

Abstract
Over twenty different missense mutations in the α-subunit of the adult skeletal muscle Na+ channel (hSkM1) have been identified as a cause of myotonia or periodic paralysis. We examined state-dependent mexiletine block for mutations involving the putative binding site in S6 segments (V445M, S804F, V1293I, V1589M and M1592V). Whole-cell Na+ currents were measured from wild-type (WT) and mutant channels transiently expressed in HEK cells.
Use-dependent block (10 ms pulses to −10 mV, at 20 Hz) in 100 μm mexiletine was reduced modestly by mutations in IVS6 (V1589M, M1592V) and enhanced by the mutation in IS6 (V445M). For mutations in IIS6 (S804F) and IIIS6 (V1293I) use-dependent block was not statistically different from that of wild-type channels.
Resting-state block (10 ms pulses to −10 mV from −150 mV, at 0.1 Hz) of S6 mutants was comparable to that of WT (dissociation constant for resting channels, KR = 650 ± 40 μm, n = 9). The S6 mutant with the greatest change in KR was V445M (KR = 794 ± 45 μm, n = 5), but this difference was only marginally significant (P = 0.047).
A modified technique for estimating local anaesthetic affinity of inactivated channels was developed to reduce errors due to slow inactivation and to failure of drug binding to reach equilibrium. Mexiletine affinity for inactivated channels was reduced by mutations in IVS6 (V1589M: dissociation constant for the inactivated state (KI) = 44.7 μm; M1592V: KI = 40.0 μm) and increased by the mutation in IS6 (V445M: KI = 15.0 μm), compared to wild-type channels (KI = 28.3 μm).
We conclude that the disease-associated S6 mutations in domains I–IV cause at most a 2-fold change in inactivated state affinity and have even less of an effect on resting block. Model simulations show that the reduced use-dependent block of IVS6 mutants derives primarily from an increased off-rate at hyperpolarized potentials, whereas the enhanced use-dependent block of the IS6 mutant was due to a higher affinity for inactivated V445M channels.
Over twenty different missense mutations in the α-subunit of the adult skeletal muscle Na+ channel (hSkM1) are known to cause several heritable muscle diseases including hyperkalaemic periodic paralysis (HyperPP), paramyotonia congenita (PMC) and potassium-aggravated myotonia (PAM) (see reviews by Cannon, 1998; Lehmann-Horn & Jurkat-Rott, 1999). Functional studies of mutant channels have been performed using heterologous expression systems in order to understand the pathophysiological basis for the enhanced excitability in myotonia and the inexcitability during attacks of periodic paralysis. Fast inactivation is partially disrupted by every mutation (Cannon, 1997) and some mutations cause a hyperpolarized shift in activation (Cummins et al. 1993; Mitrovic et al. 1995; Green et al. 1998; Plassart-Schiess et al. 1998; Takahashi & Cannon, 1999). Model simulations have shown that these functional defects are sufficient to cause repetitive discharges in myotonia and, in combination with defects in slow inactivation, may cause paralysis for mutants with more severely disrupted fast inactivation by a depolarization-induced loss of excitability (Cannon et al. 1993; Hayward et al. 1997).
The antiarrhythmic drug mexiletine, a lidocaine (lignocaine) derivative, is used clinically to reduce or prevent myotonia (Rüdel et al. 1980; Jackson et al. 1994; Hudson et al. 1995). Class I antiarrhythmic agents and local anaesthetics, including mexiletine and lidocaine, exert their therapeutic effects by blocking voltage-gated sodium channels in a state-dependent manner. Drug affinity is low for resting channels at hyperpolarized potentials, as revealed by tonic block measured during brief infrequent depolarizations. The apparent affinity increases 20- to 100-fold for depolarized channels. Use-dependent block occurs at higher frequencies of depolarization, primarily due to a slow off-rate, and is the mechanistic basis for antiarrhythmic, antimyotonic, or antiepileptic action (Butterworth & Strichartz, 1990; Hille, 1992).
Ragsdale et al. (1994) have shown in the rat brain IIa Na+ channel that local anaesthetic binding is strongly influenced by residues lying in the mid-portion of the S6 transmembrane segment in domain IV (IVS6). Wang et al. (1997) reported similar findings for the rat skeletal muscle sodium channel. Several disease-associated mutations in hSkM1 lie in S6 segments, which led us to ask whether anecdotally reported differences in the clinical efficacy of mexiletine could be due to mutation-dependent alterations in drug affinity. To date, three studies have investigated the actions of use-dependent blockers on skeletal muscle sodium channel mutations associated with human disease. Fan et al. (1996) observed a reduction in use-dependent lidocaine block of T1313M in the III-IV linker due to a decreased affinity for inactivated channels. Use-dependent block was enhanced for R1448C in IVS4, as a result of mutation-induced alterations in channel gating. Weckbecker et al. (2000) similarly found an enhanced use-dependent block by mexiletine with another disease mutation at the same residue (R1448H), although they concluded that the enhancement was due solely to the slowness of recovery from a drug-bound state. Fleischhauer et al. (1998) observed a reduction in use-dependent block by mexiletine for G1306E in the III-IV linker and for F1473S in the IVS4-S5 loop. Based on measures of tonic block and mexiletine-induced shifts in Na+ channel availability, these authors concluded that the alteration in use dependence was produced by altered gating of mutant channels rather than a direct effect on mexiletine affinity. Two disease-associated mutations are near the proposed local anaesthetic binding site in IVS6: V1589M, a cause of potassium-aggravated myotonia, and M1592V, which causes hyperkalaemic periodic paralysis with myotonia. State-dependent block has not previously been characterized for either. Moreover, several mutations are in S6 segments of domains I, II, or III and are, therefore, potential additional candidates for altering mexiletine affinity.
In the present study, we examined state-dependent mexiletine block for mutations involving S6: V445M (IS6), S804F (IIS6), V1293I (IIIS6), V1589M (IVS6) and M1592V (IVS6). We found that use-dependent block by pulses applied at 20 Hz in the presence of mexiletine was reduced by IVS6 mutants (V1589M, M1592V), and enhanced for the IS6 mutant (V445M). Resting state block was marginally decreased for the IS6 mutant, whereas block of all other mutants was not different from that of wild-type. The affinity for inactivated channels, estimated by measuring the time course of recovery from mexiletine block, was reduced by ∼1.5-fold for V1589M and M1592V and increased by ∼2-fold for V445M, compared to wild-type. Simulations demonstrate that the enhanced use-dependent block of V445M was due to the increased affinity for block of inactivated channels. Conversely, the reduced use-dependent block of the IVS6 mutants resulted from an increased rate of unbinding from hyperpolarized channels.
METHODS
Expression of sodium channels
The adult isoform of the human skeletal muscle sodium channel α-subunit, hSkM1 (George et al. 1992), and the mutants were subcloned into the mammalian expression vector pRc/CMV. Construction of the cDNAs encoding mutant channels was described previously (Green et al. 1998; Hayward et al. 1999; Takahashi & Cannon, 1999). The human β1-subunit cDNA (McClatchey et al. 1993) was subcloned into the EcoRI site of the mammalian expression vector pcDNAI (Invitrogen, San Diego, CA, USA).
Culture of human embryonic kidney cells (HEK293t) and their transient transfection were performed as described previously (Hayward et al. 1996). In brief, plasmid DNAs encoding WT or mutant human Na+ channel α-subunits (0.8 μg per 35 mm diameter dish), the human Na+ channel β1-subunit (in 4-fold molar excess over α-subunit DNA), and a CD8 marker (0.15 μg) were co-transfected by the calcium phosphate method. At 1-3 days after transfection, the HEK cells were trypsinized briefly and passaged onto 22 mm diameter round glass coverslips for electrophysiological recording. Individual transfection-positive cells were identified by labelling with anti-CD8 antibody crosslinked to microbeads (Dynal, Great Neck, NY, USA; Jurman et al. 1994).
