA (P < 0.05). Neither the change nor the per cent change in conductance differed between propranolol and l-NMMA (P > 0.05). Sample size analysis predicted that 25 subjects would have to be studied before any difference in the change in conductance between trial 2 and trial 3 would be detectable statistically.
Figure 3. Forearm vascular conductance responses to hypoxia.
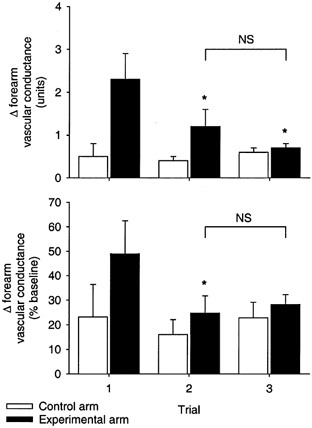
The change (upper panel) and per cent change (lower panel) in forearm vascular conductance in the control arm (□) and experimental arm (▪) are shown for the three hypoxia trials. *P < 0.05 vs. Trial 1, within arm. NS, not significantly different.
Effects of autonomic blocking agents and hypoxia on cutaneous vascular conductance
Table 4 shows the cutaneous vascular conductance values for each condition. In the six subjects in whom we measured cutaneous blood flow, administration of phentolamine (trial 1) to the experimental arm increased resting cutaneous vascular conductance (P < 0.05). The administration of propranolol (trial 2) after phentolamine did not further alter resting cutaneous vascular conductance in the experimental arm. Subsequent administration of l-NMMA (trial 3) decreased resting cutaneous vascular conductance ≈35 % in the experimental arm (P < 0.05). Throughout the study, there was a modest downward drift in cutaneous vascular conductance in the control arm (P < 0.05).
Table 4.
Effects of hypoxia on cutaneous vascular conductance
| Condition | Trial 1: phentolamine | Trial 2: phentolamine and propranolol | Trial 3: phentolamine propranolol and l-NMMA | |
|---|---|---|---|---|
| CVC in control arm | Normoxia | 10.8 ± 3.3 | 9.3 ± 2.8 † | 8.4 ± 2.5 † |
| (% maximum conductance) | Hypoxia | 12.6 ± 4.0 | 11.4 ± 4.1 † | 10.5 ± 3.8 † |
| CVC in experimental arm | Normoxia | 13.7 ± 2.2 | 14.3 ± 1.9 | 8.9 ± 1.1 † |
| (% maximum conductance) | Hypoxia | 18.6 ± 3.3 * | 24.2 ± 6.2 * | 10.0 ± 1.6 † |
CVC, cutaneous vascular conductance.
P < 0.05, Hypoxia vs. Normoxia
P < 0.05 vs. prior trial within condition.
In response to hypoxia, cutaneous vascular conductance was unchanged in the control arm (P = 0.10 vs. resting). In contrast, the experimental arm showed increased cutaneous vascular conductance during hypoxia in the first trial (P < 0.05 vs. resting) and in the second trial (P < 0.05 vs. resting). In trial three, however, the cutaneous vascular conductance response to hypoxia was abolished (P = 0.73 vs. resting). Since baseline (normoxic) conductance was altered by l-NMMA administration, we assessed the hypoxic vasodilator response in terms of the change and per cent change in conductance (Fig. 4). The rise in conductance caused by hypoxia was greatest with phentolamine alone or phentolamine plus propranolol, and least after l-NMMA (P < 0.05).
Figure 4. Cutaneous vascular conductance responses to hypoxia.
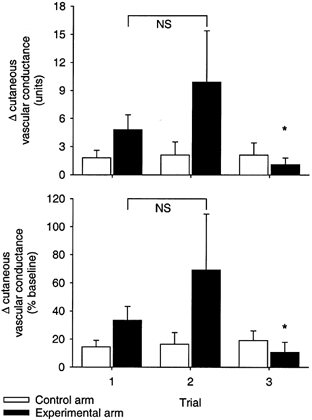
The change (upper panel) and per cent change (lower panel) in cutaneous vascular conductance in the control arm (□) and experimental arm (▪) are shown for the three hypoxia trials. *P < 0.05 vs. Trial 1, within arm. NS, not significantly different.