Whole-cell recording
Na+ currents were measured using conventional whole-cell recording techniques as described previously (Hayward et al. 1996). Recordings were made with an Axopatch 200A amplifier (Axon Instruments, Foster City, CA, USA). The output was filtered at 5 kHz and digitally sampled at 40 kHz using an LM900 interface (Dagan, Minneapolis, MN, USA). Data were stored on a Pentium-based computer under the control of a custom Axobasic (Axon Instruments) data acquisition program. Greater than 80 % of the series resistance was compensated for by the analog circuitry of the amplifier and the leakage conductance was nulled by digital scaling and subtraction of the passive current elicited by a 20 mV depolarization from the holding potential. Cells with peak currents of < 1 nA or > 20 nA upon step depolarization from -100 mV to -10 mV were excluded. After initially establishing whole-cell access, we often observed leftward shifts in the voltage dependence of gating, an increase in the size of the peak current and a decrease in the amplitude of persistent Na+ current. To minimize these effects, we waited at least 10 min for equilibration after gaining access to the cells.
Patch electrodes were fabricated from borosilicate capillary tubes with a multi-stage puller (Sutter, Novato, CA, USA). The pipette was coated with Sylgard, and the tip was heat-polished to a final tip resistance (in bath solution) of 0.5-2.0 MΩ. The pipette (internal) solution contained (mm): 105 CsF, 35 NaCl, 10 EGTA, 10 Cs-Hepes (pH 7.4). Fluoride was used in the pipette to prolong the stability of the seal. The bath solution contained (mm): 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 5 glucose, 10 Na-Hepes (pH 7.4). Recordings were made at room temperature (21-23 °C). Mexiletine was provided by Boehringer-Ingelheim (Ridgefield, CT, USA).
Data analysis
χ2 minimized curve fits were performed manually off-line using AxoBasic or Origin (Microcal, Northhampton, MA, USA). Conductance was calculated as G(V) = Ipeak(V)/(V - Vrev), where the reversal potential, Vrev, was measured experimentally for each cell. The voltage dependence of activation was quantified by fitting the conductance measures to a Boltzmann function G(V) = Gmax/{1 + exp[-(V - V1/2)/k]}. Steady-state fast inactivation was fitted to a Boltzmann function calculated as I/Ipeak = 1/{1 + exp[(V - Vh)/k]}, where Vh is the half-maximum voltage and k is the slope factor.
Symbols with error bars in the figures indicate means ±s.e.m. For statistical evaluation, Student's unpaired t test was applied and values for P < 0.02 were considered significant. All statistically derived values are given as means ±s.e.m.
RESULTS
Gating
Mexiletine block of sodium channels is highly state dependent and alterations in fast gating behaviour have been reported previously for each mutant (Mitrovic et al. 1994; Hayward et al. 1997; Green et al. 1998; Takahashi & Cannon, 1999). To assess the potential contribution of altered gating on mexiletine block, we measured the voltage dependence of activation and fast inactivation for WT and mutant Na+ channels.
The voltage dependence of Na+ channel gating was characterized by recording whole-cell sodium currents, INa, from HEK cells transiently transfected with cDNAs encoding WT or mutant hSkM1 and the human isoform of the β1-subunit. The voltage dependence of the steady-state fast inactivation, h∞(V), was measured as the peak current elicited after a 300 ms conditioning prepulse from a holding potential of -120 mV. The relative peak currents were fitted with a Boltzmann function and the estimated parameter values are listed in Table 1. The most prominent change was a 6.7 mV rightward (depolarized) shift in the midpoint for V1589M channels (P < 0.001). The voltage dependence of activation was measured by applying step depolarizations from a holding potential of -100 mV. The Na+ conductance was estimated from the peak current and the measured reversal potential, Vrev, as G(V) = Ipeak(V)/(V - Vrev). The conductance data were fitted with a Boltzmann function and the estimated parameters are listed in Table 1. The most prominent change was a 5.7 mV leftward (hyperpolarized) shift in the midpoint for the V1293I mutant (P < 0.001). The alterations in voltage-dependent gating of mutant channels, or lack thereof, shown in Table 1 are in agreement with findings from prior studies (Mitrovic et al. 1994; Hayward et al. 1997; Green et al. 1998; Takahashi & Cannon, 1999).
Table 1.
Estimated voltage-dependent gating parameters for Na+ channels
| Activation | Inactivation | |||
|---|---|---|---|---|
| V½ (mV) | k(mV) | Vh (mV) | k (mV) | |
| WT (28) | −27.6 ± 0.7 | 5.66 ± 0.2 | −73.5 ± 0.6 | 4.69 ± 0.45 |
| V445M (16) | −29.5 ± 0.9 | 6.84 ± 0.21** | −75.8 ± 0.8 | 4.66 ± 0.39 |
| S804F (16) | −30.3 ± 0.9 | 6.54 ± 0.30 | −73.9 ± 0.6 | 4.96 ± 0.38 |
| V1293I (17) | −33.3 ± 1.5** | 5.96 ± 0.27 | −73.7 ± 0.9 | 4.52 ± 0.42 |
| V1589M (20) | −27.6 ± 0.8 | 5.99 ± 0.23 | −66.8 ± 0.7** | 4.51 ± 0.09 |
| M1592V (14) | −29.1 ± 1.0 | 6.63 ± 0.21* | −75.5 ± 1.0 | 5.26 ± 0.20* |
V½, membrane potential at half-maximal inactivation; k, Boltzmann steepness coefficient; Vh, the half-maximum voltage.
P < 0.001
P < 0.02.
Mexiletine block during pulse trains
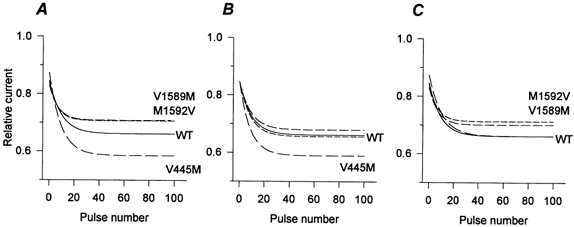
The peak current elicited during a train of depolarizing pulses provides a sensitive measure of both tonic (firstpulse) and phasic (use-dependent) block by mexiletine. To emphasize affinity-dependent differences in block of resting or inactivated channels, as opposed to gatingdependent effects on the occupancy of these states, we used holding and pulse potentials that were far from the midpoint of fast inactivation. Cells were held at -150 mV, and 10 ms depolarizations to -10 mV were applied at a frequency of 20 Hz (Fig. 1 inset). Figure 1A shows the amplitude of the peak current, normalized to the maximal current in the absence of drug. Without mexiletine, the use-dependent reduction in peak INa for both WT and each mutant was only about 5 % (Fig. 1A, cluster of responses at the top). In 100 μm mexiletine, the tonic block observed for the first-pulse was slightly reduced for the IS6 mutant V445M and yet the usedependent block after 100 pulses was greater than that seen in wild-type. In contrast, for the IVS6 mutants (V1589M and M1592V) the first-pulse block was comparable to wild-type, whereas the use-dependent block was reduced. Although there is considerable overlap in the total block (phasic and tonic together) between wild-type and mutant channels during pulse trains (Fig. 1A), the disparate effects on first-pulse versus phasic block suggests the affinity of resting versus inactivated channels may be differentially affected by missense mutations in S6 segments.
Figure 1. Mexiletine block of WT and mutant channels during pulse trains.