Effects of hypoxia on arterial catecholamines
Figure 5 shows the effects of hypoxia on arterial levels of adrenaline and noradrenaline. Adrenaline levels increased (P < 0.05) with hypoxia in trials 1 and 2, but failed to increase in trial 3 due to an elevated baseline before this trial. While all subjects showed increases in both adrenaline and forearm vascular conductance in the experimental arm in trial 1, these increases in adrenaline and vascular conductance did not correlate across subjects (r = 0.40, P = 0.32), perhaps due to individual differences in the β-adrenergic responsiveness of vessels. Noradrenaline levels did not change significantly between normoxia and hypoxia trials.
Figure 5. Arterial catecholamine responses to hypoxia.
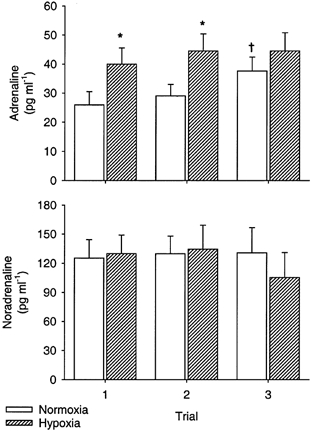
Plasma concentrations for adrenaline (upper panel) and noradrenaline (lower panel) during normoxia (□) and hypoxia ( ) are shown for the three hypoxia trials. *P < 0.05 vs. normoxia within trial. †P < 0.05 vs. Trial 1 within condition.
) are shown for the three hypoxia trials. *P < 0.05 vs. normoxia within trial. †P < 0.05 vs. Trial 1 within condition.
DISCUSSION
The degree to which changes in sympathetic nerve activity mask hypoxic vasodilatation has never been assessed in humans; the factors that mediate hypoxic dilatation are poorly understood, and the extent to which the cutaneous circulation is affected by hypoxia has been largely uninvestigated. Our results suggest that the activation of sympathetic vasoconstrictor nerves masks, to a substantial degree, the effects of hypoxia on vascular tone in the vascular beds of the human forearm. The data suggest that approximately half of the hypoxic vasodilator response is due to a rise in circulating adrenaline. While it is possible that NO contributes to this β-adrenergic receptor-mediated vasodilatation, it appears that any additional (i.e. independent of β-adrenergic pathways) NO-mediated vasodilatation is minimal. However, since we did not perform NO synthase inhibition in the absence of β-adrenergic blockade, we cannot assess the degree to which β-adrenergic vasodilatation is NO dependent. Finally, in contrast to what has been suggested previously, it appears that the cutaneous circulation is responsive to hypoxic vasodilatation, but that overt hypoxic vasodilatation is not generally observed due to over-riding sympathetic vasoconstriction.
Sympathetic responses to hypoxia
To the best of our knowledge, muscle sympathetic nerve activity to the forearm has not been measured during hypoxia. However, muscle sympathetic nerve activity to the leg is increased by hypoxia (Saito et al. 1988; Rowell et al. 1989). Furthermore, noradrenaline spillover measured in the forearm is increased by hypoxia (Leuenberger et al. 1999). Therefore, it is likely that sympathetic nerve fibres innervating the forearm are activated by hypoxia. We are not aware of any studies that have used selective regional α-adrenergic receptor blockade to study the effects of sympathetic activation on hypoxic vasodilator responses. By removing the competing influence of changes in sympathetic nerve activity, this experimental protocol: (1) provides a cleaner model for studying hypoxic vasodilatation, (2) demonstrates the extent to which sympathetic vasoconstriction normally masks hypoxic vasodilatation, and (3) documents that the sympathetic nerves can mediate vasoconstriction under hypoxic conditions. What are the implications for superimposed sympathetic vasoconstriction and hypoxia vasodilatation? It may be that hypoxic vasodilatation helps to maintain adequate blood flow at the local level, and that the sympathetic vasoconstrictor response represents a ‘safety mechanism’ preventing widespread activation of vasodilatation from outstripping cardiovascular reserves. Indeed, it is likely that blunted sympathetic vasoconstrictor responses could result in the orthostatic intolerance or hypotension that has been reported in some individuals during acute systemic hypoxia (Rowell & Blackmon, 1989).
Mechanisms of hypoxic vasodilatation
Our findings after β-adrenergic receptor blockade are in agreement with prior work by Richardson et al. (1967) and Blauw et al. (1995) in showing that a portion of the hypoxic vasodilator response is mediated via β-adrenergic receptor pathways. Accordingly, hypoxia increased circulating levels of adrenaline approximately 2-fold. It is unclear whether this is due to an increase in sympathetic nerve activity to the adrenal gland or to a direct effect of hypoxia on the adrenal gland. Nonetheless, these findings indicate that adrenaline plays a substantial role in mediating the hypoxic vasodilatation, over-riding the effects of sympathetic vasoconstriction in healthy humans. Since our observations were carried out in the presence of α-adrenergic receptor blockade, we have been able to extend prior observations by quantifying how much vasodilatation can be attributed to this mechanism (50 % of the total vasodilator response).
Our findings after NO synthase inhibition differ from those of Blitzer et al. (1996), who found that NO synthase inhibition blocked ≈55 % of the hypoxic vasodilator response. However, an important consideration is that we performed NO synthase inhibition subsequent to β-adrenergic receptor blockade. Dawes et al. (1997) found that 50-60 % of the dilatation produced by i.a. infusion of β-adrenergic agonists in humans is mediated by NO (blocked by l-NMMA). Taken together, the data in the study by Blitzer et al. (1996) and the current study are consistent with hypoxia eliciting NO-mediated vasodilatation exclusively via stimulation of β-adrenergic receptors. However, as stated above, we cannot assess the degree to which β-adrenergic vasodilatation is NO dependent.
Since considerable dilatation persists in the presence of both β-adrenergic blockade and NO synthase inhibition, it is likely that an additional vasodilator mechanism is activated by hypoxia in humans. Furthermore, animal studies and studies in isolated vessels indicate that ATP-sensitive K+ channels play an extensive role in mediating hypoxic vasodilatation (Daut et al. 1990; Spina et al. 1992) and that these channels are activated by adenosine released from the endothelium (Bryan & Marshall, 1999a, b). Adenosine levels in skeletal muscle are increased in humans during hypoxia (MacLean et al. 1998), and Leuenberger et al. (1999) recently found that aminophylline, an adenosine receptor antagonist, can reduce the hypoxic vasodilator response by approximately 80 % in humans. It is important to recognize that this study (as well as the study by Blitzer et al. 1996) did not quantify vasodilator responses in the presence of α-adrenergic blockade and that it was under these conditions that aminophylline blocked 80 % of the apparent hypoxic vasodilatation. In other words, the contribution of adenosine was likely to have been overestimated due to the effects of sympathetic vasoconstrictor activity (i.e. the apparent dilatation may have represented only 40 % of the entire hypoxic vasodilatation).
It would appear from this discussion that hypoxic vasodilatation in the forearm of humans is due to both circulating adrenaline and locally produced adenosine, but the relative contributions of these two vasodilator signals have yet to be defined. The degree to which activation of sympathetic vasoconstriction during hypoxia can mask dilator responses and obscure study results has been largely unappreciated. The current results suggest that perhaps all studies investigating the hypoxic vasodilator response should incorporate α-adrenergic blockade.
Remsburg et al. (1999) recently reported that the vascular response to hypoxia in patients with sleep apnoea is vasoconstriction, as opposed to the vasodilatation seen in healthy control subjects. An obvious question in the context of this study is whether sleep apnoea patients have a reduced vasodilator signal during hypoxia, or an augmented sympathetic vasoconstrictor response.