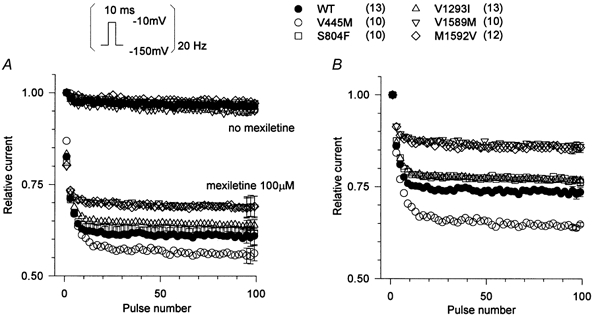
Sodium currents were elicited by 10 ms pulses to -10 mV from a holding potential of -150 mV (inset). Pulses were applied at a rate of 20 Hz with or without mexiletine. In A, the peak current amplitude during each pulse was normalized with respect to the amplitude of the first-pulse in the absence of drug and plotted against the pulse number. Without mexiletine, WT and all mutant channels showed little use-dependent decrease in current (top cluster of curves). Mexiletine (100 μm) produced first-pulse and use-dependent block for WT and mutant Na+ channels. First-pulse block was reduced for V445M but this same mutant had the greatest total block at the end of the pulse train. Conversely the IVS6 mutants had less block after 100 pulses. B, the use-dependent component of block is revealed by normalizing the mexiletine data from A by the peak of the first pulse recorded in the presence of the drug. The use-dependent component of block was significantly reduced for IVS6 mutants (P < 10−6) and increased for the IS6 mutant (P < 10−6). For clarity, every other point is shown and the error bars (s.e.m.) are shown only for the last three points.
The phasic component of block is shown more clearly in Fig. 1B, where the current amplitude of each pulse was normalized to the peak current recorded during the first-pulse in mexiletine. This normalization reveals a significant reduction in use-dependent block after 100 pulses for IVS6 mutants (V1589M: 0.87 ± 0.01, n = 10, P < 10−7; M1592V: 0.85 ± 0.01, n = 12, P < 10−6) and an increase for the IS6 mutant (V445M: 0.64 ± 0.01, n = 10, P < 10−6) compared to wild-type (0.74 ± 0.01, n = 13). These data suggest that IS6 and IVS6 mutations might differentially affect the state-dependent affinity for mexiletine. To explore this hypothesis further, we measured mexiletine block of resting and depolarized channels for WT and mutants and interpreted the data in the context of the modulated-receptor hypothesis of local anaesthetic block (Hille, 1977; Bean et al. 1983).
Affinity for resting channels
Tonic block of resting channels was measured by holding cells at a very negative potential and measuring the mexiletine-induced reduction in peak INa during infrequent pulses. Mexiletine (50 μm to 2 mm) was applied to cells held at -150 mV and 10 ms test pulses to -10 mV were applied every 10 s to assess channel availability. The peak INa was normalized to the control response without mexiletine. The relative block by mexiletine is plotted against mexiletine concentration in Fig. 2. Both WT and mutant channels showed a dose-dependent blockade of resting Na+ channels. Fitting the WT data to the logistic function gave a Hill coefficient of 1.04 ± 0.04 (n = 9). The response for mutant channels was also well fitted using a Hill coefficient of approximately 1, implying a 1:1 stoichiometry of drug bound per channel. The response of each cell was fitted using a single-site binding equation (Hill coefficient of 1) and the mean value of the estimated dissociation constant for resting channels, KR, is listed in Table 2. Mutants V445M and S804F had slightly higher KR values than WT (V445M: 790 ± 45 μm, n = 5; S804F: 780 ± 63 μm, n = 5; WT: 650 ± 40 μm, n = 9), but this 20 % difference was only marginally significant for V445M (P = 0.047) and not at all significant for S804F (P = 0.094). The slightly lower resting state affinity of V445M evident in the dose-response curve (Fig. 2) is consistent with the larger fraction of unblocked channels observed for the first-pulse of a train in 100 μm mexiletine (Fig. 1). On the other hand, the similarity in the estimated KR values is illustrated by the curves in Fig. 2, which are computed based on the mean value of KR for WT (continuous) and for V445M (dotted) channels. We interpret these data as evidence that there was no significant difference in mexiletine affinity for the resting state of these mutant channels.
Figure 2. Mexiletine block of resting channels.
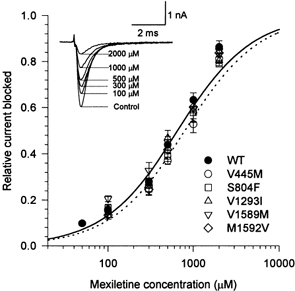
Mexiletine (50 μm to 2 mm) was applied to cells that were held at -150 mV and pulsed to -10 mV for 10 ms at a frequency of 0.1 Hz. Traces (inset) show examples of currents for WT channels without (control) and with mexiletine at concentrations of 100-2000 μm. The peak INa was normalized by the response without mexiletine and the fractional current blocked is plotted against mexiletine concentration. Mutant V445M channels showed less block by mexiletine. The smooth curve shows a fit to the WT data with a Hill coefficient of 1 and dissociation constant KR = 650 μm; the dotted curve shows the fit for V445M, KR = 790 μm. Symbols show means ±s.e.m. for n = 5-9 cells.
Table 2.
Estimated mexiletine affinity
| KR (μM) | KI (μM) | |
|---|---|---|
| WT | 653 ± 40.3 (9) | 28.3 |
| V445M | 794 ± 45.2 (5) | 15.0 |
| S804F | 782 ± 63.3 (5) | — |
| V1293I | 710 ± 97.8 (5) | — |
| V1589M | 589 ± 63.1 (5) | 44.7 |
| M1592V | 631 ± 57.7 (7) | 40.0 |
KR, dissociation constant for the resting state; KI, dissociation constant for the inactivated state.
Affinity for inactivated channels
Local anaesthetics bind preferentially to depolarized sodium channels. The high-affinity conformation probably encompasses several kinetically defined states of the channel, e.g. open, closed pre-open and fast-inactivated. For simplicity, and in keeping with the original nomenclature of modulated receptor hypothesis, we will refer to the high-affinity conformation as ‘inactivated’.
Affinity for inactivated channels was estimated by measuring recovery from mexiletine block. Cells were held at -150 mV and conditioning pulses of 300-1500 ms duration were applied at a potential at which approximately half the channels were fast inactivated (the measured Vh for each channel type). The conditioning pulse was followed by a recovery interval at -150 mV for 0.05-10 000 ms (Fig. 3 inset). Peak Na+ current was measured in response to a subsequent test depolarization to -10 mV. Channel availability was measured as the ratio of the peak current during the test depolarization to that during a reference pulse from -150 mV to -10 mV in the absence of mexiletine. The time course of recovery for WT channels in the absence of mexiletine and in the presence of 100 μm mexiletine are shown in Fig. 3.
Figure 3. Recovery from inactivation and mexiletine block of WT channels.
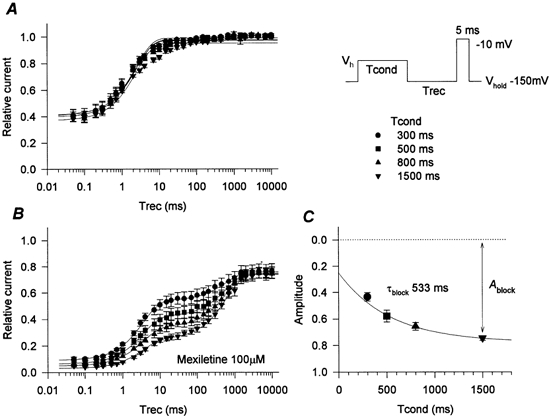
A two-pulse protocol (inset) was used to measure recovery from inactivation and from mexiletine block enhanced by depolarization. Relative availability was normalized to the peak current during the test depolarization to -10 mV without a conditioning pulse in the absence of mexiletine. Symbols denote the duration of the conditioning pulse (300-1500 ms). A, without mexiletine, most of the channels recovered within 20 ms, even after the 1500 ms conditioning pulse. The smooth curves were generated with mean parameter values from fitting the data set of each cell to a single exponential. B, in the presence of 100 μm mexiletine, a slowly recovering component became more prominent with progressively longer conditioning pulses. This slowly recovering component corresponded to recovery from mexiletine block. The extent of maximum recovery was about 80 % of the control due to the block of resting channels by mexiletine. The recovery data were fitted to a two-exponential function, using the time constant of recovery obtained without mexiletine for the fast component. Smooth curves were generated using mean parameter values for each cell. C, the estimated amplitude of the slow component for each cell was plotted against conditioning pulse duration and fitted by a single exponential to estimate the time constant for the onset of block (τblock). The smooth curve was generated with τblock = 533 ms. Means ±s.e.m. for n = 5 cells are shown.