Cutaneous circulation
In addition to measuring whole-limb blood flow responses, we applied laser Doppler flowmetry to quantify the cutaneous blood flow response to hypoxia. Our results are in contrast to previous studies in this area, many relying on plethysmography, that have concluded that skin circulation in humans is unresponsive to a hypoxic stimulus (Heistad & Wheeler, 1970; Sagawa et al. 1986). Our results suggest that the changes in sympathetic vasoconstrictor nerve activity to the skin largely offset hypoxic vasodilator signals such that there is little if any discernable change in cutaneous vascular conductance during hypoxia in humans (Leuenberger et al. 1999). However, in the absence of α-adrenergic vasoconstrictor responses, overt hypoxic vasodilatation was unmasked. Furthermore, although functional β-adrenergic receptors have been documented in human skin (Crandall et al. 1997), this hypoxic vasodilator response is insensitive to β-adrenergic blockade, suggesting that it is not a response to circulating adrenaline. Cutaneous hypoxic vasodilator responses were absent following NO synthase inhibition, suggesting that NO is involved in this response. It is well established that NO plays a ‘permissive’ role in some cutaneous vascular responses (Farrell & Bishop, 1997) via interactions between cAMP and cGMP pathways. Therefore, at present we are unable to ascertain whether hypoxic vasodilatation in the skin is mediated by NO or is merely NO dependent via a similar interaction. We would be negligent to not point out that our observations in the skin were based on a limited number of subjects (n = 6) and responses were variable. Nonetheless, we believe that these observations are novel and informative, although perhaps not completely conclusive.
Limitations
Our goal was to selectively block β-adrenergic receptors in the forearm of the experimental arm without interfering with β-adrenergic receptors in the contralateral arm, which was serving as a time control. We did this by administering propranolol i.a. in a dose sufficient to block the forearm vasodilator response to the β-adrenergic agonist isoproterenol (Johnsson, 1967; Eklund & Kaijser, 1976). However, throughout the protocol, we observed a small but consistent decline in heart rate that could be due to systemic spillover of propranolol, as well as some subtle changes in resting ventilation, which might be consistent with a central neural effect of propranolol on respiration. It is unclear whether a mild systemic effect of propranolol could also account for elevated baseline adrenaline levels prior to the third trial. Thus, the question arises as to whether or not the systemic effects of the infused propranolol are important confounding factors that must be considered when interpreting the results. However, vascular conductance in the control arm does not appear to have been affected by propranolol, as normoxic and hypoxic values and responses were consistent throughout the three trials. Therefore, it does not appear that propranolol spillover, if it occurred, undermines the interpretation and conclusions of this study.
Conclusions
In the human forearm, activation of sympathetic vasoconstrictor nerves masks most of the effects of hypoxia on vascular tone. It appears that circulating adrenaline is a major factor in the hypoxic vasodilator response, accounting for perhaps half of the vasodilatation that occurs in the forearm. NO-mediated vasodilatation, independent of what may be activated by adrenaline, is minimal. Finally, it appears that the cutaneous circulation is responsive to hypoxic vasodilatation, but that overt hypoxic vasodilatation is not generally observed due to over-riding sympathetic vasoconstriction.
Acknowledgments
We thank Sarah Joy Carlson, Erin Clark, Eric Cornidez, Annette Nelson, Isaac Keiffer and Karen Krucker for their technical assistance. We especially thank the subjects who volunteered for this study. Cara J. Weisbrod was supported by an Undergraduate Summer Research Fellowship from the American Physiological Society. This research was supported in part by American Heart Association, Northland Affiliate, Scientist Development grant 30403Z; National Institutes of Health (NIH) grants DK-09826, NS-32352, HL-46493 and AG07004; NIH General Clinical Research Center grant RR-00585 (to the Mayo Clinic, Rochester, MN, USA); and a grant from the Wilderness Medical Society. Further support was provided by the Glen L. and Lyra M. Ebling Cardiology Research Endowment and the Mayo Foundation.