We employed a conditioning voltage near Vh (about -75 mV) to minimize the development of slow inactivation, which is difficult to distinguish kinetically from mexiletine block. Indeed, the fractions of WT channels which were slow-inactivated (defined as the component not recovered at -150 mV within 20 ms) were 6 % and 10 % with 800 and 1500 ms conditioning pulses, respectively (Fig. 3A). Of V445M channels, which have enhanced slow inactivation (Takahashi & Cannon, 1999), only 13 % were slow inactivated, even with a 1500 ms conditioning pulse (data not shown). For all other S6 mutants the fraction of channels slow inactivated by a 1500 ms conditioning pulse was less than or equal to that of WT channels.
In the absence of mexiletine, about 60 % of channels were fast-inactivated by all of the conditioning pulse durations tested, as evidenced by an initial relative peak INa of 0.4 and nearly full recovery within 20 ms (Fig. 3A). Thus, the conditioning voltages employed were, on average, ∼2.5 mV more depolarized than Vh. The time course of recovery was fitted to a single exponential for each cell, with the constraint that a common time constant was used to fit the recovery from the entire range of conditioning pulse durations. The smooth curves in Fig. 3A were generated with mean parameter values obtained from fitting the data for each cell individually. This single-exponential approximation for recovery was suboptimal, especially for longer conditioning pulse durations, because of slow inactivation and a two- or three-exponential approximation was more accurate. We employed a single-exponential approximation, however, because it is representative of the majority of the current recovery.
In the presence of 100 μm mexiletine (Fig. 3B), the time course of recovery showed two distinct components and reached maximum recovery of around 80 %, due to tonic block (see Fig. 2). The time course of the fast component was similar to that observed without mexiletine and we interpret this component to be the fraction of unblocked channels. Conversely, the slow component corresponds to recovery from the use-dependent mexiletine block that occurred during the conditioning pulse. The time course of recovery was fitted to a two-exponential function for each cell. The time constant for the fast component was fixed to the value obtained without mexiletine and the slow time constant was constrained to fit the combined recovery data for all conditioning pulse durations. The relative amplitude of the slow component was allowed to vary for each conditioning pulse duration. The smooth curves in Fig. 3B were generated with mean parameter values obtained from fitting the data from each cell to a two-exponential function. The amplitude of the slowly recovering component increased with increasing duration of the conditioning pulse; it approached a steady-state level after a conditioning pulse of 1500 ms duration (note that the series of conditioning pulse durations shown in Fig. 3 increases exponentially). The fraction of mexiletine-blocked channels (Ablock) was estimated as the sum of the slowly recovering component (use-dependent block) plus the residual fraction that remained unavailable even after a 10 s recovery period (tonic block). This computation assumes that slow inactivation during the conditioning pulse was negligible. The development of block as a result of progressively longer conditioning pulse durations is shown in Fig. 3C, with greater block plotted in the downward direction to facilitate a direct comparison to the raw data in Fig. 3B. The time course of block was estimated by fitting the data in Fig. 3C to a single exponential. For WT channels the time constant for development of block, τblock, was 530 ± 50 ms and the maximal block (use-dependent and tonic) was 0.75 ± 0.018 (n = 5).
The depolarization-induced block in 100 μm mexiletine is compared in WT and mutant Na+ channels in Fig. 4. The development of use-dependent block is shown in Fig. 4A, where the data are plotted in the same format as those in Fig. 3C. After a 1500 ms conditioning pulse to Vh, the IVS6 mutants displayed less block (V1589M: Ablock = 0.66 ± 0.033, n = 6, P < 0.001; M1592V Ablock = 0.66 ± 0.018, n = 6, P < 0.0005) than WT channels (0.75 ± 0.018, n = 5). In contrast, block was slightly increased for the IS6 mutant (V445M: Ablock = 0.82 ± 0.011, n = 5, P < 0.05). The time constant for the development of block in 100 μm mexiletine was comparable for WT and mutant channels, with a value of about 500 ms (Fig. 4B).
Figure 4. Development of mexiletine block for depolarized channels.
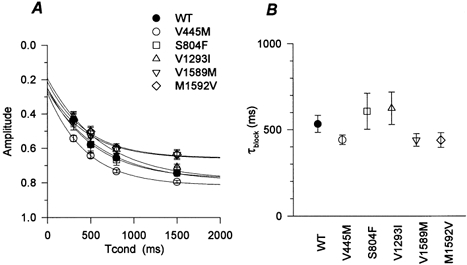
For each cell, the data set in the presence of 100 μm mexiletine was simultaneously fitted to a two-exponential function (one component for fast inactivation obtained without mexiletine and the other for recovery from mexiletine block; see Fig. 3). A, for each mutant the estimated amplitude of the slow component was plotted against the duration of the conditioning pulse. The fraction blocked by mexiletine with a 1500 ms conditioning pulse (Ablock) was significantly smaller for V1589M and M1592V, and larger for V445M, than that for WT. The smooth curves were generated with mean parameter values from fits to a single-exponential curve. B, the estimated time constant of block development (τblock) is plotted for each channel type. There was no difference in τblock for each channel type.
To provide a more direct comparison between the usedependent components of block in WT and mutant channels, the recovery data after the 1500 ms conditioning pulse are superimposed in Fig. 5. The data for each cell were normalized to the maximal recovery after 10 s at -150 mV, while in 100 μm mexiletine. This normalization compensates for small mutation-dependent differences in tonic block. Figure 5A shows that, compared to WT, IVS6 mutant (V1589M, M1592V) channels had less use-dependent block (i.e. showed greater recovery within 20 ms) and the IS6 mutant (V445M) had more. Thus, independently of the quality of the exponential fits required to estimate Ablock, the superimposed recovery data directly show mutation-dependent differences in mexiletine block of depolarized channels. Figure 5A also shows that the time course of recovery from block varied among the different mutants. Unblock was faster for the IVS6 mutants, as shown by the leftward shift in the recovery curves (Fig. 5A) and this was quantified as the time constant for the slower component of recovery, τoff, in Fig. 5B.
Figure 5. Recovery from inactivation and mexiletine block in WT and mutant channels.
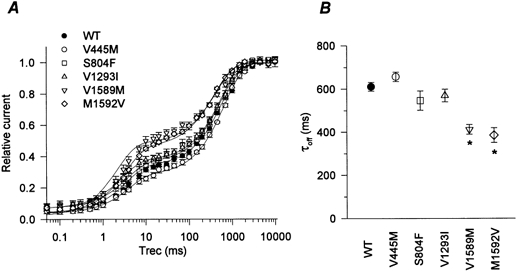
A, the recovery recorded after a 1500 ms conditioning pulse is superimposed for each Na+ channel type in order to provide a direct comparison of the extent and kinetics of the slow component. To compensate for small differences in tonic block, the current amplitudes have been normalized to maximal available current after a 30 s recovery at -150 mV in 100 μm mexiletine. The recovery curves for mutants V1589M and M1592V are shifted upward (fewer channels are blocked) and to the left (recovery from mexiletine block is faster). Smooth curves were generated using mean parameter values obtained by fitting with a two-exponential function for each cell (see Fig. 3). Symbols show means ±s.e.m. for n = 4-7 cells. B, the time constant for the slow component of recovery is compared in WT and mutant channels. Recovery was significantly slower for the IVS6 mutants (*P < 0.02).
Since use-dependent block by 100 μm mexiletine differed from WT in three mutant channels (V445M, V1589M, M1592V), we repeated the recovery experiments for this group of channels at different drug concentrations in an effort to estimate the dissociation constant of the inactivated state. The time constant for the development of block during the conditioning pulse increased as the [mexiletine] was decreased (data not shown). This placed a practical limit of 50 μm on the minimal [mexiletine], because the time constant for block became too long (∼1000 ms) compared to the conditioning pulse duration (1500 ms). On the other hand, the time constant for recovery of the drug-bound fraction (∼600 ms) was independent of mexiletine concentration (within the 50-200 μm range). The lack of a concentration effect on recovery rate is consistent with the notion that unbinding upon repolarization occurs primarily from the resting state, for which the dissociation constant is much greater than 200 μm (see Scheme 1, below). The fractional amplitude of block at 1500 ms is plotted against mexiletine concentration in Fig. 6. Both WT and mutant channels showed dose-dependent block by mexiletine. The apparent dissociation constant (KApp) at the conditioning pulse potential was estimated by fitting the data with the single-site binding relation:
The dissociation constant for the inactivated state, KI, was subsequently calculated from KApp, KR and the measured value of h∞ at the conditioning potential (the asymptote for brief recovery intervals in Fig. 3A), using the modulated receptor model of Bean et al. (1983): 1/KApp = h∞/KR+ (1 - h∞)/KI. Rearranging terms:
Estimated KI values for WT and for each mutant are listed in Table 2. For WT channels, KI was 28 μm, which is a 23-fold lower dissociation constant than for resting channels (Table 2). The estimated dissociation constants for V1589M and M1592V were increased ∼1.5-fold compared with that for WT whereas, for V445M, KI was about half that of WT channels. The response from each cell yielded only a single point on the dose-response curve (Fig. 6), because the protocol to estimate Ablock required the measurement of a full recovery time course (30 different recovery intervals, for a total acquisition time of nearly 15 min). Hence, it was not possible to compute a meaningful s.e.m. value for KI because pooling of data from multiple cells was required for a single estimate. Moreover the propagation of uncertainties in the estimates of KR and h∞ further complicates the interpretation of a standard error.
Figure 6. Dose-response curve for mexiletine block at a membrane potential equal toVh.
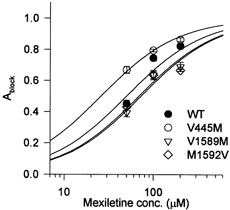
The Ablock for the WT and for each mutant is plotted against the concentration of mexiletine. The relationship was shifted to the left for mutant V445M and to the right for mutants V1589M and M1592V. The smooth curves show fits to the data with a single-site binding relation using the apparent dissociation constant (KApp). Symbols show means ±s.e.m. for n = 5-6 cells.
Simulation of use-dependent block
We performed simulations to test whether the measured changes in mexiletine affinity were sufficient to produce the differences in use-dependent block observed for mutant channels in Fig. 1. Block was simulated with the modulated receptor model (Scheme 1), in which the affinity is greater for inactivated channels than for resting closed channels (Bean et al. 1983).
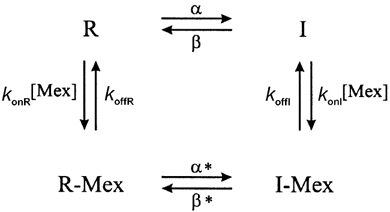 |
In this scheme, R is the fraction of closed resting channels, R-Mex is the fraction of drug-bound resting channels, I is the fast-inactivated fraction and I-Mex is the fraction of fast-inactivated mexiletine-bound channels. α and β are the voltage-dependent rates of inactivating and recovering from inactivation, respectively. This model does not explicitly show an open state. The peak current that could be elicited is proportional to the fraction of channels that reside in R and therefore are available for opening. For convenience, we have designated the high-affinity depolarized state as equivalent to fast-inactivated channels, although recent data from our laboratory (Vedantham & Cannon, 1999) have shown that the high-affinity conformation does not require closure of the fast-inactivation gate (the interdomain III-IV linker). This study also showed that the off-rate of the fast-inactivation gate is not slowed by the presence of bound drug. Hence, we assume that β*=β. Conservation of microscopic reversibility requires that α*=α(KR/KI). Notice that since KR/KI > 1, then α* > α.
The voltage-dependent rates, α and β, were computed from the steady-state availability, h∞, in Table 1, and from the macroscopic time constant for fast inactivation, τh, as:
τh at -10 mV was measured from the time constant of the decay in INa. Because h∞ was approximately 0 at -10 mV, the estimate for α(-10) from τh is accurate, whereas β(-10) is essentially indistinguishable from 0. At -150 mV, τh was determined by the time course of recovery from inactivation in the absence of mexiletine. Now h∞ was approximately 1.0, so β(-150) was well defined by τh, but α(-150) was indistinguishable from 0.
The on- and off-rates for mexiletine were estimated from the rates of the development of and recovery from block and the dissociation constants KR and KI. The off-rate from resting channels was approximated as, koffR = 1/τoff (see Appendix). The corresponding on-rate was then computed as konR = koffR/KR. The on-rate for inactivated channels was estimated from the rate of block onset at -75 mV (Fig. 4) and KI as:
(see Appendix for details). Finally, we set koffI = KIkonI. Values for α, β, konR, koffR, konI and koffI are listed for the WT and for each mutant in Table 3. Using these rate constants, the time course of use-dependent block was computed by digital integration with a time step of 0.1 ms.
Table 3.
Estimated rate constants for the modulated receptor model in Scheme 1
| α(−150) (s−1) | β(−150) (s−1) | α(−10) (s−1) | β(−10) (s−1) | konR ((M s)−1) | koffR (s−1) | konI ((M s)−1) | koffI (s−1) | |
|---|---|---|---|---|---|---|---|---|
| WT | 0.000047 | 400 | 1800 | 0.010 | 2500 | 1.6 | 22000 | 0.63 |
| V445M | 0.000064 | 550 | 1900 | 0.011 | 1900 | 1.5 | 33000 | 0.49 |
| V1589M | 0.0000074 | 760 | 1800 | 0.093 | 4000 | 2.4 | 22000 | 0.99 |
| M1592V | 0.00039 | 550 | 1600 | 0.035 | 4100 | 2.9 | 25000 | 1.00 |
α(−150) and β(−150), rates of entry and recovery from inactivation at −150 mV; α(−10) and β(−10), rate constants at −10 mV; konR and konI, second-order rate constants for mexiletine binding to resting and inactivated channels, respectively; koffR and koffI, drug unbinding rates for resting and inactivated channels.
The simulated responses to a 20 Hz train of 10 ms pulses to -10 mV in the absence of mexiletine did not show use dependency for WT or for any of the simulated mutants (relative INa > 99 %; data not shown), in agreement with the experimental data in Fig. 1A. Use dependence was prominent for simulations in the presence of 100 μm mexiletine (Fig. 7A). The simulated responses of mutant channels differed from those of the WT. As in the experimental data in Fig. 1, the simulated IS6 mutant (V445M) responses had less first-pulse block, but more use-dependent block with subsequent pulses. The simulated IVS6 mutant responses had normal first-pulse block but less use dependence, in agreement with the experimental observations.
Figure 7. Simulation of use-dependent block by mexiletine.
Use-dependent block (10 ms pulses to -10 mV, at 20 Hz) was simulated with the modulated receptor model shown in Scheme 1. A, the rate constants listed in Table 3 were used. Use-dependent block of simulated channels was reduced for IVS6 mutants and increased for the IS6 mutant. B, setting the on- and off-rates to WT values for mexiletine binding to the resting state abolishes the reduced use dependence of IVS6 mutants but does not remove the enhanced block for the IS6 mutant. C, with on- and off-rates for block of the inactivated state set to WT values, but rates for block of the resting state set to measured values, the enhanced use dependence of the IS6 mutant is removed, but not the decreased use dependence for the IVS6 mutants.