References
- Banzett RB, Garcia RT, Moosavi SH. Simple contrivance “clamps” end-tidal PCO2 and PO2 despite rapid changes in ventilation. Journal of Applied Physiology. 2000;88:1597–1600. doi: 10.1152/jappl.2000.88.5.1597. [DOI] [PubMed] [Google Scholar]
- Blauw GJ, Westendorp RGJ, Simons M, Chang, P C, Frölich M, Meinders AE. β-adrenergic receptors contribute to hypoxaemia induced vasodilatation in man. British Journal of Clinical Pharmacology. 1995;40:453–458. [PMC free article] [PubMed] [Google Scholar]
- Blitzer ML, Lee SD, Creager MA. Endothelium-derived nitric oxide mediates hypoxic vasodilation of resistance vessels in humans. American Journal of Physiology. 1996;271:H1182–1185. doi: 10.1152/ajpheart.1996.271.3.H1182. [DOI] [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Adenosine receptor subtypes and vasodilatation in rat skeletal muscle during systemic hypoxia: a role for A1 receptors. Journal of Physiology. 1999a;514:151–162. doi: 10.1111/j.1469-7793.1999.151af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Cellular mechanisms by which adenosine induces vasodilatation in rat skeletal muscle: significance for systemic hypoxia. Journal of Physiology. 1999b;514:163–175. doi: 10.1111/j.1469-7793.1999.163af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall CG, Etzel RA, Johnson JM. Evidence of functional β-adrenoceptors in the cutaneous vasculature. American Journal of Physiology. 1997;273:H1038–1043. doi: 10.1152/ajpheart.1997.273.2.H1038. [DOI] [PubMed] [Google Scholar]
- Daut J, Maier-Rudolph W, Vonbeckerath N, Mehrke G, Günther K, Goedel-Meinen L. Hypoxic dilation of coronary arteries is mediated by ATP-sensitive potassium channels. Science. 1990;247:1341–1344. doi: 10.1126/science.2107575. [DOI] [PubMed] [Google Scholar]
- Dawes M, Chowienczyk PJ, Ritter JM. Effects of inhibition of the l-arginine/nitric oxide pathway on vasodilation caused by β-adrenergic agonists in human forearm. Circulation. 1997;95:2293–2297. doi: 10.1161/01.cir.95.9.2293. [DOI] [PubMed] [Google Scholar]
- Dietz NM, Rivera JM, Eggener SE, Fix RT, Warner DO, Joyner MJ. Nitric oxide contributes to the rise in forearm blood flow during mental stress in humans. Journal of Physiology. 1994;480:361–368. doi: 10.1113/jphysiol.1994.sp020366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund B, Kaijser L. Effect of regional α- and β-adrenergic blockade on blood flow in the resting forearm during contralateral isometric handgrip. Journal of Physiology. 1976;262:39–50. doi: 10.1113/jphysiol.1976.sp011584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelke KA, Halliwill JR, Proctor DN, Dietz NM, Joyner MJ. Contribution of nitric oxide and prostaglandins to reactive hyperemia in the human forearm. Journal of Applied Physiology. 1996;81:1807–1814. doi: 10.1152/jappl.1996.81.4.1807. [DOI] [PubMed] [Google Scholar]
- Farrell DM, Bishop VS. The roles of cGMP and cAMP in active thermoregulatory vasodilation. American Journal of Physiology. 1997;272:R975–981. doi: 10.1152/ajpregu.1997.272.3.R975. [DOI] [PubMed] [Google Scholar]
- Greenfield ADM, Whitney RJ, Mowbray JF. Methods for the investigation of peripheral blood flow. British Medical Bulletin. 1963;19:101–109. doi: 10.1093/oxfordjournals.bmb.a070026. [DOI] [PubMed] [Google Scholar]
- Halliwill JR, Lawler LA, Eickhoff TJ, Dietz NM, Nauss LA, Joyner MJ. Forearm sympathetic withdrawal and vasodilatation during mental stress in humans. Journal of Physiology. 1997;504:211–220. doi: 10.1111/j.1469-7793.1997.211bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heistad DD, Abboud FM. Circulatory adjustments to hypoxia. Dickinson W. Richards Lecture. Circulation. 1980;61:463–470. doi: 10.1161/01.cir.61.3.463. [DOI] [PubMed] [Google Scholar]