The mechanisms for the enhanced use-dependent block of V445M, and for the reduced block in the IVS6 mutants, are suggested by the rate constants in Table 3. The higher on-rate of mexiletine for inactivated V445M channels, coupled with a modestly slowed off-rate from the resting state, is expected to enhance use dependence. In contrast, the reduced use dependence for IVS6 mutants (V1589M and M1592V) appears to derive from a nearly 2-fold increase in the off-rate from the resting state. Parameter sensitivity studies were performed to test these hypotheses. Figure 7B shows the simulated use dependence if mexiletine on- and off-rates for the resting state were fixed to be identical to those of WT, whereas the corresponding rates for the inactivated state assumed their mutation-specific values. The simulated IVS6 mutants are now comparable to WT, whereas the simulated IS6 mutant still exhibits enhanced use dependence. This example shows that the altered use dependence of IVS6 mutants must originate primarily from changes in binding to the resting state, whereas alterations in block of the inactive state alone are sufficient to reproduce the enhanced use dependence of V445M. The converse parameter simulation is shown in Fig. 7C; on- and off-rates for block of inactivated channels retain WT values while the corresponding rates for the resting state keep their mutation-specific values. Now the enhanced use dependence of simulated V445M is entirely lost, while the simulated IVS6 mutants still show reduced block. This second example shows that, for the model, the altered block of the resting state alone is sufficient to reduce use-dependent block of IVS6 mutants, whereas the small changes in resting state block for V445M have no effect on use-dependent block during a 20 Hz train of pulses. Interestingly, the resting state dissociation constants for IVS6 mutants were nearly identical to those of WT. The major difference from WT was not in the equilibrium behaviour (Fig. 2), but in the slower kinetics of unblock for mutants (Fig. 5). The simulation for IVS6 mutants in Fig. 7C illustrates the danger of the common assumption that altered use dependence during a pulse train necessarily implies a change in binding to the high-affinity inactivated state. Even when equilibrium binding to the resting state is normal, changes in use-dependent block may occur as a result of alterations in the kinetics of resting state block alone.
For both WT and mutant channels, the magnitude of the simulated use-dependent block is smaller (Fig. 7A) than that which was observed experimentally (Fig. 1). This discrepancy is partially due to additional inactivated states of the channel. For example, even in the absence of mexiletine, availability is reduced by ∼5 % experimentally (Fig. 1A), whereas no reduction is predicted for a 20 Hz train of 10 ms depolarizations in a model that has only fast inactivation. Despite these shortcomings of the model, the simulation reproduces the general phenomenon of use-dependent block fairly well, especially in view of the fact that none of the model parameter values were derived from data measured during pulse trains. The rank order in the degree of simulated use-dependent block parallels that observed experimentally (simulated: V1589M ≈ M1592V < WT < V445M; experimental: V1589M ≈ M1592V < WT < V445M).
DISCUSSION
We have examined state-dependent mexiletine block of WT and mutant human skeletal muscle sodium channels that contain disease-associated missense substitutions in S6: V445M (IS6), S804F (IIS6), V1293I (IIIS6), V1589M (IVS6) and M1592V (IVS6). None of these mutants produced a dramatic change in mexiletine block. On the other hand, use-dependent block during a train of depolarizing pulses was unequivocally altered by missense mutations in IS6 and IVS6. The pulse protocol employed for the use dependence assay was optimized for the detection of differences in state-dependent mexiletine affinity, as opposed to secondary effects arising from altered gating, because the strongly hyperpolarized holding potential and depolarized test pulses were far from the steep voltage-dependent range of fast inactivation (-100 to -40 mV). Other protocols designed to specifically measure affinity of resting or of inactivated channels revealed the mechanism of altered use dependence. The enhanced use dependence for V445M was associated with higher affinity block of inactivated channels (primarily due to increased konI). Reduced use dependence of IVS6 mutants was primarily due to an increased off-rate at -150 mV (Fig. 5). It was proposed that this unbinding occurred almost entirely from the resting state because prior work has shown that the inactivation gate (III-IV linker) recovers within milliseconds despite the slow return of channel availability over hundreds of milliseconds (Vedantham & Cannon, 1999). Interestingly, there was no difference in the equilibrium binding of mexiletine to resting channels at -150 mV between IVS6 mutants and WT. For the model, this implies a balanced increase in on- and off-rates for the resting state so that the dissociation constant is unaffected.
Measures of mexiletine affinity
In this study, the affinity of mexiletine for resting channels was determined by holding cells at a very negative potential (-150 mV) and measuring the mexiletine-induced reduction in peak INa during infrequent pulses to -10 mV. Several recent studies have emphasized the importance of using strongly negative holding potentials to minimize binding to high-affinity inactivated states (Fan et al. 1996; Wright et al. 1997) which would cause an apparent increase in the resting state affinity. Our measured affinity of mexiletine for resting channels (650 ± 40 μm for WT) is weaker than reported in prior studies. Sah et al. (1998) reported an IC50 of 368 μm for the rat skeletal muscle sodium channel expressed in Xenopus oocytes. Using frog semitendinosus muscle fibres, De Luca et al. (1997) measured an IC50 of 83 μm for resting channels. Since the holding potential was -100 mV in both studies, it is likely that these higher apparent affinities of tonic block derive from a substantial fraction of inactivated drug-bound channels at the holding potential. Indeed, if the affinity for inactivated channels is 30-fold higher than for resting ones, then the modulated receptor model predicts that as little as 3 % of channels being inactivated at the holding potential (i.e. h∞ = 0.97) causes an apparent affinity 2-fold higher than that of the true resting state. Of course, other possible explanations include differences in channel isoform (rat or frog versus human), preparations, and recording methods.
Several strategies have been used to estimate the affinity of local anaesthetics for inactivated sodium channels (Bean et al. 1983). A commonly used measure is the concentration dependence of the drug-induced negative shift in steady-state availability (h∞ curve). Long conditioning pulses, of the order of seconds, have usually been used to ensure that steady-state binding is reached (Bean et al. 1983). While this approach has been used successfully for cardiac sodium channels, the protocol is problematic when studying the adult skeletal muscle isoform due to its greater tendency to slow inactivate compared to the cardiac isoform (Richmond et al. 1998). Fan et al. (1996) attempted to minimize the effects of slow inactivation by using conditioning pulses of 500 ms duration. With this technique, these authors found a reduced affinity for inactivated-state lidocaine block in the paramyotonia congenita mutant T1313M (KI = 63 μm) compared to that in WT (KI = 11 μm). An alternative method for estimating KI is based on a twopulse recovery protocol in which the slowly recovering component is interpreted as the fraction of drug-bound channels (Bean et al. 1983). Of course this method is also potentially limited by the development of slow inactivation from which recovery is slow and indistinguishable from drug-induced changes. Sah et al. (1998) attempted to minimize the effects of slow inactivation by using a 500 ms conditioning pulse to -10 mV. We used an alternative approach. By using a less depolarized conditioning voltage near Vh (typically -75 mV), we were able to use durations of up to 1500 ms with much less contamination by slow inactivation. Note that our estimate of KI from KApp is much less sensitive to uncertainties in h∞ than the common practice of estimating KR from KApp at hyperpolarized potentials where h∞≈ 1. The difference is due to the very steep dependence of KApp on h∞, as h∞ approaches 1 because KR/KI is typically 20-100. For example, given KR/KI = 25, if h∞ is actually 0.96 at the holding potential, the measured KApp underestimates the resting state dissociation constant (KR) by 200 %. For our computation of KI from KApp when h∞≈ 0.5, this same error of 0.04 in h∞ would result in only a 7% underestimate of KI.
Our attempt to separate the effects of mexiletine block from slow inactivation was no panacea. Measurements made at low drug concentrations were still problematic. Below 50 μm mexiletine, the kinetics of block were comparable to those of slow inactivation onset. Even worse, the ‘confounding’ slow inactivated component became a larger fraction of the total current reduction (block + slow inactivated) and thereby introduced a greater relative error. Despite these limitations, Fig. 4A shows how much better our protocol (1500 ms) was at estimating ‘steady-state’ block of depolarized channels, compared to the frequent practice of using short conditioning pulses of only 300 or 500 ms. We found an inactivated-state dissociation constant for mexiletine of 28 μm which is smaller than the value of 44 μm reported by Sah et al. (1998). The discrepancy is probably due to an underestimate of the fraction of drug-bound channels by Sah et al. (1998) when using a 500 ms conditioning pulse.
Structural implications for a mexiletine binding site
Mutational analyses have identified several regions in the sodium channel α-subunit that affect the affinity and/or efficacy of local anaesthetics and anti-arrhythmic drugs. Ragsdale et al. (1994) identified several residues critical for local anaesthetic binding by alanine-scanning mutagenesis throughout IVS6 in rat brain IIa channels. At sites homologous to the disease mutations studied here, alanine substitutions caused varied effects on local anaesthetic binding. V1767A (residue V1589 in hSkM1) increased the affinity of resting channels and reduced use-dependent block. We found V1589M also reduced use-dependent block, but there was no change in resting state affinity. M1770A in RBIIa channels (residue M1592 in hSkM1) did not alter resting or use-dependent block. In hSkM1, we found M1592V did not alter resting block, but decreased use-dependent block.
Other S6 segments in the primary structure of the α-subunit have been reported to affect binding of local anaesthetics. Wang et al. (1998) reported that two mutations in IS6 of the rat skeletal muscle sodium channel, N434K and L437K (corresponding to N440K and L443K in hSkM1) reduced both the resting and the inactivated state affinity for etidocaine. In this study, we found increased depolarized state affinity for the IS6 mutant V445M. Mutations in the rat SkM1 IIIS6 segment, S1276 and L1280 (corresponding to 1283 and 1287 in hSkM1), reduced the affinity of the inactivated state for bupivacaine (Wang et al. 2000). In our study, however, no change in affinity for either the resting or the depolarized state was observed for the neighbouring mutation, V1293I.
A third region important for local anaesthetic block is the selectivity filter. Sunami et al. (1997) reported that charge-reversing substitutions in the selectivity filter segment of domain III (K1237E in rat SkM1) increased the resting block by lidocaine, but not for neutral analogues. Although the structural implication is unclear, a disease mutant in III-IV linker (T1313M) has been shown to have reduced affinity for inactivated channel states (Fan et al. 1996).
Clinical implications
Mexiletine and the related compounds tocainide and fleccanide have been used to reduce or prevent myotonia associated with hyperkalaemic periodic paralysis, paramyotonia congenita and potassium-aggravated myotonia (Rüdel et al. 1980; Jackson et al. 1994). The clinical response to mexiletine has been published as anecdotal case reports in which the efficacy of mexiletine has been variable (Trudell et al. 1987; Rosenfeld et al. 1997). Mutation-specific changes in use-dependent block are potential sources of this variability. These differences may, in turn, result from altered affinity or from altered gating. Heterologous expression of mutant sodium channels offers the opportunity to test these hypotheses and may impact on the choice of therapy in a mutation-specific manner. To date, the efficacy of local anaesthetic block has been examined for eleven missense mutations found in heritable diseases of skeletal muscle. Three mutations have been found to decrease the affinity of local anaesthetics for the inactivated state: the III-IV loop mutation T1313M caused a 5-fold reduction (Fan et al. 1996), whereas the two IVS6 mutants V1589M and M1592V (this study) produced only a 50 % reduction. Use-dependent block was also reduced for G1306E and F1473S but there was no discernible change in drug affinity, from which the authors conclude that alterations in gating must be the source of the reduced use-dependent block (Fleischhauer et al. 1998). An increase in use-dependent block has been observed for R1448C (Fan et al. 1996), R1448H (Weckbecker et al. 2000) and V445M (this study). Consistent with this change in phasic block, the inactivated state affinity was 2-fold higher for V445M than for WT channels. However, a difference in the affinity for the inactivated state was not noted for R1448 mutations. These examples illustrate the combined importance of both state-dependent affinity and gating in determining the efficacy of block by local anaesthetic agents.
Caution should be used in applying the results from our heterologous expression studies to the clinical management of patients. First, the therapeutic plasma concentration of mexiletine (2.8-11 μm) is much lower than the concentration used in these studies (100 μm) (Monk & Brogden, 1990). Second, the resting membrane potential of skeletal muscle is more depolarized than the holding potentials used in these studies, which will tend to increase block. Third, temperature will affect mexiletine block. As discussed above, it is likely that not only the affinity of mexiletine but also the gating behaviour of each mutant channel might define clinical effects of mexiletine.
Acknowledgments
We thank Vasanth Vadantham and Jim Morrill for comments on the manuscript and Miwako Takahashi for her technical assistance. This work was supported by the National Institutes of Arthritis and Musculoskeletal Diseases (R01-AR42703 to S.C.C.), the Sumitomo Life Insurance Welfare Services Foundation (to M.P.T.) and the Massachusetts General Hospital Fund for Medical Discovery (to M.P.T.).
APPENDIX
The rate constants for the modulated receptor model shown in Scheme 1 were estimated from the measured rates of the onset of, and recovery from, block and from the dissociation constants. The time constant of recovery at -150 mV (τoff in Fig. 5B) provides a good estimate for the off-rate of mexiletine from resting channels, koffR≈ 1/τoff. This approximation holds for two reasons. First, at -150 mV virtually all the drug-bound channels recover by a pathway through the R-Mex state, i.e. I-Mex → R-Mex is much faster than I-Mex → I (Vedantham & Cannon, 1999). Second, when the mexiletine concentration is much less than KR, the rate of blocking of resting channels is negligible compared to the unblocking rate. The on-rate for resting channels was then calculated as konR = koffR/KR. For inactivated channels, the on-rate of mexiletine was computed from the time course of the additional block produced by depolarizing the membrane from -150 to -70 mV. Because the affinity of resting channels is more than an order of magnitude lower than that of inactivated ones, τblock at -70 mV primarily depends on the on- and off-rates from the inactivated state. The equilibration rate between R and I is much faster than drug binding/unbinding and therefore the effect of protecting channels from high-affinity block in the resting state can be incorporated by scaling the on-rate by the fraction of inactivated channels, α/(α+β):
Solving this equation for konI, recalling that KI = koffI/konI, yields the relation used to estimate the on-rate for inactivated channels:
Finally, the off-rate was computed as koffI = KIkonI. The estimated values for the rate constants are listed in Table 3.