- Heistad DD, Wheeler RC. Effect of acute hypoxia on vascular responsiveness in man. Journal of Clinical Investigation. 1970;49:1252–1265. doi: 10.1172/JCI106338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsson G. The effects of intra-arterially administered propranolol and H 56/28 on blood flow in the forearm – a comparative study of two β-adrenergic receptor antagonists. Acta Pharmacologica et Toxicologica. 1967;25:63–74. doi: 10.1111/j.1600-0773.1967.tb02997.x. [DOI] [PubMed] [Google Scholar]
- Kellogg DL, Jr, Crandall CG, Liu Y, Charkoudian N, Johnson JM. Nitric oxide and cutaneous active vasodilation during heat stress in humans. Journal of Applied Physiology. 1998;85:824–829. doi: 10.1152/jappl.1998.85.3.824. [DOI] [PubMed] [Google Scholar]
- Leuenberger U, Glesson K, Wroblewski K, Prophet S, Zelis R, Zwillich C, Sinoway L. Norepinephrine clearance is increased during acute hypoxemia in humans. American Journal of Physiology. 1991;261:H1659–1664. doi: 10.1152/ajpheart.1991.261.5.H1659. [DOI] [PubMed] [Google Scholar]
- Leuenberger UA, Gray K, Herr MD. Adenosine contributes to hypoxia-induced forearm vasodilation in humans. Journal of Applied Physiology. 1999;87:2218–2224. doi: 10.1152/jappl.1999.87.6.2218. [DOI] [PubMed] [Google Scholar]
- MacLean DA, Sinoway LI, Leuenberger U. Systemic hypoxia elevates skeletal muscle interstitial adenosine levels in humans. Circulation. 1998;98:1990–1992. doi: 10.1161/01.cir.98.19.1990. [DOI] [PubMed] [Google Scholar]
- Mancia G. Influence of carotid baroreceptors on vascular responses to carotid chemoreceptor stimulation in the dog. Circulation Research. 1975;36:270–276. doi: 10.1161/01.res.36.2.270. [DOI] [PubMed] [Google Scholar]
- Reed AS, Tschakovsky ME, Minson CT, Halliwill JR, Torp KD, Nauss LA, Joyner MJ. Skeletal muscle vasodilatation during sympathoexcitation is not neurally mediated in humans. Journal of Physiology. 2000;525:253–262. doi: 10.1111/j.1469-7793.2000.t01-1-00253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remsburg S, Launois SH, Weiss JW. Patients with obstructive sleep apnea have an abnormal peripheral vascular response to hypoxia. Journal of Applied Physiology. 1999;87:1148–1153. doi: 10.1152/jappl.1999.87.3.1148. [DOI] [PubMed] [Google Scholar]
- Richardson DW, Kontos HA, Raper AJ, Patterson JL., Jr Modification by beta-adrenergic blockade of the circulatory responses to acute hypoxia in man. Journal of Clinical Investigation. 1967;46:77–85. doi: 10.1172/JCI105513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowell LB, Blackmon JR. Hypotension induced by central hypovolaemia and hypoxaemia. Clinical Physiology. 1989;9:269–277. doi: 10.1111/j.1475-097x.1989.tb00979.x. [DOI] [PubMed] [Google Scholar]
- Rowell LB, Johnson DG, Chase PB, Comess KA, Seals DR. Hypoxemia raises muscle sympathetic activity but not norepinephrine in resting humans. Journal of Applied Physiology. 1989;66:1736–1743. doi: 10.1152/jappl.1989.66.4.1736. [DOI] [PubMed] [Google Scholar]
- Sagawa S, Shiraki K, Konda N. Cutaneous vascular responses to heat simulated at a high altitude of 5,600 m. Journal of Applied Physiology. 1986;60:1150–1154. doi: 10.1152/jappl.1986.60.4.1150. [DOI] [PubMed] [Google Scholar]
- Saito M, Mano T, Iwase S, Koga K, Abe H, Yamazaki Y. Responses in muscle sympathetic activity to acute hypoxia in humans. Journal of Applied Physiology. 1988;65:1548–1552. doi: 10.1152/jappl.1988.65.4.1548. [DOI] [PubMed] [Google Scholar]
- Spina D, Fernandes LB, Preuss JMH, Hay DWP, Muccitelli RM, Page CP, Goldie RG. Evidence that epithelium-dependent relaxation of vascular smooth muscle detected by co-axial bioassays is not attributable to hypoxia. British Journal of Pharmacology. 1992;105:799–804. doi: 10.1111/j.1476-5381.1992.tb09060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]