References
- Bean BP, Cohen CJ, Tsien RW. Lidocaine block of cardiac sodium channels. Journal of General Physiology. 1983;81:613–642. doi: 10.1085/jgp.81.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterworth JF, Strichartz GR. Molecular mechanisms of local anesthesia: a review. Anesthesiology. 1990;72:711–734. doi: 10.1097/00000542-199004000-00022. [DOI] [PubMed] [Google Scholar]
- Cannon SC. From mutation to myotonia in sodium channel disorders. Neuromuscular Disorders. 1997;7:241–249. doi: 10.1016/s0960-8966(97)00430-6. [DOI] [PubMed] [Google Scholar]
- Cannon SC. Ion channel defects in the hereditary myotonias and periodic paralyses. In: Martin JB, editor. Molecular Neurology. New York: Scientific American; 1998. pp. 257–277. [Google Scholar]
- Cannon SC, Brown RH, Jr, Corey DP. Theoretical reconstruction of myotonia and paralysis caused by incomplete inactivation of sodium channels. Biophysical Journal. 1993;65:270–288. doi: 10.1016/S0006-3495(93)81045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Zhou J, Sigworth FJ, Ukomadu C, Stephan M, Ptácek LJ, Agnew WS. Functional consequences of a Na+ channel mutation causing hyperkalemic periodic paralysis. Neuron. 1993;10:667–678. doi: 10.1016/0896-6273(93)90168-q. [DOI] [PubMed] [Google Scholar]
- De Luca A, Pierno S, Natuzzi F, Franchini C, Duranti A, Lentini G, Tortorella V, Jockusch H, Camerino DC. Evaluation of the antimyotonic activity of mexiletine and some new analogs on sodium currents of single muscle fibers and on the abnormal excitability of the myotonic ADR mouse. Journal of Pharmacology and Experimental Therapeutics. 1997;282:93–100. [PubMed] [Google Scholar]
- Fan Z, George AL, Jr, Kyle JW, Makielski JC. Two human paramyotonia congenita mutations have opposite effects on lidocaine block of Na+ channels expressed in a mammalian cell line. Journal of Physiology. 1996;496:275–286. doi: 10.1113/jphysiol.1996.sp021684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischhauer R, Mitrovic N, Deymeer F, Lehmann-Horn F, Lerche H. Effects of temperature and mexiletine on the F1473S Na+ channel mutation causing paramyotonia congenita. Pflügers Archiv. 1998;436:757–765. doi: 10.1007/s004240050699. [DOI] [PubMed] [Google Scholar]
- George AL, Jr, Komisarof J, Kallen RG, Barchi RL. Primary structure of the adult human skeletal muscle voltage-dependent sodium channel. Annals of Neurology. 1992;31:131–137. doi: 10.1002/ana.410310203. [DOI] [PubMed] [Google Scholar]
- Green DS, George AL, Jr, Cannon SC. Human sodium channel gating defects caused by missense mutations in S6 segments associated with myotonia: S804F and V1293I. Journal of Physiology. 1998;510:685–694. doi: 10.1111/j.1469-7793.1998.685bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward LJ, Brown RH, Jr, Cannon SC. Inactivation defects caused by myotonia-associated mutations in the sodium channel III-IV linker. Journal of General Physiology. 1996;107:559–576. doi: 10.1085/jgp.107.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward LJ, Brown RH, Jr, Cannon SC. Slow inactivation differs among mutant Na channels associated with myotonia and periodic paralysis. Biophysical Journal. 1997;72:1204–1219. doi: 10.1016/S0006-3495(97)78768-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward LJ, Sandoval GM, Cannon SC. Defective slow inactivation of sodium channels contributes to familial periodic paralysis. Neurology. 1999;52:1447–1453. doi: 10.1212/wnl.52.7.1447. [DOI] [PubMed] [Google Scholar]
- Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. Journal of General Physiology. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. USA: Sinauer Associates; 1992. [Google Scholar]
- Hudson AJ, Ebers GC, Bulman DE. The skeletal muscle sodium and chloride channel diseases. Brain. 1995;118:547–563. doi: 10.1093/brain/118.2.547. [DOI] [PubMed] [Google Scholar]
- Jackson CE, Barohn RJ, Ptácek LJ. Paramyotonia congenita: abnormal short exercise test, and improvement after mexiletine therapy. Muscle and Nerve. 1994;17:763–768. doi: 10.1002/mus.880170710. [DOI] [PubMed] [Google Scholar]
- Jurman ME, Boland LM, Liu Y, Yellen G. Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. Biotechniques. 1994;17:874–881. [PubMed] [Google Scholar]
- Lehmann-Horn F, Jurkat-Rott K. Voltage-gated ion channels and hereditary disease. Physiological Reviews. 1999;79:1317–1372. doi: 10.1152/physrev.1999.79.4.1317. [DOI] [PubMed] [Google Scholar]
- McClatchey AI, Cannon SC, Slaugenhaupt SA, Gusella JF. The cloning and expression of a sodium channel β1-subunit cDNA from human brain. Human Molecular Genetics. 1993;2:745–749. doi: 10.1093/hmg/2.6.745. [DOI] [PubMed] [Google Scholar]
- Mitrovic N, George AL, Jr, Heine R, Wagner S, Pika U, Hartlaub U, Zhou M, Lerche H, Fahlke C, LehmannHorn F. K+-aggravated myotonia: destabilization of the inactivated state of the human muscle Na+ channel by the V1589M mutation. Journal of Physiology. 1994;478:395–402. doi: 10.1113/jphysiol.1994.sp020260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovic N, George AL, Jr, Lerche H, Wagner S, Fahlke C, Lehmann-Horn F. Different effects on gating of three myotonia-causing mutations in the inactivation gate of the human muscle sodium channel. Journal of Physiology. 1995;487:107–114. doi: 10.1113/jphysiol.1995.sp020864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk JP, Brogden RN. Mexiletine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in the treatment of arrhythmias. Drugs. 1990;40:374–411. doi: 10.2165/00003495-199040030-00005. [DOI] [PubMed] [Google Scholar]
- Plassart-Schiess E, Lhuillier L, George AL, Jr, Fontaine B, Tabti N. Functional expression of the Ile693Thr Na+ channel mutation associated with paramyotonia congenita in a human cell line. Journal of Physiology. 1998;507:721–727. doi: 10.1111/j.1469-7793.1998.721bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- Richmond JE, Featherstone DE, Hartmann HA, Ruben PC. Slow inactivation in human cardiac sodium channels. Biophysical Journal. 1998;74:2945–2952. doi: 10.1016/S0006-3495(98)78001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld J, Sloan-Brown K, George AL. A novel muscle sodium channel mutation causes painful congenital myotonia. Annals of Neurology. 1997;42:811–814. doi: 10.1002/ana.410420520. [DOI] [PubMed] [Google Scholar]
- Rüdel R, Dengler R, Ricker K, Haass A, Emser W. Improved therapy of myotonia with the lidocaine derivative tocainide. Journal of Neurology. 1980;222:275–278. doi: 10.1007/BF00313157. [DOI] [PubMed] [Google Scholar]
- Sah RL, Tsushima RG, Backx PH. Effects of local anesthetics on Na+ channels containing the equine hyperkalemic periodic paralysis mutation. American Journal of Physiology. 1998;275:C389–400. doi: 10.1152/ajpcell.1998.275.2.C389. [DOI] [PubMed] [Google Scholar]
- Sunami A, Dudley SC, Jr, Fozzard HA. Sodium channel selectivity filter regulates antiarrhythmic drug binding. Proceedings of the National Academy of Sciences of the USA. 1997;94:14126–14131. doi: 10.1073/pnas.94.25.14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi MP, Cannon SC. Enhanced slow inactivation by V445M: a sodium channel mutation associated with myotonia. Biophysical Journal. 1999;76:861–868. doi: 10.1016/S0006-3495(99)77249-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudell RG, Kaiser KK, Griggs RC. Acetazolamide-responsive myotonia congenita. Neurology. 1987;37:488–491. doi: 10.1212/wnl.37.3.488. [DOI] [PubMed] [Google Scholar]
- Vedantham V, Cannon SC. The position of the fast-inactivation gate during lidocaine block of voltage-gated Na+ channels. Journal of General Physiology. 1999;113:7–16. doi: 10.1085/jgp.113.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GK, Quan C, Wang S. A common local anesthetic receptor for benzocaine and etidocaine in voltage-gated mu1 Na+ channels. Pflügers Archiv. 1997;435:293–302. doi: 10.1007/s004240050515. [DOI] [PubMed] [Google Scholar]
- Wang GK, Quan C, Wang SY. Local anesthetic block of batrachotoxin-resistant muscle Na+ channels. Molecular Pharmacology. 1998;54:389–396. doi: 10.1124/mol.54.2.389. [DOI] [PubMed] [Google Scholar]
- Wang SY, Nau C, Wang GK. Residues in Na+ channel D3-S6 segment modulate both batrachotoxin and local anesthetic affinities. Biophysical Journal. 2000;79:1379–1387. doi: 10.1016/S0006-3495(00)76390-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckbecker K, Würz A, Mohammadi B, Mansuroglu T, George AL, Jr, Lerche H, Dengler R, Lehmann-Horn F, Mitrovic N. Different effects of mexiletine on two mutant sodium channels causing paramyotonia congenita and hyperkalemic periodic paralysis. Neuromuscular Disorders. 2000;10:31–39. doi: 10.1016/s0960-8966(99)00060-7. [DOI] [PubMed] [Google Scholar]
- Wright SN, Wang SY, Kallen RG, Wang GK. Differences in steady-state inactivation between Na channel isoforms affect local anesthetic binding affinity. Biophysical Journal. 1997;73:779–788. doi: 10.1016/S0006-3495(97)78110-4. [DOI] [PMC free article] [PubMed] [Google Scholar]